0022-538X/10/$12.00 doi:10.1128/JVI.01506-10
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Physical Interaction between the Herpes Simplex Virus Type 1
Exonuclease, UL12, and the DNA Double-Strand
Break-Sensing MRN Complex
䌤
Nandakumar Balasubramanian, Ping Bai, Gregory Buchek, George Korza, and Sandra K. Weller*
Department of Molecular, Microbial and Structural Biology and The Molecular Biology and Biochemistry Graduate Program, The University of Connecticut Health Center, Farmington, Connecticut 06030
Received 19 July 2010/Accepted 5 October 2010
The herpes simplex virus type 1 (HSV-1) alkaline nuclease, encoded by the UL12 gene, plays an important role in HSV-1 replication, as a UL12 null mutant displays a severe growth defect. The HSV-1 alkaline exonuclease UL12 interacts with the viral single-stranded DNA binding protein ICP8 and promotes strand exchangein vitroin conjunction with ICP8. We proposed that UL12 and ICP8 form a two-subunit recombinase reminiscent of the phage lambda Red␣/recombination system and that the viral and cellular recombinases contribute to viral genome replication through a homologous recombination-dependent DNA replication mechanism. To test this hypothesis, we identified cellular interaction partners of UL12 by using coimmuno-precipitation. We report for the first time a specific interaction between UL12 and components of the cellular MRN complex, an important factor in the ATM-mediated homologous recombination repair (HRR) pathway. This interaction is detected early during infection and does not require viral DNA or other viral or cellular proteins. The region of UL12 responsible for the interaction has been mapped to the first 125 residues, and coimmunoprecipitation can be abolished by deletion of residues 100 to 126. These observations support the hypothesis that cellular and viral recombination factors work together to promote efficient HSV-1 growth.
The herpes simplex virus (HSV) genome replicates in the nucleus of an infected host cell, resulting in the production of longer-than-unit-length head-to-tail concatemers of viral DNA. Production of infectious virus requires the processing of concatemeric DNA into unit-length genomes by the packaging machinery followed by encapsidation into preassembled cap-sids (63). Several lines of evidence suggest that recombination plays a role in concatemer formation (69). Genomic inversions, proposed to occur through recombination, can be detected very early during infection, and these inversions require se-quence homology (2, 23, 56). In addition, replication interme-diates in HSV type 1 (HSV-1)-infected cells adopt a complex nonlinear structure that does not migrate in a pulsed-field gel, even after digestion with an enzyme that cuts once per unit length of the genome (1, 33, 51, 71). Electron micrographs have revealed that replication intermediates are branched and contain Y- and X-shaped junctions (19, 52). Furthermore, high levels of recombination have been reported not only between coinfecting viral strains (4, 17, 50, 62, 66) but also in plasmids containing repeated sequence elements (10, 11). These obser-vations, taken together, are not consistent with a simple rolling circle mechanism of replication and suggest that, reminiscent of the bacteriophages T4 and lambda, HSV-1 utilizes a recom-bination-dependent replication mechanism to generate con-catemeric viral DNA.
We have previously reported that virus-encoded proteins are capable of participating in recombination eventsin vitro. The
viral 5⬘–3⬘alkaline exonuclease (UL12) and the single-strand binding protein (ICP8) together mediate strand exchangein
vitro, and we have proposed that UL12 and ICP8 function as a
two-component recombinase similar to the well-studied phage lambda Red␣/ recombination system (48). ICP8 and UL12 also interactin vivoduring HSV-1 infection (61). The lambda exonuclease (Red ␣) and the single-strand binding protein (Red) mediate strand exchange and recombination (57). In the absence of host recombination proteins, Red␣/is capable of generating head-to-tail concatemers by strand annealing, and in the presence of host recombination factors, the viral recombinase participates in strand invasion reactions (44, 57). The phenotype of AN-1, a UL12 null virus, is complex (see Discussion for more detail), suggesting that UL12 plays a role either in the initiation of recombination or in processing of recombination intermediates (or both) (33, 43, 53, 64). By analogy with lambda, the viral recombinase may also function in conjunction with components of the cellular DNA damage response machinery responsible for the homologous recombi-nation repair (HRR) pathway.
During HSV-1 infection, the nucleus is dramatically reorga-nized, resulting in the formation of globular replication com-partments (RCs) in which gene expression, DNA replication, and encapsidation occur (7, 23, 45). Several cellular proteins known to participate in host damage responses are recruited to replication compartments (15, 30, 55, 60, 65, 68) and are re-quired for efficient virus production (29, 30, 37, 60). The MRN complex, consisting of Mre11, Rad50, and Nbs1, is particularly important in the normal cellular damage response, as it acts as a sensor of double-strand breaks (DSBs) and is recruited to the site of DNA damage, thereby signaling activation of at least one of the phosphatidylinositol 3-kinase (PI3 kinase)-like ki-nases (PIKKs), ATM (ataxia-telangiectasia mutated) (24, 30,
* Corresponding author. Mailing address: Department of Molecu-lar, Microbial and Structural Biology, The University of Connecticut Health Center, Farmington, CT 06030. Phone: (860) 679-2310. Fax: (860) 679-1239. E-mail: [email protected].
䌤Published ahead of print on 13 October 2010.
12504
on November 8, 2019 by guest
http://jvi.asm.org/
41, 42, 70). In 2004, we first demonstrated that HSV-1 infection results in the phosphorylation of Nbs1 (68), and other ATM targets were also identified, including Chk2, P53, 53BP1, and phospho-ATM (30, 55, 59). The activation of MRN and the ATM pathway in infected cells is consistent with a role for HRR during HSV-1 infection.
To test the hypothesis that viral and cellular proteins par-ticipate in recombination-dependent DNA synthesis in HSV-1-infected cells, we examined interactions between the viral recombinase and cellular HRR pathway proteins. It has been previously reported that the viral single-strand binding protein ICP8 interacts with various cellular DNA repair proteins, in-cluding Rad50, Mre11, DNA-PKcs, RPA, Ku86, BLM, BRG1, and WRN (60); however, the question of whether some of these interactions are direct or are mediated through other viral and cellular proteins has never been addressed. In this study we focus on identification of cellular proteins that inter-act with UL12, the nuclease component of the viral recombi-nase. We report a direct and specific interaction between UL12 and components of the MRN complex.
MATERIALS AND METHODS
Cell culture and viruses. African green monkey kidney (Vero) cells were purchased from the American Type Culture Collection and were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum. Cells were grown in the presence of 0.1 mg/ml of streptomycin and 100 U/ml of penicillin at 37°C and 5% CO2. The KOS strain of HSV-1 was used as the wild-type virus, and it produces both full-length UL12 (residues 1 to 626) and a subgenic protein, UL12.5 (residues 127 to 626). The UL12 null virus AN-1 does not express either UL12 or UL12.5, while the UL12 mutant virus ANF-1 produces only UL12.5. These were propagated in UL12-complementing 6-5 cells (53). ICP8 mutant virus HD-2 was provided by David Knipe (Harvard Medical School) (12). Baculoviruses expressing Mre11, His-Rad50, and glutathioneS-transferase (GST)–Nbs1 were provided by Tanya Paull (University of Texas at Austin) (27).
Antibodies. Anti-Rad50 (13B3) and Mre11 (12D7) monoclonal anti-bodies were purchased from Genetex. Anti-Nbs1 monoclonal antibody (1C3) was purchased from Novus Biologicals. Antitubulin antibody (T6557) was pur-chased from Sigma. Anti-UL12 antibody (BWpUL12), which detects both UL12 and UL12.5, was a gift from Joel Bronstein and Peter Weber (Parke-Davis Pharmaceutical Research). Anti-ICP8 monoclonal antibody (39-S) was provided by David Knipe (Harvard Medical School), and the polyclonal antibody (367) was provided by William Ruyechan (The State University of New York) (54).
Plasmids. pSAKUL12/12.5 expresses both full-length UL12 and UL12.5, pSAKUL12.5 expresses only UL12.5, the pSAK N terminal expresses only the first 126 amino acids of UL12, and pSAK was used as the empty vector (47). The N-terminal truncations of UL12 were a gift from James Smiley (6). The
C-terminal truncations of UL12 were constructed by cloning PCR-amplified frag-ments of UL12 using pSAKUL12/12.5 as the template into EcoRI- and BamHI-double-digested pSAK vector to derive pBN104, pBN271, pBN390, pBN421, and pBN490. Table 1 lists the oligonucleotides used in the construction of the various constructs in this report. FP1 was used as the forward primer along with the reverse primers RP104, RP271, RP330, RP421, and RP490 to PCR amplify the corresponding UL12 fragments. To construct the internal deletions of UL12, the fragments corresponding to residues 127 to 626 of UL12 were PCR amplified using FP2 as the forward primer and RP2 as the reverse primer. The PCR product was digested with EcoRI and BamHI and cloned into pSAK to derive pBNUL12.5. The fragments corresponding to the first 50, 75, and 100 residues of UL12 were PCR amplified using the forward primer FP3 and the respective reverse primers RP50, RP75, and RP100. The PCR fragments were digested with HindIII and EcoRI and cloned into pBNUL12.5 to yield pBN(⌬50–125)UL12, pBN(⌬75–125)UL12, and pBN(⌬100–125) UL12. This method generated the desired truncations and also introduced an EcoRI restriction site to GAATTC between the two fragments, resulting in an in-frame addition of amino acids E and F to the sequence between the fragments. The UL12.5 fragment with a nuclear localization signal (NLS) was constructed by PCR amplification of pBNUL12.5 using the forward primer FP4, which encodes a simian virus 40 (SV40) large T antigen NLS, PKKKRKV (20), after the start site of UL12.5 and the reverse primer RP2. The PCR-amplified constructs were double digested with EcoRI and BamHI and cloned into pSAK, creating pBN-NLS-UL12.5.
Transfection and infection.Vero cells at 70 to 80% confluence plated the previous day were transfected with plasmids expressing the indicated proteins and using Lipofectamine Plus reagent (Invitrogen) according to the manufac-turer’s instructions. For immunoprecipitation (IP), cells in 100-mm2
dishes were transfected with 4g of DNA, and for immunofluorescence cells in 12-well dishes were transfected with 0.5g DNA. At 20 h posttransfection the cell monolayer was washed three times in phosphate-buffered saline (PBS) with 1 mM MgCl2and 0.1 mM CaCl2and processed either for immunofluorescence or immunoprecipitation as described below. Infections and viral growth analyses were carried out as described in reference 53 with minor modifications.
[image:2.585.45.546.77.216.2]Immunofluorescence.Infected or transfected cells grown on glass coverslips were washed three times in PBS and fixed in 4% paraformaldehyde in PBS for 10 min. Fixed cells were washed three times in PBS and were permeabilized for 10 min in 1% Triton X-100 in PBS. Fixed and permeabilized cells were washed three times in PBS and blocked overnight at 4°C using 3% normal goat serum (NGS) in PBS. Soluble proteins were removed with cold Cytoskeleton (CSK) buffer (10 mM HEPES-KOH [pH 7.4], 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and protease inhibitors) containing 0.5% Triton X-100 as described in reference 9. Preextracted cells were washed with PBS and then fixed with 4% paraformaldehyde. The fixed cells were processed for immunofluorescence as described in reference 32 and using the primary antibodies (anti-ICP8 antibody 39-S, 1:300; anti-UL12 antibody BWpUL12, 1:300; anti-Rad50 antibody 13B3, 1:100) diluted in 3% NGS for a period from 2 h to overnight. AlexaFluor anti-mouse and anti-rabbit antibodies (Molecular Probes) conjugated to fluoro-phores that were excited at wavelengths of 488 and 647 nm and diluted in 3% NGS were used as the secondary antibodies. TO-PRO-3 (Molecular Probes) was used as a nuclear marker at a 1:500 dilution, along with the secondary antibody mix. Mounting of the coverslips, imaging on a Zeiss LSM 510 Meta confocal
TABLE 1. Sequences of oligonucleotides used in the construction of the various truncation mutants of UL12
Oligonucleotide
name Sequence (5⬘–3⬘)
a
FP1 ...AAGCTTCGAATTCCGCCACCATGGAGT
RP104...GCAGATGGATCCTCACGGAATGTCCGGGGTCGGAGGC RP271...GCAGATGGATCCTCACGAAGACTGTACATCCGGCCG RP330...GCAGATGGATCCTCAACCGTCCATGAGGACCCCACA RP421...GCAGATGGATCCTCACGGGCCGGGGACGCGCCCGGGCG RP490...GCAGATGGATCCTCAGGGCTCGCGGCGGACCAGATCC FP2 ...TAAGAATTCATGTGGTCGGCGTCGGTGATCCCCAACGCGCT RP2...ATTGGATCCTCAGCGAGACGACCTCCCCGT
FP3 ...ATAAGCTTATGGAGTCCACGGGAGGCCCAGCA RP50...ATTGAATTCGGGACGGAAGGTGGTTGTCAGCGG RP75...ATTGAATTCCTGATCACGTGGGGGGTTAACGGG RP100...ATTGAATTCGGTCGGAGGCCCGGAGGCGTCAGA
a
The restriction sites in the primer sequences are indicated in bold.
on November 8, 2019 by guest
http://jvi.asm.org/
microscope, and processing of images were performed as described in refer-ence 32.
Immunoprecipitation.Infected or transfected cells were washed twice with cold PBS at the indicated times. The cells were scraped into cold PBS containing protease inhibitors and transferred into 15-ml tubes. Scraped cells were pelleted for 10 min at 1,500 rpm at 4°C and frozen at⫺80°C. Frozen cells were thawed on ice and resuspended in 450l of the IP buffer (1⫻PBS with 1% NP-40, 0.5% sodium deoxycholate [SOC], and 0.1% SDS) with protease inhibitors and trans-ferred to 1.5-ml tubes. The cells were further disrupted using a 1-ml syringe with a 25-gauge 5/8 needle and incubated on ice for 1.5 h for complete lysis. Cell extracts were clarified by centrifugation at 10,000 rpm for 10 min at 4°C, and the supernatant was collected as the cell lysate. The following steps were carried out at 4°C with constant agitation in the presence of protease inhibitors. Cell lysates were precleared with 5l normal rabbit IgG (Santa Cruz Biotechnology) for 1.5 h, followed by the addition of 40l protein A-agarose beads (Santa Cruz Biotechnology) for 1.5 h. Precleared lysates were collected after centrifugation at 2,500 rpm for 10 min. A small portion of the precleared lysates (10l) was mixed with SDS sample loading buffer and saved as the input sample. The remaining precleared lysate was immunoprecipitated with 5 l of UL12 antibody (BWpUL12) overnight followed by the addition of 40l protein A-agarose beads. After 24 h beads were collected by centrifugation at 2,500 rpm for 10 min and washed in PBS with protease inhibitors. For samples that were subjected to DNase treatment, the beads were incubated in a 50l buffer containing 5U of DNase (Invitrogen) and 10 mM MgCl2in PBS with protease inhibitors for 15 min at room temperature. The DNase activity was tested on an identical preparation to that used in the IP experiment to verify that the DNase was active (data not shown). The beads were washed five times with 500l IP buffer for 15 min followed by centrifugation for 8 min at 2,500 rpm. After washing, the beads were resuspended in 40l SDS sample buffer and stored at⫺20°C until SDS-PAGE separation and immunoblotting.
Immunoblotting.Cell monolayers washed in PBS were lysed in buffer con-taining 0.5% (wt/vol) SDS and 0.05 M Tris-Cl, pH 8.0. Lysates were freeze-thawed once and sonicated, and the cell debris was removed by centrifugation before adding SDS sample buffer and processing for immunoblotting. Samples boiled in SDS sample buffer were separated on a 4-to-20% SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane. Membranes were cut into horizontal strips according to the molecular weight of the protein of interest before probing with primary antibody. The membranes were immunoblotted and developed as described in reference 46. The membranes were probed with primary antibodies (anti-ICP8 antibody [367] at 1:1,000; anti-UL12 antibody [BWpUL12] at 1:5,000; antitubulin antibody [BWpUL12] at 1:5,000; anti-Mre11 antibody [12D7] at 1:500; anti-Rad50 [13B3]; anti-Nbs1 at 1:800) diluted in Tris-buffered saline with Tween 20 (TBST). Appropriate horseradish peroxidase-conjugated secondary antibodies were used, and Super Signal West Pico chemi-luminescence (Pierce) was used to detect the antigen-antibody complexes. For probing proteins of similar molecular weights, membranes were either stripped (25 mM glycine, 1% SDS with HCl; pH 2.0) or quenched (10 mM sodium azide in TBST) prior to probing with a different antibody.
Protein purification.UL12 was expressed in insect cells infected with recom-binant baculovirus and purified to over 90% purity as described in reference 47. The MRN complex was expressed in insect cells infected with recombinant baculoviruses encoding His-Mre11, GST-Nbs1, or His-Rad50 and purified as described in reference 27. The MRN protein complex purified by nickel chro-matography was subjected to SDS-PAGE. All three subunits were detected by Coomassie staining (data not shown).
Surface plasmon resonance (SPR).Protein-protein interactions were moni-tored in real time using the Biacore T100 optical biosensor (GE Healthcare). Purified UL12 was covalently immobilized on a CM5 sensor chip. Protein ana-lytes in HSB buffer (10 mM HEPES [pH 7.4] containing 3 mM EDTA, 0.15 M NaCl, and 0.05% surfactant P20) were injected over the sensor surface at a flow rate of 30l/min for 180 s followed by HSB buffer without analyte. The surface was regenerated between injections by using 10 mM NaOH at a flow rate of 50 l/min for 30 s. Purified bovine serum albumin (BSA; Sigma) was used as a negative control. Sensorgrams were analyzed using the Biacore T100 evaluation software, and the data were fitted onto the 1:1 Langmuir binding model.
RESULTS
A subpopulation of UL12 is detergent resistant and colocal-izes with ICP8 in viral replication compartments.UL12 and ICP8 have been shown to function as a two-subunit
recombi-nasein vitro(48).An interaction between ICP8 and UL12 was
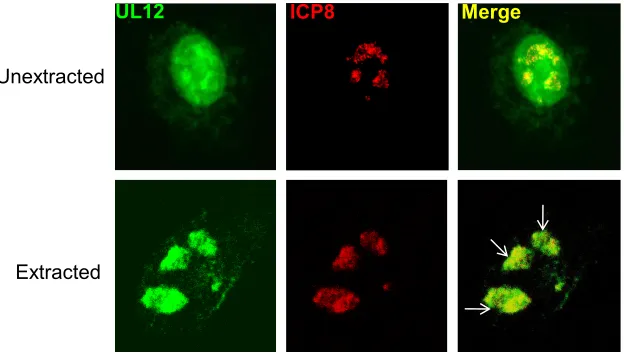
first reported in 1992 (61) and confirmed by coimmunoprecipi-tation in lysates of insect cells coinfected with baculoviruses expressing ICP8 and UL12 (47). It has long been recognized that ICP8 localizes to replication compartments in infected cells; however, initial immunofluorescence (IF) studies indi-cated that UL12 localizes diffusely in the infected cell nucleus (47, 61). In these experiments, the diffuse staining made it difficult to determine whether some UL12 colocalized with ICP8. In order to more easily visualize UL12 in replication compartments, we preextracted infected cells with detergent prior to fixation, as described previously (9, 67). Detergent extraction has been reported to remove soluble proteins and proteins that are tethered by protein-protein interactions while leaving insoluble any chromatin-associated or matrix-associ-ated proteins (9, 67). It has been reported that UL12 binds to viral DNA (5), and we hypothesized that a subpopulation of UL12 would be resistant to detergent extraction. In the top panel of Fig. 1 (unextracted), infected cells were fixed and permeabilized, and we observed that UL12 localized diffusely throughout the nucleus, although some staining could be de-tected in RCs stained with ICP8 antibodies. The bottom panel shows infected cells that were preextracted with a cytoskeletal buffer prior to fixation, and we observed that most of the pan-nuclear UL12 staining was removed, leaving behind a sub-population of UL12 that localized in replication compartments stained with ICP8 antibodies.
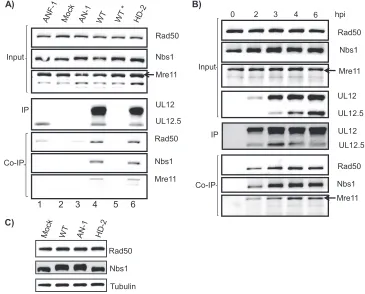
The HSV-1 alkaline nuclease UL12 interacts with compo-nents of the cellular MRN complex during infection.Several HRR proteins, including Rad51, RPA, Ku86, ATM, and MRN, have been reported to localize to RCs (15, 60, 65, 68); however, their precise roles in the virus life cycle have not been defined, nor is it clear how they are recruited. To address these questions, we sought to identify cellular interaction partners of UL12. UL12 was immunoprecipitated from mock-infected and wild-type- and mutant HSV-1-infected Vero cells, and copre-cipitating proteins were analyzed by immunoblotting for DNA repair and recombination factors. Figure 2A (input panel) demonstrates that approximately equal levels of cellular pro-teins were present in all the infected cell lysates. In lane 4, coimmunoprecipitation with anti-UL12 antibody shows that MRN components Mre11, Rad50, and Nbs1 associate with UL12 in KOS-infected cell extracts. Immunoblots were incu-bated with antisera specific for other cellular factors involved in HRR, such as Brca2, Rad51, ATM, and RPA32; however, these proteins did not appear to coimmunoprecipitate with UL12 under these conditions (data not shown) and are not considered further in this paper.
ICP8 has been reported to interact with Rad50 and Mre11 during HSV-1 infection (60). In order to determine whether the interaction between the MRN components and UL12 seen in Fig. 2 reflected a direct association with UL12 or were mediated by ICP8, cells were infected with the ICP8 null virus (HD-2). Figure 2A, lane 6, shows that Mre11, Rad50, and Nbs1 still precipitated with UL12 even in lysates from cells infected with the ICP8 null virus (HD-2), indicating that the interaction between UL12 and MRN is independent of ICP8. MRN components were not precipitated from cell lysates of mock-infected or UL12 null virus (AN-1)-infected control samples (Fig. 2A, lanes 2 and 3). Furthermore, lysates from cells infected with the wild-type virus incubated with a
on November 8, 2019 by guest
http://jvi.asm.org/
cific rabbit IgG antibody were used to rule out nonspecific precipitation of the MRN components (Fig. 2A, lane 5). Under these conditions a small amount of Rad50 was detected in all samples, indicating some nonspecific background precipita-tion. However, the amount of Rad50 coimmunoprecipitated in lanes 4 and 6, where UL12 was present, was significantly higher, suggesting that Rad50 is specifically precipitated when antibody to UL12 is used. Figure 2B shows that MRN compo-nents coimmunoprecipitated with UL12 as early as 2 h postin-fection, as soon as UL12 was detected, and this was maintained as late as 10 h postinfection (hpi) (data not shown).
The UL12 gene is encoded by a 2.3-kb mRNA, and embed-ded within this RNA is a subgenic 1.9-kb mRNA encoding an N-terminally truncated version of UL12, designated UL12.5 (34). In order to map the MRN interaction domain within UL12, we first asked whether MRN proteins could be precip-itated by full-length UL12 and UL12.5. We previously re-ported the isolation of a mutant virus, ANF-1, which produces only UL12.5 (34). Immunoprecipitation of lysates from ANF-1-infected cells indicated that UL12.5 alone does not appear to interact with MRN (Fig. 2A, lane 1). It is possible that UL12.5 does not interact with MRN because of its cytoplasmic local-ization; however, experiments presented below indicate that UL12.5 that has been directed to the nucleus is also unable to interact with MRN components.
HSV-1 infection results in the activation of the ATM kinase and its downstream targets, such as Nbs1 and Chk2 (30, 55, 68). In Fig. 2A and 3A, Nbs1 mobility is shifted (indicating phosphorylation) only in cells infected with wild-type and UL12 mutant viruses but not in mock- or HD-2-infected cells. The Nbs1 mobility shift in cells infected with wild-type or UL12 mutant virus can be more clearly seen in Fig. 2C, for which cell lysates were resolved for a longer time by SDS-PAGE. These results are consistent with the observation that Nbs1 is not phosphorylated in cells infected with viruses that fail to synthesize viral DNA, such as the viral polymerase null
virus (HP66) (15, 68). Viral DNA synthesis would be expected in cells infected with wild-type and UL12 mutant viruses but not in mock- or HD-2-infected cells.
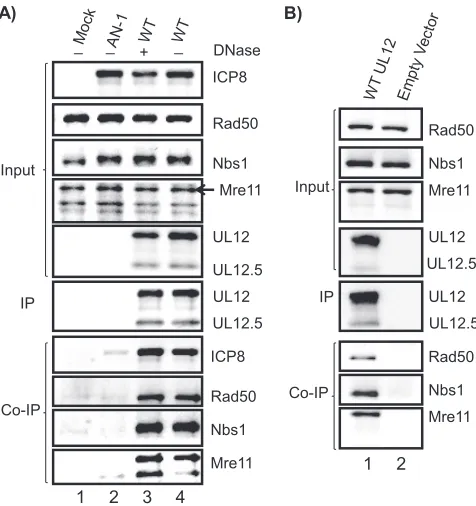
The UL12- MRN complex interaction is independent of DNA and does not require other viral proteins.Both UL12 and the MRN complex are known to interact with DNA, and we next asked whether the observed interaction is mediated through protein-protein interactions or indirectly through DNA. In Fig. 3A, UL12 was precipitated from KOS-infected cell lysates as described above except that the sample shown in lane 3 was subjected to DNase treatment as described in Ma-terials and Methods. Beads containing the immunoprecipitates were incubated in the presence of DNase prior to the final washes. If MRN components were precipitated with the UL12 antibody by virtue of their ability to interact with DNA, they would be removed during the washing steps. The results shown in Fig. 3A confirm that MRN proteins did not precipitate in the absence of UL12, as MRN components were not present in immunoprecipitates from mock-infected (lane 1) or AN-1-in-fected (lane 2) cell lysates. Figure 3A, lane 3, demonstrates that the interaction between UL12 and components of the MRN complex were not affected by DNase treatment, indicat-ing that this interaction is not mediated by DNA. UL12 has also been reported to interact with ICP8 (47, 60, 61); however, the question of whether this interaction is dependent on DNA has never been addressed. Figure 3A, lane 3, demonstrates that UL12 interacts with ICP8 in a DNA-independent fashion.
[image:4.585.137.449.69.246.2]Next we asked whether ICP8 or any other viral protein is required for UL12 to interact with the components of the MRN complex. Vero cells were transfected with plasmids that express full-length UL12 and UL12.5 (pSAK UL12/UL12.5). Transfected cell lysates were immunoprecipitated with anti-UL12 antibody. Figure 3B (lane 1) demonstrates that expres-sion of UL12 and UL12.5 is sufficient to coimmunoprecipitate the components of the MRN complex. MRN proteins were not observed in the precipitates from cells transfected with an
FIG. 1. A subpopulation of UL12 is detergent resistant and localizes to replication compartments. (Top panels) KOS-infected Vero cells were fixed and permeabilized as described in Materials and Methods. (Bottom panels) KOS-infected Vero cells were extracted with CSK buffer containing Triton X-100 prior to fixation to remove nucleosolic and cytosolic fractions, followed by fixation and permeabilization. The cells were labeled with the polyclonal anti-UL12 antibody BWpUL12 (green), and the monoclonal anti-ICP8 antibody 39S (red) was used as the marker for viral replication compartments. Merged pixels are shown in yellow. In this experiment the majority of cells (34 out of 38) showed that UL12 localized in replication compartments in preextracted cells. Arrows indicate replication compartments within a single nucleus.
on November 8, 2019 by guest
http://jvi.asm.org/
empty vector (Fig. 3B, lane2). Taken together, these observa-tions indicate that the interaction between UL12 and MRN is not mediated by other viral proteins or DNA.
UL12 directly interacts with the MRN complexin vitro.The experiments described above indicate that the UL12-MRN interaction is independent of DNA and viral proteins, but the experiments did not address whether the interaction could be mediated through other cellular proteins. We next asked if this interaction could be demonstratedin vitrowith purified recom-binant proteins in SPR assays. In Fig. 4, purified UL12 was immobilized on a biosensor chip and protein analytes of known concentrations were injected over the sensor as described in Materials and Methods. The binding of MRN at multiple con-centrations (6, 12.5, 25, 50, and 100 nM) to immobilized UL12 is shown. Purified BSA at 100 nM did not bind to the MRN complex (data not shown), suggesting that the MRN-UL12 interaction is specific. These observations show that UL12 can bind directly to the MRN components even in the absence of other viral and cellular factors. The kinetics of the MRN-UL12 interaction at various concentrations was analyzed, and the rate of association (Ka,4.266E⫹4 per ms) and the rate of
dis-sociation (Kd, 0.001323 per s) was determined. Based on these values the apparent equilibrium dissociation constant (KD) for the UL12-MRN interaction was determined to be 31.1 nM, which represents a fairly high-affinity interaction. To put this number in perspective, it is similar to other reported interac-tions between cellular DNA repair factors MutS and the WRN complex (8.8 nM) and between MutS␣and the WRN complex (35 nM) (49). InSaccharomyces cerevisiae, the inter-action between MutS␣and the yeast ortholog of WRN, Sgs1, is required for Sgs1 function during DNA repair in vivo (13, 58).
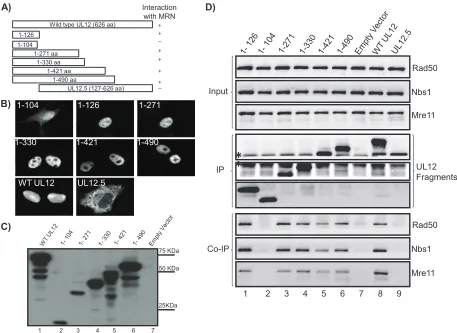
[image:5.585.108.473.70.364.2]Residues in the N terminus of UL12 are responsible for the interaction with the MRN complex.In order to more finely map the region of UL12 responsible for the MRN interaction, cells were transfected with a series of C-terminal truncation mutants of UL12, generated as described in Materials and Methods. To minimize problems associated with expression of globally misfolded proteins from truncation mutants, the end points of the truncations were selected based on secondary structure predictions and were chosen at positions predicted to be either unstructured or in loop regions between domains
FIG. 2. UL12 interacts with components of the cellular MRN complex in HSV-1-infected cell lysates. (A) Vero cells were either mock infected (lane 2) or infected with UL12.5 virus ANF-1 (lane 1), UL12 null virus AN-1 (lane 3), wild-type (WT) KOS virus (lanes 4 and 5), or ICP8 null virus HD-2 (lane 6). Anti-UL12 antibody was used to immunoprecipitate cell lysates of infected cells collected 6 hpi as described in Materials and Methods, except for lane 5, in which normal rabbit IgG was used as a control (indicated by the*). Input samples contained 3% of the precleared cell lysate collected prior to immunoprecipitation. UL12 fragments that were immunoprecipitated with anti-UL12 antibody are shown in the panel labeled IP. Proteins that coimmunoprecipitated with UL12 are shown in the panels labeled Co-IP. The arrow indicates the position of the band corresponding to full-length Mre11. (B) Vero cells were infected with KOS and collected at the indicated time points postinfection. Immuno-precipitation was performed, and results are displayed as described for panel A. (C) Vero cells were mock infected (lane 1) or infected with WT KOS virus (lane 2), UL12 null virus AN-1 (lane 3), or ICP8 null virus HD-2 (lane 4). Infected cells were collected 6 hpi, and cell lysates were prepared for immunoblotting as described in Materials and Methods. For the experiment shown in this panel, the proteins were resolved for a longer period of time to highlight the mobility shift of Nbs1 upon HSV-1 infection. Tubulin was used as the loading control.
on November 8, 2019 by guest
http://jvi.asm.org/
predicted to have structure. Figure 5A shows a diagram of the full-length UL12, UL12.5, and the series of truncations used in this experiment. In Fig. 5B, the truncations were tested for their expression and localization by IF. The fragment 1-104 was localized primarily to the nucleus with some diffuse cyto-plasmic localization. All other C-terminal truncations, includ-ing 1-126, showed predominantly nuclear localization, confirm-ing the presence of an NLS within the first 126 residues of UL12 (47).
In Fig. 5C, the immunoblot results with whole-cell lysates of Vero cells transfected with the various C-terminal truncations created in this study indicate that UL12 fragments of the ex-pected size were synthesized from the respective truncation plasmids. The shortest fragment of 1-104 was detected around the expected size of 11.5 kDa (lane 2), and the full-length UL12 was detected around 75 kDa (lane 1). The other C-terminal truncations also produced fragments of expected sizes (lanes 3 to 6). Vero cells were transfected with the various constructs described above, and lysates were immunoprecipi-tated using anti-UL12 antibody and probed with antibodies for each of the components of the MRN complex. The input panel shows the presence of equal levels of the cellular MRN com-ponents in all lysates. Figure 5D confirms that proteins of the expected sizes were expressed by each of the truncations. A
band of around 50 kDa was detected in all the lanes, likely due to nonspecific cross-reactions between IgG and the secondary antibody. The three bottom panels of Fig. 5D demonstrate that all constructs, with the exception of fragment 1-104, UL12.5, and the empty vector (lanes 2, 7, and 9) produced a fragment that could interact with components of the MRN complex. The observation that fragment 1-126 and larger fragments, but not fragment 1-104, interact with MRN components indicates that the interaction domain resides within the first 125 amino acids of UL12.
To map the interaction more finely, we used a series of N-terminal truncations provided by James Smiley (Fig. 6A) (6). In Fig. 6B, cell lysates of Vero cells transfected with the full-length and mutant constructs were precipitated using the anti-UL12 antibody. The input panel shows the presence of equal levels of the cellular MRN components in all lanes. The IP panel shows that the truncation mutants expressed proteins of the expected sizes (Fig. 6B, lanes 1 to 6) (6). As seen in Fig. 5D, a nonspecific band resulting from cross-reaction of the secondary antibody with the IgG was observed in all lanes. MRN components were not precipitated from lysates of cells transfected with the empty vector or plasmids expressing UL12.5,⌬100,⌬125, or M185 (Fig. 6B, lanes 3 to 6 and 8). On the other hand, full-length UL12 or fragments lacking the first 25 or first 50 residues of UL12 were able to precipitate similar levels of MRN (Fig. 6B, lanes 1, 2, and 7), indicating that the first 50 residues of UL12 are dispensable for the MRN inter-action. The lack of apparent interaction between MRN and UL12.5 (lane 8) could be due to the fact that UL12.5 resides primarily in the cytoplasm and therefore may not be in direct contact with the nuclear MRN complex (47). A UL12.5 frag-ment fused with the SV40 NLS still failed to interact with the MRN complex (Fig. 7C), confirming that specific residues re-quired for interaction reside within the first 126 residues of UL12.
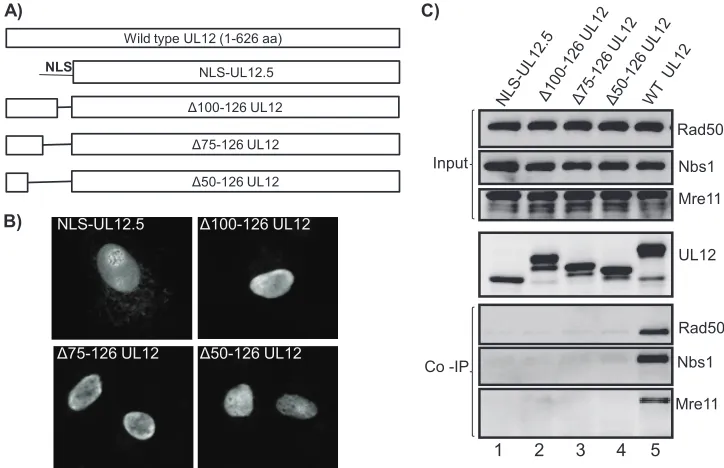
[image:6.585.44.282.67.320.2]The UL12-MRN interaction domain thus appears to reside between residues 50 and 125 of UL12. To more finely map the required residues, three UL12 constructs with internal dele-tions in the region between residues 50 and 125 (Fig. 7A) were constructed, and they all localized efficiently to the nucleus (Fig. 7B), indicating that residues 1 to 50 of UL12 are sufficient
[image:6.585.300.542.68.185.2]FIG. 4. UL12 directly binds to the MRN complex. SPR sensor-grams of the UL12 interaction with the MRN complex at increasing concentrations of 6.25, 12.5, 25, 50, and 100 nM were performed using a Biacore T100 at room temperature. The black lines represent the binding curves of the analytes at specific concentrations, and the gray lines represent the 1:1 binding model curves which were used to fit the data and calculate the kinetics of the interaction.
FIG. 3. The UL12-MRN interaction does not require DNA or other viral proteins. (A) Vero cells mock infected or infected with UL12 null virus AN-1 or the wild-type (WT) KOS virus, as indicated, were collected 6 hpi. Cell lysates were immunoprecipitated with anti-UL12 antibody. In lane 3 (⫹), the sample was subjected to DNase treatment during immunoprecipitation, while in lanes 1, 2, and 4 (⫺), immunoprecipitation was carried out in the absence of DNase. (B) Vero cells were transfected with either pSAK UL12/12.5 express-ing the wild-type UL12 under the cytomegalovirus promoter (lane1) or with pSAK empty vector (lane 2). Cells were collected 20 h posttrans-fection, and cell lysates were immunoprecipitated using anti-UL12 antibody and subjected to DNase treatment as described in Materials and Methods.
on November 8, 2019 by guest
http://jvi.asm.org/
for nuclear localization. Vero cell lysates transfected with the indicated constructs were immunoprecipitated using anti-UL12 antibody, and the precipitates were immunoblotted with antibodies to each of the MRN components. The input panels demonstrate that equal amounts of the cellular MRN compo-nents were present in all lanes. The IP panel shows that equal levels of UL12 or the various fragments of UL12 were precip-itated, and the bottom three panels indicate that the MRN complex can only be precipitated from cells transfected with full-length UL12 (lane 5). These results indicate that the re-gion between residues 50 and 126 of UL12 contains the inter-action domain for MRN, and even a relatively small deletion of 25 residues between residues 100 and 126 is sufficient to dis-rupt this interaction.
DISCUSSION
In this paper we report a direct interaction between UL12 and the components of the MRN complex as early as 2 hpi, and
we have identified the region between residues 50 and 125 of UL12 to be necessary for this interaction. This observation suggests that viral and cellular recombination factors may act together during HSV-1 replication. Furthermore, we show that this interaction is not dependent on the presence of ICP8 or any other viral or cellular proteins, nor is it mediated by DNA, suggesting that this is a direct protein-protein interaction. Us-ing SPR analysis we determined the apparent equilibrium dis-sociation constant (KD) to be 31.1 nM.
[image:7.585.63.520.67.400.2]Potential role of UL12 in the HSV-1 life cycle.The precise role of UL12 in viral infection is not known, and the analysis of UL12 mutant viruses has revealed a complex phenotype. UL12 null virus (AN-1) yield is decreased significantly (100- to 1,000-fold) in Vero cells, but viral DNA synthesis is only moderately affected (64). In AN-1-infected cells, some DNA is encapsi-dated; however, these capsids show a tendency to disgorge their contents, forming “A” or empty capsids, and are defective in nuclear egress (33, 53). DNA packaged in cells infected with UL12 mutant viruses is not infectious (43). One model that is
FIG. 5. UL12 interacts with components of the MRN complex through the N-terminal region of UL12. (A) Schematic representation of the UL12 constructs used in panels B and C. (B) Vero cells were transfected with plasmids expressing C-terminal truncations of UL12, wild-type (WT) UL12, and UL12.5 as indicated. Transfected cells were fixed 20 h posttransfection and tested for expression and localization of the UL12 fragments by immunofluorescence. (C) Whole-cell lysates of Vero cells transfected with full-length UL12 (lane 1), C-terminal truncations (lanes 2 to 6), or empty vector (lane 7) were immunoblotted and probed with UL12 antibody. Full-length UL12 and truncations were able to express UL12 fragments of the expected sizes. (D) Vero cells transfected with plasmids expressing C-terminal truncations of UL12 as indicated (lanes 1 to 6), empty vector (lane 7), wild-type UL12 (lane 8), or UL12.5 (lane 9) were collected at 20 h posttransfection. Cell lysates were immunoprecipitated using anti-UL12 antibody and probed for the indicated proteins to detect interactions. The immunoprecipitation reaction was subjected to DNase treatment as described in Materials and Methods. The asterisks mark nonspecific bands arising due to the cross-reaction between the secondary antibody and IgG.
on November 8, 2019 by guest
http://jvi.asm.org/
consistent with this complex phenotype is that UL12 is essen-tial for the production of viral DNA that can be packaged productively into virions. Thus, accumulation of fragile genomes and capsids that fail to leave the nucleus in AN-1-infected cells
[image:8.585.92.500.73.308.2]could result from improperly initiated or improperly processed viral replication products (14, 33, 53). Interestingly, baculovirus encodes a related exonuclease, and null mutants in this gene display similar defects to those of AN-1 in DNA processing,
FIG. 6. The N-terminal 50 amino acids of UL12 are not required for the UL12-MRN interaction. (A) Schematic representation of the N-terminal deletion constructs used for the experiment shown in panel B. (B) Vero cells were transfected with plasmids expressing N-terminal truncations of UL12 as indicated (lanes 1 to 5), empty vector (lane 6), full-length UL12 (lane 7), or UL12.5 (lane 8). Cells were collected at 20 h posttransfection, and cell lysates were immunoprecipitated using anti-UL12 antibody as described in the legend for Fig. 5. The asterisk marks a nonspecific band arising due to the cross-reaction between the secondary antibody and IgG.
FIG. 7. The N-terminal amino acids 100 to 126 of UL12 are essential for the UL12 interaction with the components of the MRN complex. (A) Schematic representation of UL12 constructs used in the experiments shown in panels B and C. (B) Vero cells were transfected with plasmids expressing the N-terminal internal deletions of UL12 as indicated. Cells were fixed at 20 h posttransfection and tested for UL12 expression and localization by immunofluorescence. (C) Vero cells were transfected with plasmids expressing the indicated constructs described for B (lanes 1 to 4) or full-length UL12 (lane 5). Cells were collected at 20 h posttransfection and immunoprecipitated as described in the legend for Fig. 5.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.110.472.443.677.2]resulting in aberrant nuclear egress and encapsidation (39). Thus, the role of viral exonucleases in DNA replication may be evolu-tionarily conserved among different dsDNA viruses. In addition, UL12 has been reported to play a role in resolving adeno-asso-ciated virus replication intermediates generated when HSV-1 is used as a helper virus (38).
Viral and cellular recombination factors may collaborate to mediate viral recombination-dependent replication.The over-all mechanism of HSV-1 DNA replication and the formation of larger-than-unit-length concatemers are poorly understood. Although it was previously proposed that linear virion DNA circularizes in infected cells and that concatemers are gener-ated by rolling circle replication (3, 18), several lines of evi-dence suggest that the fate of the viral genome in the infected cell and the mechanism of DNA replication is more complex than predicted by this model. It has been recognized for some time that the packaged HSV-1 genome contains nicks and gaps (69). If these nicks and gaps are left unrepaired, the passing of a replication fork would be expected to produce a DSB. Since DSBs are known to be highly recombinogenic in all other systems studied (21), it is possible that DSBs generated during HSV-1 replication would stimulate recombination.
The two major kinases that sense DSBs are ATM and the DNA-protein kinase catalytic subunit (DNA-PKcs), stimulat-ing the HRR and nonhomologous end-joinstimulat-ing (NHEJ) path-ways, respectively. DSBs are sensed by the MRN complex to signal ATM activation or by Ku70/Ku86 to activate DNA-PKcs. As mentioned above, ATM targets are phosphorylated in infected cells (30, 55, 55, 68), and virus yields are decreased in cells deficient in ATM, WRN, Chk2, Mre11, and Rad51 (29, 30, 37, 60), suggesting that these proteins may play a positive role in HSV infection. On the other hand, HSV-1 infection results in the degradation of DNA-PKcs in many cell types (28, 40), and viral yields increase severalfold in cells deficient for DNA-PKcs and Ku70 (40, 60), indicating that at least some components of NHEJ may be inhibitory for viral growth. In-terestingly, cellular ubiquitin ligases RNF8 and RNF168 (com-ponents of the ATM pathway) are degraded during infection (31). Thus, HSV has apparently evolved a very complex inter-action with the host DNA damage response pathways, activat-ing some components and inactivatactivat-ing others.
The MRN complex, acts as a sensor of DSBs by binding to the sites of DNA damage and acting to recruit and activate the ATM kinase and its downstream targets (25, 26). Rad50 un-winds the DNA ends (36), enabling the recruitment of cellular nucleases such as Exo1 and Sae2, which play a role in resecting the broken ends (35). The 3⬘-single-stranded tail arising after resection serves as a substrate for the sequential recruitment RPA and Brca2, ultimately resulting in the formation of Rad51 nucleofilaments on the 3⬘-single-stranded tails. Rad51 nucleo-filaments are capable of detecting homology in duplex DNA and participating in strand invasion to initiate HRR. It is in-teresting that, in addition to its role in the ATM-mediated HRR pathways, MRN has also been shown to participate in end resection in the classical NHEJ pathway (16) and the alternative (A-NHEJ) pathway (8). MRN also associates with telomeres and participates in the maintenance of normal telo-mere length (22, 72).
In summary, this paper provides evidence that the viral exonuclease UL12 can interact directly with the MRN
com-ponents, important players in the DNA damage response of the cell. Taken together with our previous observation that UL12 and ICP8 can function as a two-subunit recombinase
in vitro(48), these results may indicate that UL12 and ICP8
along with host recombination factors act to promote con-catemer formation by stimulating homologous recombina-tion. According to this scenario, UL12 in collaboration with ICP8 may steer the MRN complex toward the viral genome, resulting in end resection and the generation of 3⬘tails that can participate in strand invasion or strand annealing. Al-ternatively, since the MRN complex is known to play roles in multiple repair pathways, it is possible that the UL12-MRN interaction acts to regulate MRN, perhaps by influencing the repair pathway choice most beneficial for HSV. The pathway activated by HSV may influence the production of viral DNA concatemers that can be accurately processed and packaged to produce infectious virus. Current efforts are under way to prove the biological consequences of the UL12-MRN interaction.
ACKNOWLEDGMENTS
We thank John Carson for his help and comments with the SPR experiment, Betty Eipper and Richard Mains for use of the instru-ments, and Tanya Paull for providing the MRN constructs. We also thank the members of our laboratory for their helpful comments and suggestions.
This work was supported by Public Health Service grant AI069136.
REFERENCES
1.Bataille, D., and A. Epstein.1994. Herpes simplex virus replicative concate-mers contain L components in inverted orientation. Virology203:384–388. 2.Bataille, D., and A. L. Epstein.1997. Equimolar generation of the four
possible arrangements of adjacent L components in herpes simplex virus type 1 replicative intermediates. J. Virol.71:7736–7743.
3.Boehmer, P. E., and I. R. Lehman.1997. Herpes simplex virus DNA repli-cation. Annu. Rev. Biochem.66:347–384.
4.Brown, S. M., J. H. Subak-Sharpe, J. Harland, and A. R. MacLean.1992. Analysis of intrastrain recombination in herpes simplex virus type 1 strain 17 and herpes simplex virus type 2 strain HG52 using restriction endonuclease sites as unselected markers and temperature-sensitive lesions as selected markers. J. Gen. Virol.73:293–301.
5.Chou, J., and B. Roizman.1989. Characterization of DNA sequence-com-mon and sequence-specific proteins binding to cis-acting sites for cleavage of the terminal a sequence of the herpes simplex virus 1 genome. J. Virol.
63:1059–1068.
6.Corcoran, J. A., H. A. Saffran, B. A. Duguay, and J. R. Smiley.2009. Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial local-ization sequence proximal to the N terminus. J. Virol.83:2601–2610. 7.de Bruyn Kops, A., and D. M. Knipe.1988. Formation of DNA replication
structures in herpes virus-infected cells requires a viral DNA binding protein. Cell55:857–868.
8.Deriano, L., T. H. Stracker, A. Baker, J. H. Petrini, and D. B. Roth.2009. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J. recom-bination intermediates. Mol. Cell34:13–25.
9.Dimitrova, D. S., and D. M. Gilbert.2000. Stability and nuclear distribution of mammalian replication protein A heterotrimeric complex. Exp. Cell Res.
254:321–327.
10.Dutch, R. E., V. Bianchi, and I. R. Lehman.1995. Herpes simplex virus type 1 DNA replication is specifically required for high-frequency homologous recombination between repeated sequences. J. Virol.69:3084–3089. 11.Dutch, R. E., R. C. Bruckner, E. S. Mocarski, and I. R. Lehman.1992.
Herpes simplex virus type 1 recombination: role of DNA replication and viral a sequences. J. Virol.66:277–285.
12.Gao, M., and D. M. Knipe.1989. Genetic evidence for multiple nuclear functions of the herpes simplex virus ICP8 DNA-binding protein. J. Virol.
63:5258–5267.
13.Goldfarb, T., and E. Alani.2005. Distinct roles for the Saccharomyces cer-evisiae mismatch repair proteins in heteroduplex rejection, mismatch repair and nonhomologous tail removal. Genetics169:563–574.
14.Goldstein, J. N., and S. K. Weller.1998. The exonuclease activity of HSV-1 UL12 is required for in vivo function. Virology244:442–457.
15.Gregory, D. A., and S. L. Bachenheimer.2008. Characterization of mre11 loss following HSV-1 infection. Virology373:124–136.
on November 8, 2019 by guest
http://jvi.asm.org/
16.Helmink, B. A., A. L. Bredemeyer, B. S. Lee, C. Y. Huang, G. G. Sharma, L. M. Walker, J. J. Bednarski, W. L. Lee, T. K. Pandita, C. H. Bassing, and B. P. Sleckman.2009. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J. Exp. Med.206:669–679. 17.Honess, R. W., A. Buchan, I. W. Halliburton, and D. H. Watson.1980.
Recombination and linkage between structural and regulatory genes of her-pes simplex virus type 1: study of the functional organization of the genome. J. Virol.34:716–742.
18.Jacob, R. J., L. S. Morse, and B. Roizman.1979. Anatomy of herpes simplex virus DNA. XII. Accumulation of head-to-tail concatemers in nuclei of infected cells and their role in the generation of the four isomeric arrange-ments of viral DNA. J. Virol.29:448–457.
19.Jacob, R. J., and B. Roizman.1977. Anatomy of herpes simplex virus DNA VIII. Properties of the replicating DNA. J. Virol.23:394–411.
20.Kalderon, D., B. L. Roberts, W. D. Richardson, and A. E. Smith.1984. A short amino acid sequence able to specify nuclear location. Cell39:499–509. 21.Kowalczykowski, S. C.2000. Initiation of genetic recombination and
recom-bination-dependent replication. Trends Biochem. Sci.25:156–165. 22.Lamarche, B. J., N. I. Orazio, and M. D. Weitzman. 2010. The MRN
complex in double-strand break repair and telomere maintenance. FEBS Lett.584:3682–3695.
23.Lamberti, C., and S. K. Weller.1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology226:403–407.
24.Lavin, M. F., and S. Kozlov.2007. ATM activation and DNA damage response. Cell Cycle6:931–942.
25.Lee, J. H., and T. T. Paull.2007. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene26:7741–7748. 26.Lee, J. H., and T. T. Paull.2005. ATM activation by DNA double-strand
breaks through the Mre11-Rad50-Nbs1 complex. Science308:551–554. 27.Lee, J. H., and T. T. Paull.2006. Purification and biochemical
characteriza-tion of ataxia-telangiectasia mutated and Mre11/Rad50/Nbs1. Methods En-zymol.408:529–539.
28.Lees-Miller, S. P., M. C. Long, M. A. Kilvert, V. Lam, S. A. Rice, and C. A. Spencer.1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol.70:7471–7477.
29.Li, H., R. Baskaran, D. M. Krisky, K. Bein, P. Grandi, J. B. Cohen, and J. C. Glorioso.2008. Chk2 is required for HSV-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology375:13–23.
30.Lilley, C. E., C. T. Carson, A. R. Muotri, F. H. Gage, and M. D. Weitzman.
2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A.102:5844–5849.
31.Lilley, C. E., M. S. Chaurushiya, C. Boutell, S. Landry, J. Suh, S. Panier, R. D. Everett, G. S. Stewart, D. Durocher, and M. D. Weitzman.2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J.29:943–955.
32.Livingston, C. M., N. A. DeLuca, D. E. Wilkinson, and S. K. Weller.2008. Oligomerization of ICP4 and rearrangement of heat shock proteins may be important for herpes simplex virus type 1 prereplicative site formation. J. Virol.82:6324–6336.
33.Martinez, R., R. T. Sarisky, P. C. Weber, and S. K. Weller.1996. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J. Virol.70:2075–2085.
34.Martinez, R., L. Shao, J. C. Bronstein, P. C. Weber, and S. K. Weller.1996. The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology
215:152–164.
35.Mimitou, E. P., and L. S. Symington.2009. DNA end resection: many nucleases make light work. DNA Repair (Amst.)8:983–995.
36.Moncalian, G., B. Lengsfeld, V. Bhaskara, K. P. Hopfner, A. Karcher, E. Alden, J. A. Tainer, and T. T. Paull. 2004. The rad50 signature motif: essential to ATP binding and biological function. J. Mol. Biol.335:937–951. 37.Muylaert, I., and P. Elias.2010. Contributions of nucleotide-excision repair, DNA polymeraseand homologous recombination to replication of UV-irradiated Herpes simplex virus type I. J. Biol. Chem.285:13761–13768. 38.Nicolas, A., N. azard-Dany, C. Biollay, L. Arata, N. Jolinon, L. Kuhn, M. Ferro,
S. K. Weller, A. L. Epstein, A. Salvetti, and A. Greco.2010. Identification of Rep-associated factors in herpes simplex virus type 1-induced adeno-associated virus type 2 replication compartments. J. Virol.84:8871–8887.
39.Okano, K., A. L. Vanarsdall, and G. F. Rohrmann.2007. A baculovirus alkaline nuclease knockout construct produces fragmented DNA and aber-rant capsids. Virology359:46–54.
40.Parkinson, J., S. P. Lees-Miller, and R. D. Everett.1999. Herpes simplex virus type 1 immediate-early protein Vmw110 induces the proteasome-de-pendent degradation of the catalytic subunit of DNA-deproteasome-de-pendent protein kinase. J. Virol.73:650–657.
41.Paull, T. T., and J. H. Lee.2005. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle4:737–740. 42.Petrini, J. H., and T. H. Stracker.2003. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol.
13:458–462.
43.Porter, I. M., and N. D. Stow.2004. Virus particles produced by the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12 contain abnor-mal genomes. J. Gen. Virol.85:583–591.
44.Poteete, A. R.2001. What makes the bacteriophage lambda Red system useful for genetic engineering: molecular mechanism and biological func-tion. FEMS Microbiol. Lett.201:9–14.
45.Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell36:857–868.
46.Rajagopal, C., K. L. Stone, V. P. Francone, R. E. Mains, and B. A. Eipper.
2009. Secretory granule to the nucleus: role of a multiply phosphorylated intrinsically unstructured domain. J. Biol. Chem.284:25723–25734. 47.Reuven, N. B., S. Antoku, and S. K. Weller.2004. The UL12.5 gene product
of herpes simplex virus type 1 exhibits nuclease and strand exchange activ-ities but does not localize to the nucleus. J. Virol.78:4599–4608. 48.Reuven, N. B., A. E. Staire, R. S. Myers, and S. K. Weller.2003. The herpes
simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol.77:7425–7433. 49.Saydam, N., R. Kanagaraj, T. Dietschy, P. L. Garcia, J. Pena-Diaz, I.
Shev-elev, I. Stagljar, and P. Janscak.2007. Physical and functional interactions between Werner syndrome helicase and mismatch-repair initiation factors. Nucleic Acids Res.35:5706–5716.
50.Schaffer, P. A., M. J. Tevethia, and M. Yesh-Melnick.1974. Recombination between temperature-sensitive mutants of herpes simplex virus type 1. Vi-rology58:219–228.
51.Severini, A., A. R. Morgan, D. R. Tovell, and D. L. Tyrrell.1994. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology200:428–435.
52.Severini, A., D. G. Scraba, and D. L. Tyrrell.1996. Branched structures in the intracellular DNA of herpes simplex virus type 1. J. Virol.70:3169–3175. 53.Shao, L., L. M. Rapp, and S. K. Weller.1993. Herpes simplex virus 1 alkaline
nuclease is required for efficient egress of capsids from the nucleus. Virology
196:146–162.
54.Shelton, L. S., A. G. Albright, W. T. Ruyechan, and F. J. Jenkins.1994. Reten-tion of the herpes simplex virus type 1 (HSV-1) UL37 protein on single-stranded DNA columns requires the HSV-1 ICP8 protein. J. Virol.68:521–525. 55.Shirata, N., A. Kudoh, T. Daikoku, Y. Tatsumi, M. Fujita, T. Kiyono, Y.
Sugaya, H. Isomura, K. Ishizaki, and T. Tsurumi.2005. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduc-tion elicited by herpes simplex virus infectransduc-tion. J. Biol. Chem.280:30336– 30341.
56.Smiley, J. R., J. Duncan, and M. Howes.1990. Sequence requirements for DNA rearrangements induced by the terminal repeat of herpes simplex virus type 1 KOS DNA. J. Virol.64:5036–5050.
57.Stahl, M. M., L. Thomason, A. R. Poteete, T. Tarkowski, A. Kuzminov, and F. W. Stahl. 1997. Annealing invasion in phage lambda recombination. Genetics147:961–977.
58.Sugawara, N., T. Goldfarb, B. Studamire, E. Alani, and J. E. Haber.2004. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. U. S. A.101:9315–9320.
59.Takaoka, A., S. Hayakawa, H. Yanai, D. Stoiber, H. Negishi, H. Kikuchi, S. Sasaki, K. Imai, T. Shibue, K. Honda, and T. Taniguchi.2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature424:516–523.
60.Taylor, T. J., and D. M. Knipe.2004. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol.78:
5856–5866.
61.Thomas, M. S., M. Gao, D. M. Knipe, and K. L. Powell.1992. Association between the herpes simplex virus major DNA-binding protein and alkaline nuclease. J. Virol.66:1152–1161.
62.Umene, K.1985. Intermolecular recombination of the herpes simplex virus type 1 genome analysed using two strains differing in restriction enzyme cleavage sites. J. Gen. Virol.66:2659–2670.
63.Vlazny, D. A., A. Kwong, and N. Frenkel.1982. Site-specific cleavage/pack-aging of herpes simplex virus DNA and the selective maturation of nucleo-capsids containing full-length viral DNA. Proc. Natl. Acad. Sci. U. S. A.
79:1423–1427.
64.Weller, S. K., M. R. Seghatoleslami, L. Shao, D. Rowse, and E. P. Car-michael.1990. The herpes simplex virus type 1 alkaline nuclease is not essential for viral DNA synthesis: isolation and characterization of a lacZ insertion mutant. J. Gen. Virol.71:2941–2952.
65.Wilcock, D., and D. P. Lane.1991. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature349:429–431.
66.Wildy, P.1955. Recombination with herpes simplex virus. J. Gen. Microbiol.
13:346–360.
67.Wilkinson, D. E., and S. K. Weller.2006. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci.119:2695–2703.
on November 8, 2019 by guest
http://jvi.asm.org/
68.Wilkinson, D. E., and S. K. Weller.2004. Recruitment of cellular recombi-nation and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prerep-licative sites and correlates with the induction of the DNA damage response. J. Virol.78:4783–4796.
69.Wilkinson, D. E., and S. K. Weller.2003. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life55:451–458. 70.Williams, R. S., J. S. Williams, and J. A. Tainer.2007. Mre11-Rad50-Nbs1
is a keystone complex connecting DNA repair machinery, double-strand
break signaling, and the chromatin template. Biochem. Cell Biol.85:509– 520.
71.Zhang, X., S. Efstathiou, and A. Simmons. 1994. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electro-phoresis: implications for viral DNA amplification strategies. Virology202:
530–539.
72.Zhu, X. D., B. Kuster, M. Mann, J. H. Petrini, and T. de Lange.2000. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat. Genet.25:347–352.