Characterization of Elements Regulating the
Nuclear-to-Cytoplasmic Translocation of ICP0 in Late Herpes Simplex
Virus 1 Infection
Subodh Kumar Samrat,aBinh L. Ha,a*Yi Zheng,aHaidong Gua
aDepartment of Biological Sciences, Wayne State University, Detroit, Michigan, USA
ABSTRACT Infected cell protein 0 (ICP0) of herpes simplex virus 1 (HSV-1) is an im-mediate early protein containing a RING-type E3 ubiquitin ligase. It targets several host factors for proteasomal degradation and subsequently activates viral expres-sion. ICP0 has a nuclear localization sequence and functions in the nucleus early during infection. However, later in infection, ICP0 is found solely in the cytoplasm. The molecular mechanism and biological function of the ICP0 nuclear-to-cytoplasmic translocation are not well understood. In this study, we sought to characterize ele-ments important for this translocation. We found that (i) in human embryonic lung fibroblast (HEL) cells, ICP0 C-terminal residues 741 to 775 were necessary but not sufficient for the nuclear-to-cytoplasmic translocation; (ii) the loss of ICP0 E3 ubiqui-tin ligase activity, which led to defective viral replication in nonpermissive cells, also caused mutant ICP0 to be retained in the nucleus of HEL cells; (iii) in permissive U2OS cells, however, ICP0 lacking E3 ligase activity was translocated to the cytoplasm at a pace faster than that of wild-type ICP0, suggesting that nuclear retention of ICP0 occurs in an ICP0 E3 ligase-dependent manner; and (iv) the ICP0 C terminus and late viral pro-teins cooperate in order to overcome nuclear retention and stimulate ICP0 cytoplasmic translocation. Taken together, less ICP0 nuclear retention may contribute to the permis-siveness of U2OS cells to HSV-1 in the absence of functional ICP0.
IMPORTANCE A distinct characteristic for eukaryotes is the compartmentalization of cell metabolic pathways, which allows greater efficiency and specificity of cellular functions. ICP0 of HSV-1 is a multifunctional viral protein that travels through differ-ent compartmdiffer-ents as infection progresses. Its main regulatory functions are carried out in the nucleus, but it is translocated to the cytoplasm late during HSV-1 tion. To understand the biological significance of cytoplasmic ICP0 in HSV-1 infec-tion, we investigated the potential players involved in this nuclear-to-cytoplasmic translocation. We found that there is a nuclear retention force in an ICP0 E3 ubiqui-tin ligase-dependent manner. In addition, we identified the C terminus of ICP0 as a
cis element cooperating with late viral proteins to overcome the nuclear retention
and stimulate the nuclear-to-cytoplasmic translocation of ICP0.
KEYWORDS E3 ubiquitin ligase, HSV-1, ICP0, nuclear retention, nuclear-to-cytoplasmic translocation, virus-host interactions
H
erpes simplex virus 1 (HSV-1) is the etiological cause of a wide range of mild tosevere diseases such as cold cores, genital herpes, keratitis, and encephalitis. The virus establishes lifelong latency after primary infection. The sporadic reactivation of latently infected virus can be triggered by many incidents such as fever, stress, immune suppression, and other unknown reasons. Infected cell protein 0 (ICP0) is an immediate early viral protein crucial for both lytic and latent HSV-1 infection. The main function of ICP0 is to offset the cellular frontline antiviral defenses and, consequently, to enhance
Received20 September 2017Accepted24 October 2017
Accepted manuscript posted online1 November 2017
CitationSamrat SK, Ha BL, Zheng Y, Gu H. 2018. Characterization of elements regulating the nuclear-to-cytoplasmic translocation of ICP0 in late herpes simplex virus 1 infection. J Virol 92:e01673-17.https://doi.org/10.1128/ JVI.01673-17.
EditorRozanne M. Sandri-Goldin, University of California, Irvine
Copyright© 2018 American Society for Microbiology.All Rights Reserved. Address correspondence to Haidong Gu, [email protected].
*Present address: Binh L. Ha, University of Detroit Mercy School of Dentistry, Detroit, Michigan, USA.
VIRUS-CELL INTERACTIONS
crossm
on November 6, 2019 by guest
http://jvi.asm.org/
downstream virus expression (1). The most important functional domain of ICP0 is a RING-type E3 ubiquitin ligase located in its second exon, which targets several host factors for proteasome-dependent degradation. Some of the ICP0 E3 substrates are part of host intrinsic defenses. Their degradation contributes to the augmentation of viral gene expression and DNA replication (for reviews, see references 2–4).
ICP0 is known to traffic through subcellular compartments at different infection phases. This protein contains a canonical nuclear localization signal (NLS) located in
lysine/arginine-rich residues 500 to 506. Uponde novosynthesis early during infection,
ICP0 is immediately found in the nucleus and localized to a dynamic nuclear structure termed nuclear domain 10 (ND10) (5). The discrete ND10 nuclear bodies are involved in many regulatory pathways such as apoptosis, DNA damage and repair, tumor suppres-sion, and antiviral defense (for reviews, see references 6–8). Two of the ND10 organizers, promyelocytic leukemia (PML) protein and Sp100, are substrates for the ICP0 E3 ligase, which leads to the ubiquitination and the subsequent proteosomal degradation of both of them (9). After the loss of organizers, ND10 bodies are dispersed, and ICP0 is diffused to fill the entire nucleus. Interestingly, later in HSV-1 infection, ICP0 disappears from the nucleus and accumulates solely in the cytoplasm (10). This nuclear-to-cytoplasmic translocation requires the onset of viral DNA replication, suggesting the potential involvement of a late viral protein(s) in facilitating translocation (10). A tegument protein, VP22, has been shown to affect the translocation of several viral and cellular proteins, including ICP0 (11).
Along its path of subcellular trafficking, ICP0 carries out multiple functions through-out infection. On the molecular level, there are two major actions for ICP0: (i) degrading cellular restrictive factors by its E3 ubiquitin ligase and (ii) interacting with various binding partners to modify cell pathways (3). Both the E3 enzyme activity and protein-protein interactions of ICP0 contribute to its ability to counteract host defenses and subsequently to enhance downstream viral expression (2). For example, the conver-gence of ND10 components at the incoming viral DNA is part of the cell’s attempts to prevent the viral genome from establishing transcription and replication (12, 13). As a counteraction, HSV-1 deploys ICP0 to target PML and Sp100 for proteosomal degra-dation, which leads to the dispersal of ND10 bodies and the derepression of viral genes (2, 8). Another example is the formation of the naked incoming HSV-1 genome into the nucleosome-like structure by associating it with host histones and chromatin remod-elers (14, 15). ICP0 is also known to interact with host factors such as CoREST and CLOCK to modulate chromatin-associated gene regulation (16, 17). The complex interactions between ICP0 and its cellular binding partners or its E3 substrates are likely regulated when ICP0 navigates the subcellular compartments. To better understand ICP0 multi-functionality and the coordination of ICP0 functional domains throughout HSV-1 infection, we carefully dissected the nuclear trafficking of ICP0 around ND10. We reported previously that ICP0 requires different domains to accomplish a dynamic interaction with ND10 nuclear bodies (18, 19). Although many of the ICP0 functions, such as the degradation of PML and interaction with CoREST, occur in the nucleus (16), cytoplasmic ICP0 may also have independent functions. In the present study, we focused on the nuclear-to-cytoplasmic translocation of ICP0 occurring late during infection. We investigated the domains or factors involved in determining ICP0 trans-location. We found that both the RING-type E3 ubiquitin ligase and the C-terminal residues are involved in cytoplasmic translocation but with different mechanisms.
RESULTS
The ICP0 C terminus is required for the nuclear-to-cytoplasmic translocation of ICP0. The NLS of ICP0 is located at residues 500 to 506, which drives the nuclear localization of ICP0 upon its synthesis. The presence of the nuclear-to-cytoplasmic translocation of ICP0 late during infection suggests the potential masking of the NLS or unmasking of a nuclear export sequence (NES) that may be related to the HSV-1 infection process. To elucidate the molecular mechanism of ICP0 translocation, we sought to first identify the domain(s) necessary for the event. For this purpose, we examined the subcellular localization of ICP0 mutants that lack either the N terminus,
Samrat et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
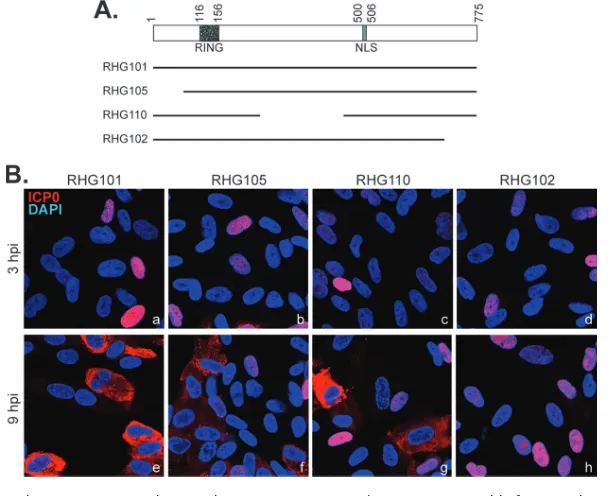
the central region, or the C terminus. As shown in Fig. 1A, human embryonic lung fibroblast (HEL) cells were infected with recombinant viruses RHG101, RHG105, RHG110 (18), and RHG102, in which the respective ICP0 gene either remains the wild type or lacks N-terminal residues 1 to 83, central region residues 245 to 474, or C-terminal residues 719 to 775. At 3 h postinfection (hpi), various ICP0 proteins were localized in the nucleus (Fig. 1Ba to d). Consistent with data from previous reports (10, 18, 20), at 9 hpi, wild-type ICP0 was found mainly in the cytoplasm (Fig. 1Be). Interestingly, while the deletion of the N terminus or central region of ICP0 did not affect the nuclear-to-cytoplasmic translocation late during infection (Fig. 1Bf and g), we found that the deletion of the C-terminal 57 amino acids completely abolished translocation (Fig. 1Bh),
suggesting that the sequence required for ICP0 translocation is aciselement located in
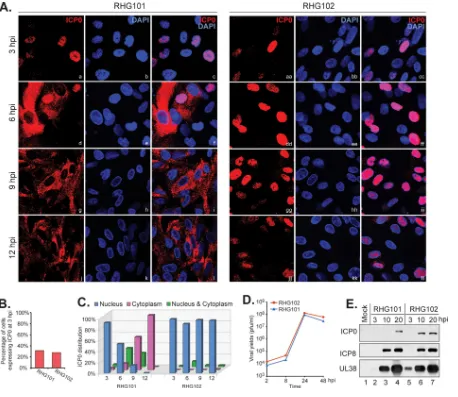
the C terminus of ICP0. We further performed a time course comparison between wild-type ICP0 and the mutant with a C-terminal truncation and tabulated data for about 400 cells to quantitate the ICP0 subcellular distribution. The results showed that at 3 hpi, about 30% of RHG101- or RHG102-infected cells expressed ICP0 at detectable levels (Fig. 2Aa to c and aa to cc and B), indicating that the initial infection rates for the two viruses were at comparable levels. Furthermore, at 3 hpi, 92% of RHG101-infected cells that expressed ICP0 and 98% of the RHG102-infected counterparts had the protein completely localized in the nucleus (Fig. 2C). As infection progressed, ICP0 in RHG101-infected cells was steadily translocated into the cytoplasm, with 54%, 14%, and 0% of the protein located in the nucleus and 12%, 60%, and 100% located in the cytoplasm at 6, 9, and 12 hpi, respectively (Fig. 2Ad to l and C). In the meantime, ICP0 in about 90% of the RHG102-infected cells remained in the nucleus at all time points (Fig. 2Add to ll and C). We further examined viral growth and viral protein expression and found no substantial differences between RHG101 and RHG102 (Fig. 2D and E), suggesting that in HEL cells, the loss of the C-terminal 57 residues in ICP0 led to a failure in the ICP0 nuclear-to-cytoplasmic translocation, but it did not affect the main functions of ICP0 in enhancing gene expression and viral replication.
FIG 1The ICP0 C terminus, but not the N terminus or central region, is responsible for its nucleus-to-cytoplasm translocation. (A) Schematic illustration of domain structures in ICP0 cDNA and the ICP0 truncations used to map the domain required for ICP0 translocation. (B) HEL cells grown on 4-well slides were infected by the indicated viruses at 4 PFU/cell. At 3 and 9 hpi, cells were fixed, permeabilized, and reacted with rabbit anti-ICP0 antibody and then Alexa 594-conjugated anti-rabbit secondary antibody. Confocal images of representative cells were taken with a 40⫻objective on a Leica SP8 microscope.
Elements Regulating ICP0 Translocation Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
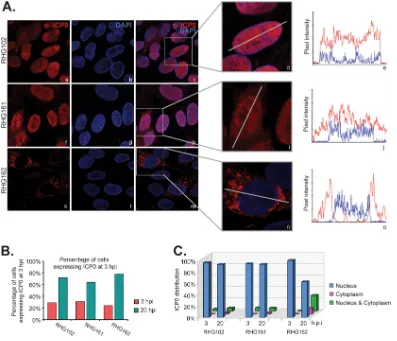
[image:3.585.52.356.72.320.2]The last 35 amino acids of ICP0 are the minimum sequence necessary, but not sufficient, for cytoplasmic translocation of ICP0.To identify the minimum sequence in the ICP0 C terminus that is important for the nuclear-to-cytoplasmic translocation, we constructed the recombinant viruses RHG161 and RHG162, which contain deletions of ICP0 residues 741 to 769 and 758 to 775, respectively, and examined their ICP0 distribution during infection. About 30% of HEL cells infected with RHG102, RHG161, and RHG162 at 4 PFU/cell expressed ICP0 at 3 hpi. This percentage increased to about 80% for all three infections at 20 hpi (Fig. 3B), indicating that the three different infections started at comparable levels and progressed at similar speeds. We extended the infection period to 20 h in this experiment to further preclude a possible delayed effect that may come with the mutation in ICP0. In line with the above-described observations, we found that 91% of RHG102-infected cells had ICP0 confined within the nucleus this late during infection (Fig. 3Aa to c and C). Further deletion of ICP0 showed that 93% of the ICP0 protein lacking residues 741 to 769 (RHG161) remained in the
FIG 2ICP0 lacking the C terminus is confined within the nucleus after prolonged infection. (A to C) HEL cells infected by RHG101 or RHG102 at 4 PFU/cell for 3, 6, 9, and 12 h were fixed and stained as described above. Confocal images were taken with a 40⫻objective on a Zeiss LSM780 microscope. (A) Representative images. (B) Tabulation of the percentage of cells expressing ICP0 at 3 hpi. (C) Tabulation of the percentage of ICP0 distributed in the nucleus, cytoplasm, or both. (D) One-step growth curves for RHG101 and RHG102. HEL cells were infected with RHG101 or RHG102 at 0.1 PFU/cell. At the indicated times postinfection, cells were harvested for virus titration. The virus yields were then plotted by using Microsoft Excel. (E) Viral protein expression for RHG101 and RHG102. HEL cells were infected with RHG101 and RHG102 at 10 PFU/cell. At the indicated times postinfection, cells were washed and lysed. Total cell lysates were separated on SDS-PAGE gels, and the indicated viral proteins were probed with the corresponding antibodies on Western blots.
Samrat et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:4.585.42.500.70.467.2]nucleus at 20 hpi but with a more mixed population for ICP0 lacking the last 18 amino acids (Fig. 3Af to h and k to m). In order to carefully quantify partial effects on the distribution of ICP0, we adopted the Leica LAS X application suite to analyze the pixel intensity of ICP0. As shown in the enlarged panels in Fig. 3A (d, i, and n), we drew a straight line across individual cells and measured the fluorescence intensity along the
line for both ICP0 and 4=,6-diamidino-2-phenylindole (DAPI). Figure 3A (e, j, and o)
shows examples of intensity curves for three categories of cells, those located in the nucleus (Fig. 3Ae), those located in both the nucleus and cytoplasm (Fig. 3Aj), and those located predominantly in the cytoplasm (Fig. 3Ao). The blue line represents DAPI pixels and marks the boundary of the nucleus, which was used as a reference to quantify the intensity of ICP0 inside versus outside the nucleus. With this tool, we tabulated data for about 400 cells for each infection and found that at 20 hpi, ICP0 lacking residues 758 to 775 was partially translocated to the cytoplasm, with 28% of infected cells having ICP0 evenly distributed in both the nucleus and the cytoplasm, 10% having ICP0 located predominantly in the cytoplasm, and only 62% having ICP0 retained mainly in the nucleus (Fig. 3C). These results suggest that the last 35 amino acids of ICP0 are the minimum sequence necessary for nuclear-to-cytoplasmic translocation. The presence of residues 741 to 757 at the C terminus can only partially restore translocation late during infection.
We then investigated whether the last 35 residues can work as an NES to drive the nuclear export of ICP0. We took an existing recombinant virus, RHG103, in which ICP0
FIG 3The C-terminal 35 amino acids of ICP0 comprise the minimum sequence required for ICP0 nuclear-to-cytoplasmic translocation. (A) HEL cells infected by RHG102, RHG161, and RHG162 at 4 PFU/cell for 3 and 20 h were fixed and stained as described above. (Left) Cells infected for 20 h were photographed with a 100⫻objective on a Leica SP8 microscope. (Middle) Representative cells were enlarged to show the line drawn across the cell for the quantification of fluorescence intensity. (Right) Fluorescence pixel intensity of ICP0 and DAPI illustrated as a histogram obtained with the Leica LAS X application suite. (B) Tabulation of the percentage of cells expressing ICP0 at 3 and 20 hpi. (C) Tabulation of the distribution of ICP0.
Elements Regulating ICP0 Translocation Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
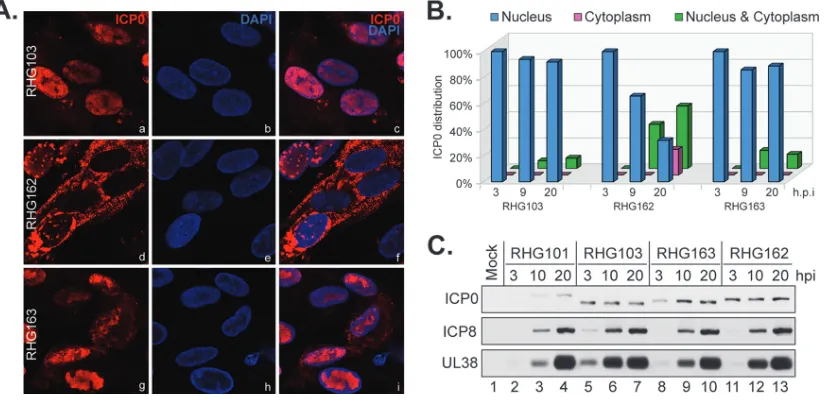
[image:5.585.40.437.71.412.2]is truncated at amino acid 668 (18), as a parental virus to construct the recombinant virus RHG163. In RHG163, we fused ICP0 residues 741 to 775 directly to the existing residues 1 to 668. Gene manipulations in these viruses did not change viral protein expression (Fig. 4C). Imaging and quantitation of the ICP0 distribution showed that in cells infected by RHG103, ICP0 lacking the entire C terminus was largely retained in the nucleus at 3, 9, and 20 hpi (Fig. 4Aa to c), whereas the insertion of the last 35 amino acids back into ICP0 (RHG163) did not restore cytoplasmic translocation for ICP0 (Fig. 4Ag to i). Throughout infection, ICP0 in RHG163-infected cells stayed mostly in the nucleus (Fig. 4B). As a comparison, in cells infected by RHG162 for 20 h, 29% of the infected cells showed ICP0 being translocated to the cytoplasm, and 27% had ICP0 redistributed in both the nucleus and cytoplasm (Fig. 4Ad to f and B). Therefore, we conclude that the sole presence of ICP0 residues 741 to 775 is not sufficient to drive nuclear-to-cytoplasmic translocation for ICP0.
ICP0 E3 ubiquitin ligase is not essential for nuclear-to-cytoplasmic transloca-tion in U2OS cells.Previously, we reported that ICP0 containing C116G/C156A sub-stitutions, which inactivate the E3 ubiquitin ligase, is sequestered at ND10 nuclear bodies in HSV-1 infection and is unable to translocate into the cytoplasm late during infection (18, 21). In Fig. 1, we show that the deletion of sequences located before and after the ICP0 RING domain did not affect the nuclear-to-cytoplasmic translocation of
ICP0. One intriguing question is whether the RING domain per se contains essential
sequences supporting translocation. Lopez et al. reported that blocking viral DNA replication prevented ICP0 from translocating into the cytoplasm, suggesting the involve-ment of an HSV-1 late protein(s) in the translocation process (10). To elucidate the function of the RING-type E3 ubiquitin ligase in nuclear-to-cytoplasmic translocation, we adopted U2OS cells, a permissive cell line that allows the ICP0-null virus to replicate to the same extent as the wild-type virus (22). The use of U2OS cells enables us to observe the nuclear-to-cytoplasmic translocation of ICP0 bearing mutated RING without affecting viral DNA replication or late protein expression.
We first took a recombinant virus, RHG120, in which ICP0 contains the C116G/C156A substitutions (ICP0 RFm) (18) and exhibits substantial defects in E3 ligase activity and viral replication during infection of nonpermissive HEL and HEp-2 cells (23). We compared the subcellular distributions of ICP0 RFm late during infection in both HEL and U2OS cells. Consistent with data from previous reports (18, 21), in HEL cells at 12
FIG 4The C-terminal 35 amino acids of ICP0 are not sufficient for nuclear-to-cytoplasmic translocation. (A and B) HEL cells infected by RHG103, RHG162, and RHG163 at 4 PFU/cell for 3, 9, and 20 h were fixed and stained as described above. (A and B) Cells infected for 20 h were photographed (A), and the percentages of ICP0 distributed in the nucleus, cytoplasm, or both in cells at all three time points were tabulated (B). (C) Viral protein expression of the indicated viruses was probed by Western blotting.
Samrat et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
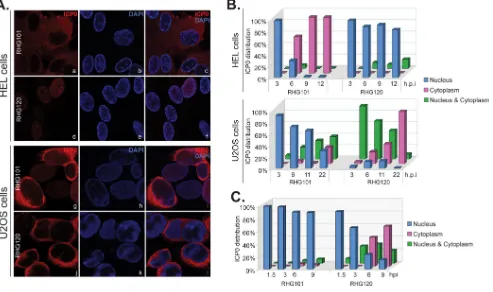
[image:6.585.45.463.72.274.2]hpi, 98% of RHG101-infected cells had wild-type ICP0 located in the cytoplasm, while 84% of RHG120-infected cells had ICP0 RFm restricted within the nucleus (Fig. 5Aa to f and B, top). However, in infected U2OS cells, both wild-type ICP0 and ICP0 RFm were found in the cytoplasm at late times during infection (Fig. 5Ag to l and B, bottom), suggesting that in permissive U2OS cells, an active E3 ubiquitin ligase is not required for nuclear-to-cytoplasmic translocation.
A nuclear retention force relies on the presence of ICP0 E3 ubiquitin ligase.We noticed that for U2OS cells infected by RHG120, ICP0 RFm was found mostly in both the nucleus and cytoplasm or was localized solely in the cytoplasm (Fig. 5B, bottom). To examine whether ICP0 RFm is still able to localize to the nucleus early during infection, we infected U2OS cells at 1 PFU/cell to slow down viral progression and quantitated the ICP0 distribution. In Fig. 5C, we show that 92% of RHG120-infected cells had ICP0 RFm localized within the nucleus at 1.5 hpi, and this value gradually decreased to 15% at 9 hpi. The percentage of cells containing ICP0 RFm in both the nucleus and cytoplasm was 8% at 1.5 hpi and peaked at between 3 and 6 hpi, whereas 46% of infected cells had ICP0 RFm solely in the cytoplasm at 6 hpi and this percentage increased to 64% at 9 hpi. As expected for a lower multiplicity of infection, the translocation of wild-type ICP0 was greatly decelerated, with 90% of cells still retaining ICP0 inside the nucleus at 9 hpi. These results confirmed that ICP0 RFm was initially localized to the nucleus of U2OS cells. We conclude that nuclear retention occurs in an ICP0 E3-dependent manner in U2OS cells. Mutations in the E3 ubiquitin ligase release this retention, and ICP0 RFm is translocated to the cytoplasm early and at a pace that is faster than that of wild-type ICP0.
Late protein expression is essential for the cytoplasmic translocation of wild-type ICP0 but not the ICP0 RFm mutant. Previously, Lopez et al. reported that phosphonoacetic acid (PAA) treatment abolished the nuclear-to-cytoplasmic
translo-FIG 5Translocation of wild-type or mutant ICP0 in nonpermissive versus permissive cells. (A and B) HEL and U2OS cells were infected by RHG101 and RHG120 at 4 PFU/cell for 3, 6, 9, and 12 h or for 3, 6, 11, and 22 h, respectively. (A) Infected cells were fixed and stained. HEL cells infected for 12 h and U2OS cells infected for 22 h were photographed as described above. (B) The percent ICP0 distributions in infected HEL and U2OS cells at all four time points were tabulated. (C) Tabulation of the percent ICP0 distribution in U2OS cells infected by RHG101 and RHG120 at 1 PFU/cell for 1.5, 3, 6, and 9 h.
Elements Regulating ICP0 Translocation Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
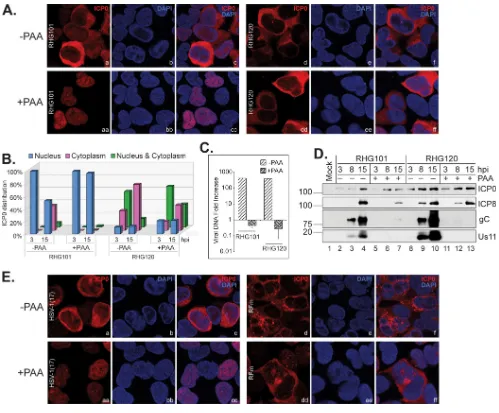
[image:7.585.46.539.73.365.2]cation of wild-type ICP0 in HEL cells, suggesting a role of late viral proteins in the translocation process (10). Our observation of ICP0 RFm being translocated faster than the wild type in U2OS cells leads to the postulation that either the kinetics of viral expression could be changed due to the C116G/C156A mutations or late protein could be irrelevant for ICP0 cytoplasmic translocation in U2OS cells. To test whether late viral proteins are involved in ICP0 translocation in U2OS cells, we compared the subcellular distributions of wild-type ICP0 and ICP0 RFm in the absence and presence of PAA. We found that PAA treatment completely blocked wild-type ICP0 from being translocated to the cytoplasm in U2OS cells (Fig. 6Aaa to cc). Cell tabulation showed that in the absence of PAA, 53% of RHG101-infected cells had ICP0 located in the nucleus, and 40% had ICP0 located solely in the cytoplasm at 15 hpi. With PAA treatment, 97% of the RHG101-infected cells had ICP0 confined within the nucleus at 15 hpi (Fig. 6B). Surprisingly, PAA treatment had a much lesser effect on ICP0 RFm translocation (Fig. 6Ad to f and dd to ff). Cell tabulation showed only a mild delay in ICP0 cytoplasmic
FIG 6In U2OS cells, ICP0 containing C116G/C156A substitutions translocates to the cytoplasm in the absence of DNA replication. (A and B) U2OS cells were infected by RHG101 or RHG120 at 4 PFU/cell for 3 and 15 h in the absence and presence of PAA. Infected cells were fixed and stained as described above. (A and B) Cells infected for 15 h were photographed (A), and percent ICP0 distributions in cells at both time points were tabulated (B). (C) U2OS cells were infected by RHG101 and RHG120 at 4 PFU/cell for 2 and 15 h in the absence and presence of PAA. Total DNA was extracted from infected cells, and qPCR was performed to quantitate the fold increase in viral DNA levels. (D) U2OS cells were infected by RHG101 and RHG120 at 4 PFU/cell for 3, 8, and 15 h. Viral protein expressions of the indicated viruses were probed in Western blotting. (E) U2OS cells were infected by HSV-1 strain 17 or RFm virus at 4 PFU/cell for 15 h in the absence and presence of PAA. The infected cells were fixed, stained, and photographed as described above.
Samrat et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.39.538.68.481.2]translocation upon PAA treatment for 15 h, with 74% having a cytoplasmic localization and 14% being distributed in both the nucleus and cytoplasm in the absence of PAA and 41% having a cytoplasmic localization and 37% being distributed in both the nucleus and cytoplasm in the presence of PAA (Fig. 6B). As a control, we showed that PAA treatment completely blocked DNA replication (Fig. 6C). Furthermore, Western blotting showed that while ICP0 and ICP8 protein levels remained constant in the
absence and presence of PAA, the expressions of the late proteins gC and US11 were
completely blocked by PAA treatment (Fig. 6D). Therefore, late proteins are required for the nuclear-to-cytoplasmic translocation of wild-type ICP0 but not the ICP0 RFm mutant.
To exclude the possibility that RHG120 has secondary mutations other than the C116G/C156A substitutions that may affect ICP0 translocation, we adopted a previously characterized RFm virus (21, 24). Both RHG120 and the RFm virus contain the same C116G/C156A substitutions in ICP0, but RHG120 was constructed by a bacterial artificial chromosome (BAC) containing the genome of HSV-1(F) (18), whereas RFm was gener-ated by a conventional recombination method with an ICP0-null virus derived from HSV-1 strain 17 (24). The results also showed effective translocation of ICP0 from the RFm virus containing the C116G/C156A mutations in the presence of PAA (Fig. 6Edd to ff), whereas wild-type ICP0 in parental HSV-1 strain 17 was restricted within the nucleus upon PAA treatment (Fig. 6Eaa to cc). Observation of the same phenomenon for two independently developed C116G/C156A mutants suggested that the successful trans-location of ICP0 RFm in the absence of DNA replication is indeed contributed by the C116G/C156A substitutions and not an adventurous secondary mutation(s).
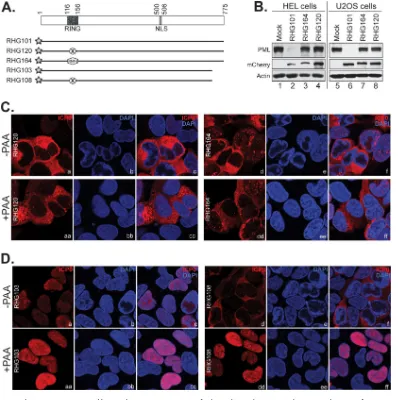
The entire RING domain sequence is dispensable for nuclear-to-cytoplasmic translocation of ICP0. We were intrigued by the fact that a late viral protein(s) is required for the translocation of wild-type ICP0 but not ICP0 RFm (Fig. 6). To examine whether the C116G/C156A substitutions in the context of other ICP0 residues have a particular sequence or structure that may alter intra- or intermolecular regulations and lead to the discrepancy in translocation regulation, we removed the rest of the ICP0 RING finger sequence and replaced it with a RING finger domain from RNF4, the cellular
E3 ubiquitin ligase responsible for PML proteasomal degradation in vivo (25). The
resulting recombinant virus is named RHG164 (Fig. 7A). Figure 7B shows that the RNF4 RING in chimeric ICP0 did not reconstitute E3 ligase activity for ICP0, and it was unable to trigger PML degradation in either HEL or U2OS cells. These results suggest that ICP0 E3 catalyzes PML ubiquitination via specific regulations in an ICP0 RING sequence-dependent fashion. Figure 7C shows that the subcellular distribution of chimeric ICP0 was similar to that of ICP0 RFm, which was readily translocated into the cytoplasm of U2OS cells in the presence of PAA. Taken together, the results shown in Fig. 5 to 7 led us to conclude that (i) in both HEL and U2OS cells, nuclear retention of ICP0 depends on the RING-type E3 ubiquitin ligase activity; (ii) the expression of late protein helps to overcome the retention of wild-type ICP0 in both HEL and U2OS cells; and (iii) this retention is absent for ICP0 mutants lacking active E3 in U2OS cells, where the ICP0 mutant is translocated to the cytoplasm at a much earlier time and even without the late protein.
The ICP0 C terminus and late viral proteins work in tandem to facilitate cytoplas-mic translocation of ICP0 in U2OS cells.We next investigated the relationship among the ICP0 C terminus, late viral proteins, and E3-dependent nuclear retention. For this purpose, we infected U2OS cells with RHG103, in which ICP0 lacks C-terminal residues 669 to 775, or with RHG108 (18), in which ICP0 lacks residues 669 to 775 and at the same time contains the C116G/C156A substitutions. In previous studies, we showed that ICP0 ΔC maintained its ability to degrade PML but that ICP0 ΔC-RFm did not (18, 23). Consistent to what we saw in RHG103-infected HEL cells (Fig. 4), ICP0 ΔC was also retained in the nucleus in RHG103-infected U2OS cells (Fig. 7Da to c). However, in RHG108-infected U2OS cells, ICP0 ΔC-RFm was successfully translocated into the cytoplasm (Fig. 7Dd to f). In duplicated slides, we examined the localization of ICP0 ΔC and ICP0 ΔC-RFm after PAA treatment. We found that while PAA treatment did not
Elements Regulating ICP0 Translocation Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
change the nuclear distribution of ICP0 ΔC (Fig. 7Daa to cc), the same treatment completely blocked ICP0 ΔC-RFm from its cytoplasmic translocation (Fig. 7Ddd to ff). From these observations, we conclude that (i) the E3 ligase-dependent nuclear reten-tion of ICP0 is the predominant force in determining the ICP0 distribureten-tion and (ii) in the absence of retention force, the ICP0 C terminus and late viral proteins work in tandem to facilitate cytoplasmic translocation. When both the C terminus and late proteins are missing, infected U2OS cells lose the ability to export ICP0 despite the absence of retention force.
DISCUSSION
HSV-1 ICP0 is a multifunctional viral protein that antagonizes cell antiviral defenses. It uses the E3 ubiquitin ligase located in its RING finger domain to target cellular restrictive proteins for proteasomal degradation or relies on its various interacting domains to directly bind and modulate cell antiviral pathways. ICP0 travels through
FIG 7The ICP0 C terminus and late viral proteins cooperate to facilitate the nuclear-to-cytoplasmic translocation of ICP0. (A) Schematic diagrams of ICP0 mutants used in this experiment. The C116G/C156A substitutions of ICP0 RFm are illustrated by a circle with an X inside, and chimeric ICP0/RNF4 RING is illustrated by a hexagon with RNF4 marked inside. The star represents the mCherry tag fused to the N terminus of ICP0. (B) PML degradation in infected HEL and U2OS cells. HEL or U2OS cells were infected by RHG101, RHG164, and RHG120 at 10 PFU/cell for 8 h. Infected cells were then harvested, washed, and subjected to Western blotting with the indicated antibodies. (C and D) U2OS cells were infected by RHG120 and RHG164 (C) or by RHG103 and RHG108 (D) at 4 PFU/cell for 15 h in the absence and presence of PAA. The infected cells were fixed, stained, and photographed as described above.
Samrat et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:10.585.40.438.71.471.2]different subcellular compartments at different infection phases. It is plausible to postulate that ICP0 trafficking promotes its regulation specificity by differentially interacting with the target proteins located in different compartments.
In the present study, we searched for elements that determine the nuclear versus cytoplasmic distribution of ICP0 so as to understand the function and significance of ICP0 trafficking in HSV-1 infection. We identified the E3 ubiquitin ligase located within ICP0 RING domain as part of a nuclear retention force that delays the cytoplasmic translocation of ICP0. In permissive U2OS cells, where HSV-1 replicates normally in the absence of ICP0 functions, ICP0 lacking the E3 ligase activity was translocated to the cytoplasm quicker and earlier than wild-type ICP0. To overcome nuclear retention, either the presence of the ICP0 C terminus or the expression of late viral protein is required. In nonpermissive HEL cells, however, both the ICP0 C terminus and late protein expression are necessary for a successful nuclear-to-cytoplasmic translocation of ICP0, suggesting that there are more layers of nuclear retention force in HEL cells than in U2OS cells.
Previously, Kalamvoki and Roizman reported that in both HEL and U2OS cells, transfection of DNA larger than 400 bp prior to HSV-1(F) infection delays the translo-cation of ICP0 into the cytoplasm (26). The introduction of foreign DNA prior to infection triggers cell defensive responses that attempt to silence exogenous DNA. ICP0 functions, such as targeting ND10 components and modulating chromatin repressors, are part of the viral efforts to antagonize the host defenses mounted against foreign DNA. ICP0 counteractions depend mainly on its E3 ubiquitin ligase (3). With the fact that both HEL and U2OS cells responded to prior DNA transfection in the same way, it is very likely that the E3 ligase-dependent nuclear retention of ICP0 involves common factors that are present in both HEL and U2OS cells. With the deletion of ICP0 E3 ligase activity, nuclear retention is obviously lost in U2OS cells (Fig. 6 and 7), indicating that nuclear retention force is probably generated from specific protein-protein interactions of the ubiquitination process. It will be interesting to see whether differential ubiquiti-nation of different ICP0 substrates (23) plays any role in regulating nuclear retention. It is known that various antiviral restrictive factors, such as ATRX (27) and STING (28), are missing in U2OS cells. The lack of these cell factors correlates with the permissive-ness of U2OS cells in supporting HSV-1 replication without functional ICP0. In nonper-missive cells, the loss of ICP0 E3 ubiquitin ligase activity renders ineffective viral replication and late protein expression, which likely is the reason for the defective translocation of ICP0 RFm in HEL cells. Currently, we cannot separate the biochemical interactions of ICP0 translocation from the ICP0 functions in HSV-1 replication in HEL cells, like what we have done with U2OS cells. Therefore, it remains unclear whether the restrictive factors that exist in HEL cells but that are missing in U2OS cells can place an additional layer of retention to further restrain ICP0 inside the nucleus.
Cell nucleocytoplasmic transport of macromolecules involves a network of interac-tions between transport receptors such as importin or exportin, nucleoporins, and a Ran cycle that generates a RanGTP gradient across the nuclear pore (29). Lengyel et al. showed that treatment with leptomycin B (LMB), a specific inhibitor of the CRM1 export pathway, did not prevent ICP0 from being translocated to the cytoplasm late during infection (30), suggesting that the cytoplasmic translocation of ICP0 is CRM1 pathway independent. Sandri-Goldin and colleagues showed that ICP27 interacted with mRNA export factors such as TAP/NXF1 and Aly/REF1 to facilitate the nuclear export of viral mRNA (31, 32), suggesting the involvement of ICP27 in manipulating cellular nuclear export. Interestingly, the deletion of ICP27 also abolishes the cytoplasmic translocation of ICP0 (33), but a single point mutation, M50T, of ICP27 causes the cytoplasmic translocation of ICP0 to be CRM1 dependent (30). These results indicate that cytoplas-mic translocation of ICP0 may be achieved via multiple pathways.
In our study, we found that in both HEL and U2OS cells, the ICP0 C terminus acts in
cisto promote cytoplasmic translocation, but the requirement for the late viral protein
differs in HEL and U2OS cells. To delineate the roles of the ICP0 C terminus and viral late proteins in ICP0 translocation, we used (i) U2OS cells and (ii) RFm to take nuclear
Elements Regulating ICP0 Translocation Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
retention out of the picture so that we could focus just on the biochemical interactions in nuclear-to-cytoplasmic transport. We found that without the E3 ubiquitin ligase activity, either ICP0 RFm or the ICP0/RNF4 RING chimera can be translocated to the cytoplasm upon PAA treatment (Fig. 6 and 7C). Late protein expression becomes essential only when the ICP0 C terminus is absent and vice versa (Fig. 7Dd to f and dd to ff), suggesting that the ICP0 C terminus and late viral proteins act in tandem to facilitate cytoplasmic translocation. This redundancy may be in line with the observa-tions that a mutation in ICP27 can alter the requirement of CRM1 for the cytoplasmic translocation of ICP0, both of which suggest the involvement of multiple export pathways.
Another viral protein that may participate in regulating cellular export is VP22. Tanaka et al. showed that mutations of the two dileucine motifs in VP22 blocked the cytoplasmic translocation of VP16, ICP0, ICP4, and ICP27 as well as a cellular protein, Hsc70 (11). In that report, the few tested proteins that undergo nuclear-to-cytoplasmic translocation during HSV-1 infection were affected in the same fashion, suggesting that VP22 may modulate the general export machinery and change the export efficiency of many proteins.
Mossman and colleagues reported the construction of cytoplasmic ICP0 by deleting the ICP0 NLS. In infected cells, this cytoplasmic ICP0 blocks the formation of the interferon (IFN) regulatory factor 3 (IRF3) dimer and consequently helps to inhibit IFN responses, therefore enhancing viral replication, in an E3 ligase-independent manner (34, 35). Interestingly, they found that ICP0 lacking both the NLS and the RING domain
stimulated more IFN-␥production in infected mice than did ICP0 lacking the NLS but
containing the RING domain (35). It will be interesting to see if the loss of E3 ligase in cytoplasmic ICP0 also leads to the inability to block IRF3 dimerization. So far, the cytoplasmic functions and the molecular basis of the cytoplasmic functions of ICP0 are not clear. More importantly, how the cytoplasmic function of ICP0 coordinates with its nuclear functions in HSV-1 infection is completely unknown. Our work to dissect the elements required for nuclear-to-cytoplasmic transport paves the road for a further understanding of the overall functionality of ICP0 in HSV-1 infection.
MATERIALS AND METHODS
Cells and viruses.Immortalized HEL fibroblasts were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Sigma). Human osteosarcoma U2OS cells were grown in McCoy’s 5A medium (Invitrogen) supplemented with 10% FBS. The recombinant viruses RHG101, RHG103, RHG105, RHG108, RHG110, and RHG120 were described previously (18).
Construction of recombinant viruses.Forward primer 5=-TCCCCCCAGTCCACGCGTCC-3=, paired with a reverse primer, 5=-GCAGTCGACTTAGGGGGGCATCACGTGGTTACC-3=, 5=-CCCGTCGACGCCATGTTC CCCACGGGGGT-3=, or 5=-GCAGTCGACTTAAGACCGCAGGCTGCGGAAGTC-3=, was used for PCR amplifica-tion. The PCR products were then digested with MluI and SalI to replace the MluI-SalI fragment in wild-type ICP0 and to generate ICP0 mutants containing residues 1 to 718, 1 to 740, and 1 to 757, named plasmids pHG102, pHG161, and pHG162, respectively.
An ICP0 mutant containing residues 741 to 775 fused with residues 1 to 668 was generated by a two-round PCR strategy. In the first round of PCR, forward primer 5=-TCCCCCCAGTCCACGCGTCC-3=was paired with mutation primer 1 (5=-CTAGGGTGCCCTGGTCGAAGGGCAGGCAGTCCCCCGTG-3=), and reserve primer 5=-CCGCTCGAGTTATTGTTTTCCCTCGTCCCGGGTC-3=was paired mutation primer 2 (5=-CACGGGG GACTGCCTGCCCTTCGACCAGGGCACCCTAG-3=), to amplify the fragments downstream and upstream of the deletion junction, respectively. In the second round of PCR, both upstream and downstream PCR products were gel purified and mixed to serve as the template to be amplified with the forward and reverse primers only. The second-round PCR product, which contained an ICP0 fragment that joined residue 668 to residue 741, was digested with MluI and XhoI to replace the MluI-SalI fragment in wild-type ICP0, which generated plasmid pHG163 that contained ICP0 mutants containing residues 1 to 668 or residues 741 to 775.
Primers 5=-GCACTCGAGAGCAGGACCATGCTGACAGC-3=and 5=-TGTACATATATAAATGGGGTGGTAC-3=
were used for PCR amplification to generate a fragment containing the RNF4 RING finger located at residues 85 to 190. The PCR product was digested with XhoI and inserted into ICP0 between the KpnI and XhoI sites, with which the KpnI-digested site was blunt ended by T4 DNA polymerase. Chimeric ICP0 had RNF4 residues 85 to 190 replacing ICP0 residues 107 to 212. The plasmid containing chimeric ICP0 was named pHG164.
The above-described ICP0 mutants were then cloned into the KO5 plasmid and electroporated into the RR1 strain containing an ICP0-null BAC, as described previously (21). The recombined BAC DNAs were then extracted and transfected into U2OS cells to generate recombinant viruses. All viruses were plaque
Samrat et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
purified at least three times on U2OS cells. Viral DNAs were isolated, and the existence of ICP0 mutations in both terminal and internal repeats was verified by Southern blotting.
Confocal microscopy.HEL and U2OS cells grown on four-well glass slides (Thermo Fisher Scientific) were exposed to recombinant viruses. After 1 h of exposure, the virus inocula were removed, and cells were incubated in growth medium (with or without 300g/ml of PAA, as indicated in Results). The cells were then fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with phosphate-buffered saline (PBS) containing 5% horse serum and 1% bovine serum albumin. The cells were reacted with anti-ICP0 polyclonal antibody at 4°C overnight, rinsed, and subjected to a reaction with Alexa 594-conjugated goat anti-rabbit secondary antibodies (Invitrogen). The slides were then mounted with Vectashield containing DAPI (Vector Laboratories), and images were taken with a confocal microscope.
Antibodies.Polyclonal anti-ICP0 antibody was raised against a glutathioneS-transferase (GST)-ICP0 exon 2 fusion protein. Plasmid pRB4994 containing ICP0 codons 20 to 241 (36) was a generous gift from Bernard Roizman (University of Chicago). The purified GST-ICP0 exon 2 fusion protein was used to immunize two rabbits at Abcam according to standard protocols. The crude serum was then subjected to protein A purification at Abcam.
Anti-mCherry polyclonal and monoclonal antibodies were purchased from Clontech. An anti-actin monoclonal antibody was purchased from Santa Cruz Biotechnology. Monoclonal anti-ICP8, monoclonal anti-US11, monoclonal anti-gC, and polyclonal anti-UL38 were generous gifts from Bernard Roizman (University of Chicago).
One-step growth curves.HEL cells were infected with viruses at 0.1 PFU/cell for 1 h. Virus inocula were then removed, and cells were incubated in DMEM supplied with 10% newborn calf serum (Fisher Thermo Scientific). At the time points indicated in Results, cells were harvested, lysed, and titrated on U2OS cells.
Western blotting. HEL or U2OS cells were mock infected or infected with 10 PFU/cell of recombinant viruses. After 1 h of exposure, the virus inocula were removed, and cells were incubated in medium with 10% newborn calf serum. At the hours indicated, the cells were harvested, washed, and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.25% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride). The total cell lysate was sonicated, electrophoretically separated on SDS-PAGE gels, and then transferred onto a polyvinylidene difluoride membrane. The membrane was blocked with 1⫻TBST (20 mM Tris [pH 7.5], 150 mM NaCl, 0.5% Tween 20) containing 5% nonfat dry milk and probed with primary antibodies. The membrane was then incubated with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Sigma) and visualized with ECL Western blotting detection reagent (GE Healthcare).
qPCR.U2OS cells were infected with recombinant viruses at 4 PFU/cell. After 1 h of exposure, the virus inocula were removed, and cells were incubated in McCoy’s 5A medium supplemented with 10% newborn calf serum, with or without 300g/ml of PAA. At 2 and 15 hpi, total DNA was extracted from infected cells and subjected to quantitative PCR (qPCR) assays using Absolute qPCR SYBR green carboxy-X-rhodamine (ROX) mix (Fisher Thermo Scientific) and a QuantStudio 3 real-time PCR system. Primers used for qPCR and the method of calculation were described previously (18).
ACKNOWLEDGMENTS
We thank financial support from an NIH grant (RO1AI118992) and a Wayne State University startup fund awarded to Haidong Gu.
We thank the Microscopy, Imaging and Cytometry Resources (MICR) Core facility at Wayne State University for technical support. We thank Amanda Rose Dame for her help in constructing recombinant virus RHG161.
REFERENCES
1. Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897.InKnipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2. Lippincott Williams & Wilkins, Philadelphia, PA.
2. Gu H, Zheng Y. 2016. Role of ND10 nuclear bodies in the chromatin repression of HSV-1. Virol J 13:62.https://doi.org/10.1186/s12985-016 -0516-4.
3. Gu H. 2016. Infected cell protein 0 functional domains and their coor-dination in herpes simplex virus replication. World J Virol 5:1–13.https:// doi.org/10.5501/wjv.v5.i1.1.
4. Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465– 481.https://doi .org/10.1099/vir.0.048900-0.
5. Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol 75(Part 6):1223–1233.https://doi.org/10.1099/0022-1317-75-6 -1223.
6. Bernardi R, Pandolfi PP. 2003. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22:9048 –9057.https:// doi.org/10.1038/sj.onc.1207106.
7. Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006 –1016.https://doi.org/10.1038/nrm2277.
8. Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158.
https://doi.org/10.1089/jir.2010.0111.
9. Chelbi-Alix MK, de The H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941.https://doi.org/10.1038/sj.onc .1202366.
10. Lopez P, Van Sant C, Roizman B. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J Virol 75:3832–3840.https://doi.org/10.1128/JVI.75.8.3832-3840.2001. 11. Tanaka M, Kato A, Satoh Y, Ide T, Sagou K, Kimura K, Hasegawa H,
Kawaguchi Y. 2012. Herpes simplex virus 1 VP22 regulates translocation
Elements Regulating ICP0 Translocation Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
of multiple viral and cellular proteins and promotes neurovirulence. J Virol 86:5264 –5277.https://doi.org/10.1128/JVI.06913-11.
12. Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75.https://doi.org/10.1006/viro.1996.0094.
13. Everett RD, Sourvinos G, Leiper C, Clements JB, Orr A. 2004. Forma-tion of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, re-cruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J Virol 78:1903–1917.https://doi.org/10.1128/ JVI.78.4.1903-1917.2004.
14. Placek BJ, Berger SL. 2010. Chromatin dynamics during herpes simplex virus-1 lytic infection. Biochim Biophys Acta 1799:223–227.https://doi .org/10.1016/j.bbagrm.2010.01.012.
15. Conn KL, Schang LM. 2013. Chromatin dynamics during lytic infection with herpes simplex virus 1. Viruses 5:1758 –1786. https://doi.org/10 .3390/v5071758.
16. Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104: 17134 –17139.https://doi.org/10.1073/pnas.0707266104.
17. Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl trans-ferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A 107:17721–17726.https://doi .org/10.1073/pnas.1012991107.
18. Gu H, Zheng Y, Roizman B. 2013. The interaction of herpes simplex virus ICP0 with ND10 bodies: a sequential process of adhesion, fusion and retention. J Virol 87:10244 –10254.https://doi.org/10.1128/JVI.01487-13. 19. Zheng Y, Gu H. 2015. Identification of three redundant segments re-sponsible for herpes simplex virus 1 ICP0 to fuse with ND10 nuclear bodies. J Virol 89:4214 – 4226.https://doi.org/10.1128/JVI.03658-14. 20. Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/
CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A 102:7571–7576.https://doi.org/10.1073/pnas.0502658102.
21. Gu H, Roizman B. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degra-dation of PML, are executed in tandem. J Virol 83:181–187.https://doi .org/10.1128/JVI.01940-08.
22. Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol 69:6249 – 6258.
23. Zheng Y, Samrat SK, Gu H. 2016. A tale of two PMLs: elements regulating a differential substrate recognition by the ICP0 E3 ubiquitin ligase of herpes simplex virus 1. J Virol 90:10875–10885.https://doi.org/10.1128/ JVI.01636-16.
24. Lium EK, Silverstein S. 1997. Mutational analysis of the herpes simplex
virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J Virol 71:8602– 8614. 25. Tatham MH, Geoffroy MC, Shen L, Plechanovova A, Hattersley N, Jaffray
EG, Palvimo JJ, Hay RT. 2008. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol 10: 538 –546.https://doi.org/10.1038/ncb1716.
26. Kalamvoki M, Roizman B. 2008. Nuclear retention of ICP0 in cells ex-posed to HDAC inhibitor or transfected with DNA before infection with herpes simplex virus 1. Proc Natl Acad Sci U S A 105:20488 –20493.
https://doi.org/10.1073/pnas.0810879105.
27. Newhart A, Rafalska-Metcalf IU, Yang T, Negorev DG, Janicki SM. 2012. Single cell analysis of Daxx and ATRX-dependent transcriptional repres-sion. J Cell Sci 125:5489 –5501.https://doi.org/10.1242/jcs.110148. 28. Deschamps T, Kalamvoki M. 2017. Impaired STING pathway in human
osteosarcoma U2OS cells contributes to the growth of ICP0-null mutant herpes simplex virus. J Virol 91:e00006-17.https://doi.org/10.1128/JVI .00006-17.
29. Cook A, Bono F, Jinek M, Conti E. 2007. Structural biology of nucleocy-toplasmic transport. Annu Rev Biochem 76:647– 671.https://doi.org/10 .1146/annurev.biochem.76.052705.161529.
30. Lengyel J, Strain AK, Perkins KD, Rice SA. 2006. ICP27-dependent resis-tance of herpes simplex virus type 1 to leptomycin B is associated with enhanced nuclear localization of ICP4 and ICP0. Virology 352:368 –379.
https://doi.org/10.1016/j.virol.2006.04.044.
31. Johnson LA, Sandri-Goldin RM. 2009. Efficient nuclear export of herpes simplex virus 1 transcripts requires both RNA binding by ICP27 and ICP27 interaction with TAP/NXF1. J Virol 83:1184 –1192.https://doi.org/ 10.1128/JVI.02010-08.
32. Tian X, Devi-Rao G, Golovanov AP, Sandri-Goldin RM. 2013. The interac-tion of the cellular export adaptor protein Aly/REF with ICP27 contrib-utes to the efficiency of herpes simplex virus 1 mRNA export. J Virol 87:7210 –7217.https://doi.org/10.1128/JVI.00738-13.
33. Zhu Z, Cai W, Schaffer PA. 1994. Cooperativity among herpes simplex virus type 1 immediate-early regulatory proteins: ICP4 and ICP27 affect the intracellular localization of ICP0. J Virol 68:3027–3040.
34. Paladino P, Collins SE, Mossman KL. 2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One 5:e10428. https://doi .org/10.1371/journal.pone.0010428.
35. Taylor KE, Chew MV, Ashkar AA, Mossman KL. 2014. Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING finger are required to block interferon-stimulated gene production but not to promote viral replication. J Virol 88:8091– 8101.https://doi.org/10.1128/ JVI.00944-14.
36. Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J Virol 71:1019 –1024.
Samrat et al. Journal of Virology