Copyright © 1997, American Society for Microbiology
Effects of Nucleotide Changes on the Ability of Hepatitis Delta
Virus To Transcribe, Process, and Accumulate
Unit-Length, Circular RNA
TING-TING WU, HANS J. NETTER,† DAVID W. LAZINSKI,‡ANDJOHN M. TAYLOR* Fox Chase Cancer Center, Philadelphia, Pennsylvania 19111-2497
Received 2 January 1997/Accepted 3 April 1997
The circular RNA genome of hepatitis delta virus (HDV) can fold into an unbranched rodlike structure. We mutagenized the two ends of this structure and assayed the effects on the ability of the genomes to replicate and accumulate processed RNA transcripts in transfected cells. The top end, defined as that nearest to the 5*end of the putative mRNA for delta antigen, was much more sensitive than the other end, defined as the bottom. Most of the 22 mutants made at the bottom were able to accumulate RNA as well as the wild type. For deletions extending as close as 2 nucleotides (nt) from the predicted domains needed for the two ribozymes, the accumulation levels dropped to <0.1%. In one mutant, 13 nt of HDV was replaced with 57 nt of non-HDV sequences, and accumulation was at 20% of the wild-type level, consistent with the potential of HDV to act as a vector. However, after replacement with a second sequence, accumulation dropped to 1%. For most of the 14 mutants made at the top of the rod, we observed dramatic inhibitory effects. For example, after removal of 3 bp from the stem adjacent to the terminal loop, accumulation dropped to <0.06% of the wild-type genome level. The top region that we considered was adjacent to both the 5*end of the putative mRNA and the domain that has been proposed to contain a promoter for RNA-directed RNA synthesis. The RNA accumulation abilities of certain mutants were tested under additional different experimental conditions. It was found that after longer times, some mutants began to catch up with the wild type. Also, it was found that certain top mutants gave much greater levels of accumulation when transfected into cells containing the small delta antigen. One interpretation of these data is that certain features at the top of the rod are needed for the accumulation of essential delta antigen mRNA species.
The genome of human hepatitis delta virus (HDV) is a small single-stranded RNA with a circular conformation (23). Nu-merous isolates from around the world have now been se-quenced and arranged according to nucleotide sequence ho-mology into three main groups, referred to as genotypes (7). Overall, isolates can differ in sequence by as much as 35%. Nevertheless, what have been largely maintained are (i) the overall length, at around 1,679 nucleotides (nt), and (ii) the ability of the RNA to fold, with about 70% of the nucleotides base paired, into what is referred to as an unbranched rodlike structure (42). In addition to these gross features, we would expect that there will be multiple essential elements on this small HDV genome. We already know several essential se-quence features: (i) the open reading frame (ORF) for the small delta antigen (dAg-S) (43); (ii) the signals for polyade-nylation (16); and (iii) the domains for the two ribozymes (22, 36). Others have begun to search for domains which might act as putative promoters for RNA-directed RNA synthesis (1, 27, 29). As with the plant viroids, which have many similarities to HDV, we expect that there will be alternative structural fea-tures that have essential transitory roles, such as metastable states occupied during RNA folding and processing (35).
Previous studies have used deliberate mutagenesis of the HDV RNA and yielded information about essential cis-acting
sequences. The studies have involved as little as only 1 or 2 nt changed (1, 6, 18, 20, 44), which in some cases was sufficient to inhibit accumulation of processed RNA transcripts. In other cases, major truncations of the rodlike structure were consid-ered. However, in all cases, such truncations completely abol-ished RNA replication and accumulation (25).
The aim of this study was to use mutagenesis to investigate what might be essential cis-acting features at the two ends of the predicted rodlike structure. As explained below, there are already data to suggest important features not at the exact end regions but adjacent to them.
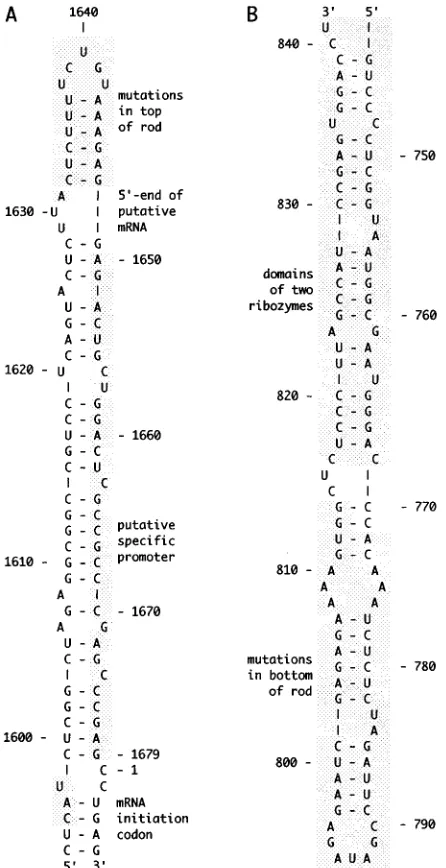
During HDV replication, there is detected, in addition to both the genome and its complement, the antigenome, rela-tively lower amounts of a third RNA. It is a polyadenylated RNA of the same polarity as the antigenome that could be the mRNA for dAg. As indicated in Fig. 1A, which shows only genomic RNA sequences, the 59 end of this RNA has been mapped by primer extension to nt 1631, 9 nt away from what is referred to here as the top of the rod (16). We do not yet know whether this 59end has a cap structure. There are data which indicate that the host RNA polymerase II is the enzyme in-volved in HDV transcription (12, 28), and it has been specu-lated that the 59end of the mRNA corresponds to a specific initiation site (39). (However, if position 1631 is truly an initi-ation site, then it is puzzling that such transcription would begin with pppU rather than with pppG or pppA.) Others have looked at this region at the top end of the rod for a specific promoter. As indicated in Fig. 1A, there is a 29-nt region, extending to within 10 nt of the top, which when expressed as a double-stranded cDNA can function as a promoter for RNA polymerase II (29). An overlapping but significantly larger region can even act to promote transcription in the opposite * Corresponding author. Mailing address: Fox Chase Cancer
Cen-ter, 7701 Burholme Ave., Philadelphia, PA 19111-2497. Phone: (215) 728-2436. Fax: (215) 728-3616. E-mail: [email protected].
† Present address: Heinrich Pette Institute, University of Hamburg, 20251 Hamburg, Germany.
‡ Present address: Tufts University School of Medicine, Boston, MA 02111.
5408
on November 9, 2019 by guest
http://jvi.asm.org/
direction (29). In a more recent study, Beard et al. have made a comparison of the top ends of several known HDV se-quences and deduced the conservation of putative promoter elements (1). Furthermore, an evaluation of nine mutants in this same region has been interpreted as providing supporting evidence (1). One of the aims of our study was to use mutagen-esis to investigate the importance of sequences at the top of the rod, adjacent to but not overlapping with the putative pro-moter element (Fig. 1A).
The cis-acting features nearest to the bottom end of the rod are the ribozymes for the genome and antigenome. As indi-cated in Fig. 1B, these ribozymes which are predicted from in vitro studies extend to about 24 nt from the bottom of the rod
(37). Such, however, are only predictions since the limits of the ribozymes have not been tested in vivo. With this limitation in mind, the second objective of our study was to use mutagenesis to investigate the bottom of the rod for possible essential cis-acting elements. Previous studies have attempted to use the bottom of the rodlike structure as a site for the insertion of non-HDV sequences (17, 32, 40). Therefore, we were also interested in testing the limits of insertions and deletions in this region.
In all of these studies, our experimental approach was to test each of the mutant genomes by transfection into cells and then quantitate the ability to replicate and thereby accumulate pro-cessed HDV RNA species. However, as will be explained, when we tested the consequences of certain mutants with spe-cific variations of this assay strategy, we sometimes obtained quite different answers. Overall, we have become more cau-tious in interpreting mutagenesis data based on an apparent loss of replication function.
MATERIALS AND METHODS
Initiation of HDV replication by using eukaryotic expression vectors contain-ing modified cDNA sequences. To evaluate the replication ability of mutant HDV genomes, we used an approach described by Lazinski and Taylor (25). Different HDV mutants were constructed from two pSVL-based expression vectors, pDL553 and pHN5. Plasmid pDL553 contained 1.2 copies of genomic cDNA, which had the redundancy of the HDV self-cleavage domains. pHN5 was produced by replacing the ampicillin resistance gene of pDL553 with the kana-mycin resistance gene from plasmid pET-28a(1) (Novagen) to eliminate the
ScaI site in the ampicillin resistance region.
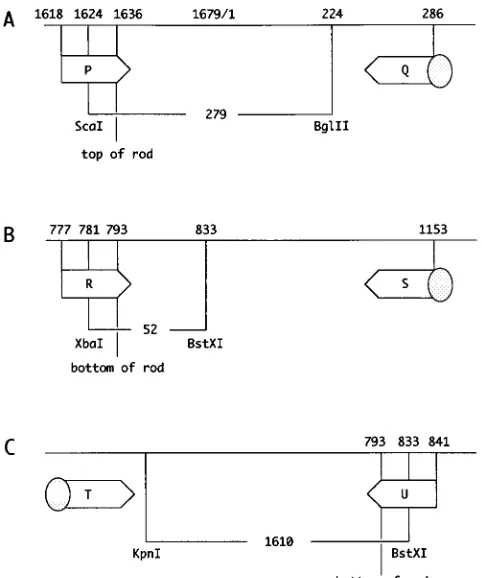
Mutations at the top and the bottom of rodlike HDV RNA (Fig. 1) were introduced by PCR, using the strategy shown in Fig. 2. A primer synthesized to contain the desired mutation, together with a fixed 59-biotin-labeled primer, was used for PCR amplification. The PCR product was immobilized to avidin-coated superparamagnetic beads (Dynal) and subjected to two consecutive restriction enzyme digestions. The second digestion released the desired fragment from the beads ready for forced cloning. For the top, mutations were cloned into plasmid pHN5 as a ScaI-BglII fragment (Fig. 2A). For the bottom, mutations were cloned into plasmid pDL553 as either an XbaI-BstXI fragment (Fig. 2B) or, for larger deletions, a KpnI-BstXI fragment (Fig. 2C). Typically, the mutant constructs were prepared by using a PerfectPrep plasmid DNA kit (5 Prime33 Prime) and verified by automated sequencing (Applied Biosystems).
To disrupt thedAg ORF, a fragment containing a 2-nt deletion at the EcoRI site (20) was excised from pDL542 (24) and cloned into individual mutant constructs.
In vitro RNA transcription.The vectors used to transcribe HDV RNA in vitro were created by using plasmid pTW101, in which a partially redundant HDV cDNA was inserted between a T7 promoter and terminator. The fragments bearing mutations were produced by digestion of pSVL-based mutant constructs with HindIII and NheI and cloned into pTW101. DNA of the pTW101-derived plasmids (1mg) was used for in vitro RNA synthesis by T7 polymerase (Promega) under standard conditions (12). Following transcription, products were treated with RQ DNase I (Promega) and extracted as previously described (12).
Transfection, RNA extraction, and Northern analysis.Huh7 (31) or COS7 (13) cells were grown on 6- or 24-well tissue culture dishes (Sigma) to approxi-mately 80% confluence. For DNA transfection, we used a calcium phosphate method (15). For RNA transfection, we used a Lipofectamine procedure (3) and COS7 cells stably transfected to expressdAg-S (8).
Cells were harvested at either 9 days or every 2 to 3 days, out to 18 days after transfection. Total RNA was extracted by a guanidine-thiocyanate-phenol-chlo-roform procedure (11). RNA samples were glyoxalated and subjected to elec-trophoresis in gels of 1.5% agarose. After electrotransfer to charged nylon membranes (Zetaprobe GT; Bio-Rad), antigenomic HDV RNA sequences were detected by using an RNA probe synthesized by transcription in the presence of [a-32P]UTP (DuPont). The levels of antigenomic RNA were quantitated with a phosphorimager (Fuji). As an internal control for the quantity and quality of the extracted RNA, the filter was stripped and rehybridized by using a 59-labeled oligonucleotide (59-GCTGAACGCCACTTGTCCCT-39) specific for human 18S rRNA.
[image:2.612.65.288.69.503.2]Cloning of HDV RNAs from transfected cells.To remove contaminating DNA from the extracted cellular RNA, the RNA was digested with RQ DNase I for 1 h and isolated as described previously (33). cDNA was synthesized by using Su-perscript II (Bethesda Research Laboratories) and primers that annealed to antigenomic RNA. The reaction product was heat inactivated and then subjected to PCR amplification using Vent polymerase (New England Biolabs). The PCR product was gel purified and cloned into a pGEM4Z vector (Promega). Recom-binant plasmid DNA was prepared by using a PerfectPrep kit, and the sequence was determined by automated sequencing.
FIG. 1. Top and bottom of the rodlike structure of HDV genomic RNA. Panels A and B show regions 1594 to 1679/1 to 6 and 744 to 841, respectively, from the nucleotide sequence of Kuo et al. (20). The two foldings were obtained by using the program RNAFOLD provided by University of Wisconsin Genetics Computer Group. The regions of the top and bottom that were mutagenized in this study are indicated with shading. The features of other shaded areas are explained in the text.
on November 9, 2019 by guest
http://jvi.asm.org/
RESULTS
Mutagenesis at the top and bottom of HDV RNA rodlike structure.Wang et al. first sequenced an entire HDV genome and predicted that it could be folded into a rodlike structure (42). Figure 1 shows the top and bottom of the structure predicted for the sequence subsequently determined by Kuo et al. (21). Using this sequence and folding, we designed top and bottom mutations, with the caveat that disruptions of the basic rodlike structure should be kept to a minimum. We made a series of insertions and deletions in the stem regions adjacent to the loops and also randomized the loop sequences. These mutant constructs were transfected into Huh7 cells, and after 9 days the RNA was harvested. Levels of antigenomic RNA accumulation were quantitated by Northern analysis and ex-pressed as a percentage of that achieved in assays using the wild-type genome.
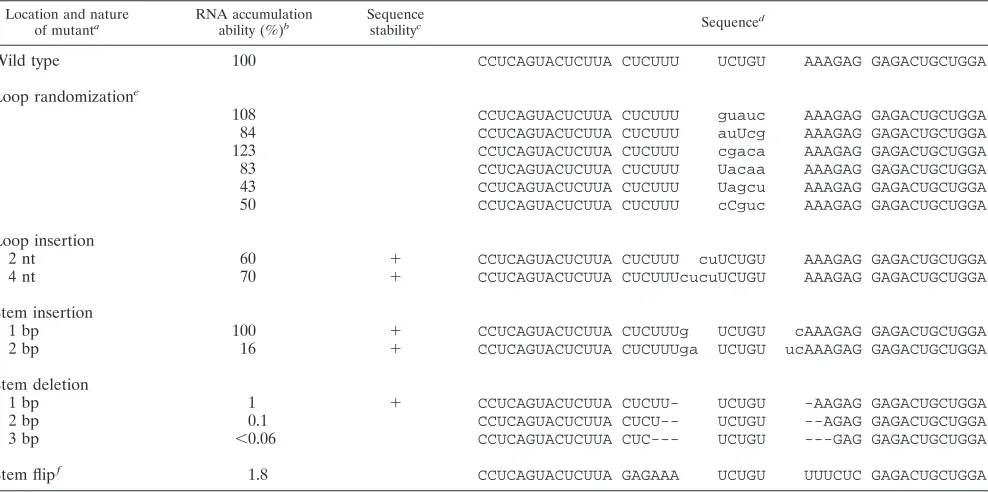
The results for 14 mutants altered at the top of the rod are summarized in Table 1. Six mutants with a randomized 5-nt loop sequence each accumulated RNA almost as well as the wild type. Similar results were obtained following the addition of 2 or 4 nt to the loop or the addition of 1 bp to the adjacent 6-bp stem. However, after insertion of 2 bp to the stem, RNA accumulation dropped sixfold. Deletion from the adjacent stem had even more dramatic effects. Removal of 1, 2, or 3 bp reduced the accumulation 100-, 1,000-, or.1,600-fold, respec-tively. A final mutation was simply to flip the nucleotides
present in the 6-bp stem. It caused a 50-fold inhibition of accumulation.
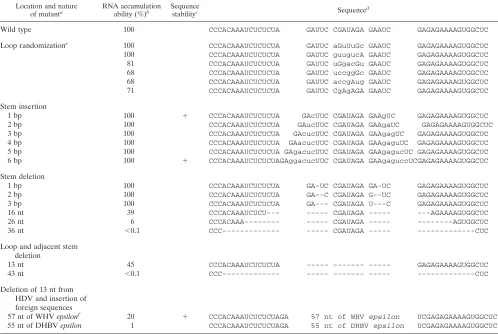
The results of a similar study of 22 mutants altered at the bottom of the rod are summarized in Table 2. Here the pre-dicted loop is larger, 7 nt, but again, each of six mutants with this sequence randomized accumulated almost as well as the wild type. Further, there was no significant effect when we added to the predicted adjacent 5-bp stem 1, 2, 3, 4, 5, or 6 bp. Moreover, in contrast to results for the top of the rod, we were able to remove 1, 2, or 3 bp from the predicted 5-bp stem without a significant effect. Surprisingly, when we removed both the 7-nt loop and an adjacent 3 bp, we saw only a reduc-tion to 45% of the wild-type level. After removal beyond this stem, we began to see dramatic inhibition of accumulation. In none of these did we remove any of the predicted ribozyme domains; however, it is possible that altered RNAs were no longer able to correctly fold the ribozyme domains. When we removed a total of 43 nt, now reaching to 2 nt from the predicted edges of known ribozyme domains, accumulation was not detectable (,0.06%).
In these studies, the bottom of the rodlike structure was found much more tolerant to changes than the top. Therefore, we considered taking the bottom structure, removing 13 nt, and then adding back foreign sequences. We show examples of adding back the epsilon sequences of woodchuck hepatitis virus (WHV) and duck hepatitis B virus (DHBV); these sequences are 57 and 55 nt long, respectively (19). Others have studied the role of these two sequences in the initiation of reverse transcription and in RNA packaging (38). One reason for considering addition of these two sequences is that their pre-dicted foldings have been previously studied and found to superficially resemble the ends of the HDV rodlike structure (19). Their predicted foldings consist of two stems, separated by a 6-nt bulge and ending with a loop of 6 to 7 nt. As summarized in Table 2, we observed that HDV containing the WHV epsilon accumulated to 20% of the wild-type level. In contrast, the insertion of DHBV epsilon reduced replication to 1%.
Sequence stability of mutant HDV.By Northern analyses, some of the top and bottom mutants accumulated RNA at levels close to the wild-type level. We therefore tested eight such mutants to determine whether this was due to sequence reversion. RNA isolated from cells at 9 days after transfection was subjected to reverse transcription (RT)-PCR, cloning, and sequencing. As summarized in Tables 1 and 2, we found no reversion. This was even true for the construct with the 57-nt WHV epsilon sequence.
Time dependence of RNA accumulation.In the studies de-scribed above, we assayed genome replication at a single time point, 9 days after transfection. For eight mutants, we next examined a time course of RNA accumulation from 0 to 18 days, with samples taken every 3 days. We determined the amount of unit-length HDV antigenomic RNA per unit mass of 18S rRNA. The results for four top and four bottom mutants are presented in Fig. 3A and B, respectively.
[image:3.612.58.299.67.356.2]Note that the RNA level for the wild type reached a peak at about day 9 and then declined. We think that two factors contribute to this apparent decline. First, the data are ex-pressed as HDV RNA per unit mass of cellular RNA, a value which itself is increasing with time. Some of this increase may be outgrowth of untransfected cells (2). Second, we note that because of RNA editing (26), increasing amounts of the large form ofdAg (dAg-L) accumulate with time and that this spe-cies acts as a potent dominant negative inhibitor of genome replication (9). In addition todAg-L, there are data to suggest that during replication other variant forms of dAg arise and FIG. 2. Three strategies used for constructing HDV genome mutations. The
nucleotide numbering and indicated restriction enzyme sites are from the se-quence of Kuo et al. (20). The predicted top and bottom of the rod are indicated. For each mutant, we used a pair of primers. One contained a 59-biotin (indicated by the shaded circle), and the other contained the mutation to be added. After PCR, the restriction fragments in each of the three panels were purified and inserted into appropriate vectors as described in Materials and Methods. P, Q, R, S, T, and U designate primers.
on November 9, 2019 by guest
http://jvi.asm.org/
that these can also contribute to the inhibition of genome replication (4, 5).
The time course for each of the four top mutants is shown in Fig. 3A. Generally they accumulated more HDV RNA over time, and they peaked either not at all or later than the wild type. Because of this, some but not all of the mutants appeared to catch up to the wild type. For example, the mutant top12bp achieved even more accumulation than wild type at days 15 and 18, while at day 9, as in Table 1, it represented only 16% of the wild-type level. Thus, the evaluation of mutant accumu-lation ability, determined at a single time point, could, depend-ing on that time point, result in an overestimate or an under-estimate.
One interpretation of the delay for such a mutant relative to the wild type is that mutant genomes can gain in replication competence by accumulating further changes, at the same site and/or at other sites. To test for such changes, we took RNAs from transfected cells at days 9 and 15 and carried out RT-PCR, cloning, and nucleotide sequencing. Three clones were partially sequenced for each time point; we found no changes compared to the original mutated sequence.
A similar time dependence of RNA accumulation was ob-tained for four mutants made at the bottom of the rod, as shown in Fig. 3B. For example, by 18 days, mutant bottom-16 almost caught up to the wild type.
dAg-S enhances replication of top mutants but not bottom mutants.In the foregoing studies,dAg-S, which is essential for genome replication, was provided by the replicating genome. Therefore, to account for possible defects indAg synthesis, we
tested two mutants by using a different strategy, in which we transfected the mutant or wild-type cDNA into COS7 cells stably transfected with a plasmid expressingdAg-S. RNA sam-ples were collected at intervals of 2 or 3 days from 0 to 18 days, and unit-length HDV RNA accumulation was assayed (Fig. 4A).
The two mutants tested, one at the top and one at the bottom, had been assayed previously as replicating at only 1 to 6% of the wild-type level (Tables 1 and 2). In this assay (Fig. 4A) with dAg provided separately, the bottom mutant still accumulated to only low levels, while the top mutant accumu-lated to levels comparable with the wild-type level. Again, we confirmed by RT-PCR, cloning, and sequencing that the se-quence of the mutant genome was stable (45). Another inter-pretation, considered in Discussion, is that the top mutant was defective in its ability to make sufficientdAg. As in Fig. 3, we note in Fig. 4A that the time-dependent accumulation curve went up to a peak and then declined; also, the top mutant achieved its peak value about 6 days after the wild type. How-ever, the results of these assays differ from those in Fig. 3 in that maximum levels of wild-type HDV accumulation are reached much faster, within only 2 to 4 days.
As a follow-up to the foregoing experiments, we tested whether the results would differ if the genome replication was initiated with RNA transcribed in vitro. We found no signifi-cant difference (Fig. 4B). Accumulation of the wild type was again very early and reached a maximum at only 2 to 4 days.
RNA accumulation when the dAg ORF is disrupted.The foregoing assays might have been complicated by the appear-TABLE 1. RNA accumulation for mutants at top of rodlike structure
Location and nature
of mutanta RNA accumulationability (%)b Sequencestabilityc Sequenced
Wild type 100 CCUCAGUACUCUUA CUCUUU UCUGU AAAGAG GAGACUGCUGGA
Loop randomizatione
108 CCUCAGUACUCUUA CUCUUU guauc AAAGAG GAGACUGCUGGA 84 CCUCAGUACUCUUA CUCUUU auUcg AAAGAG GAGACUGCUGGA 123 CCUCAGUACUCUUA CUCUUU cgaca AAAGAG GAGACUGCUGGA 83 CCUCAGUACUCUUA CUCUUU Uacaa AAAGAG GAGACUGCUGGA 43 CCUCAGUACUCUUA CUCUUU Uagcu AAAGAG GAGACUGCUGGA 50 CCUCAGUACUCUUA CUCUUU cCguc AAAGAG GAGACUGCUGGA
Loop insertion
2 nt 60 1 CCUCAGUACUCUUA CUCUUU cuUCUGU AAAGAG GAGACUGCUGGA 4 nt 70 1 CCUCAGUACUCUUA CUCUUUcucuUCUGU AAAGAG GAGACUGCUGGA Stem insertion
1 bp 100 1 CCUCAGUACUCUUA CUCUUUg UCUGU cAAAGAG GAGACUGCUGGA 2 bp 16 1 CCUCAGUACUCUUA CUCUUUga UCUGU ucAAAGAG GAGACUGCUGGA Stem deletion
1 bp 1 1 CCUCAGUACUCUUA CUCUU- UCUGU -AAGAG GAGACUGCUGGA 2 bp 0.1 CCUCAGUACUCUUA CUCU-- UCUGU --AGAG GAGACUGCUGGA
3 bp ,0.06 CCUCAGUACUCUUA CUC--- UCUGU ---GAG GAGACUGCUGGA
Stem flipf 1.8 CCUCAGUACUCUUA GAGAAA UCUGU UUUCUC GAGACUGCUGGA
aThe relevant predicted foldings of the top and bottom of the wild-type genome are presented in Fig. 1.
bBy Northern analyses, levels of antigenomic RNA were quantitated with a phosphorimager, normalized to the level for 18S rRNA, and expressed as a percentage relative to the wild-type accumulation ability.
cRNA collected at 9 days after transfection was subjected to RT-PCR and cloning. Three clones were sequenced, and1indicates that the sequences were identical to the original mutated sequence. Sequencing was not carried out for the other mutants.
dThe RNA sequences correspond to positions 1618 to 1656. Nucleotides different from the wild-type sequence are indicated in lowercase.
eTo create random mutants of the loop sequence, we used oligonucleotide primer P (Fig. 2), which was degenerate in this loop region. Six clones of unique sequence were used in transfection.
fThe 6-bp stem predicted to be adjacent to the terminal loop, as indicated in Fig. 1. The flip corresponds to exchanging the bases on one side of the stem with those
on the other.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.62.556.82.328.2]ance of dAg-L and other variants which can be dominant inhibitors of replication. This potential problem might have occurred even when somedAg-S was provided in trans from a separate plasmid. Therefore, to exclude this possibility, we introduced a deletion of 2 nt near the N terminus of the ORF fordAg into each of the constructs. Previous studies had shown that this change did not interfere with genome replication, as long asdAg-S was provided in trans (20).
[image:5.612.58.556.83.417.2]Therefore, the mutants used in Fig. 4 were modified as described above and tested by transfection of cDNA constructs into cells stably expressingdAg-S. As can be seen in Fig. 5A, the two mutants were able to accumulate antigenomic RNA at levels only,20% of the wild-type level. (The relatively high value for the wild type at day 2 was probably an experimental variation; it was not reproduced in a separate experiment.) Similar results were obtained when genome replication was initiated by using RNA synthesized in vitro (Fig. 5B). We note that the RNA accumulation reached a peak value and then tended to persist with time. We interpret this persistence as due to the fact that here all dAg is dAg-S; there are no variant forms, likedAg-L, which could inhibit HDV RNA syn-thesis.
TABLE 2. RNA accumulation for mutants at bottom of rodlike structure
Location and nature
of mutanta RNA accumulationability (%)b Sequencestabilityc Sequenced
Wild type 100 CCCACAAAUCUCUCUA GAUUC CGAUAGA GAAUC GAGAGAAAAGUGGCUC
Loop randomizatione 100 CCCACAAAUCUCUCUA GAUUC aGuUuGc GAAUC GAGAGAAAAGUGGCUC
100 CCCACAAAUCUCUCUA GAUUC guugucA GAAUC GAGAGAAAAGUGGCUC
81 CCCACAAAUCUCUCUA GAUUC uGgacGu GAAUC GAGAGAAAAGUGGCUC
68 CCCACAAAUCUCUCUA GAUUC uccggGc GAAUC GAGAGAAAAGUGGCUC
68 CCCACAAAUCUCUCUA GAUUC accgAug GAAUC GAGAGAAAAGUGGCUC
71 CCCACAAAUCUCUCUA GAUUC CgAgAGA GAAUC GAGAGAAAAGUGGCUC
Stem insertion
1 bp 100 1 CCCACAAAUCUCUCUA GAcUUC CGAUAGA GAAgUC GAGAGAAAAGUGGCUC
2 bp 100 CCCACAAAUCUCUCUA GAucUUC CGAUAGA GAAgaUC GAGAGAAAAGUGGCUC
3 bp 100 CCCACAAAUCUCUCUA GAcucUUC CGAUAGA GAAgagUC GAGAGAAAAGUGGCUC
4 bp 100 CCCACAAAUCUCUCUA GAacucUUC CGAUAGA GAAgaguUC GAGAGAAAAGUGGCUC
5 bp 100 CCCACAAAUCUCUCUA GAgacucUUC CGAUAGA GAAgagucUC GAGAGAAAAGUGGCUC
6 bp 100 1 CCCACAAAUCUCUCUAGAggacucUUC CGAUAGA GAAgaguccUCGAGAGAAAAGUGGCUC
Stem deletion
1 bp 100 CCCACAAAUCUCUCUA GA-UC CGAUAGA GA-UC GAGAGAAAAGUGGCUC
2 bp 100 CCCACAAAUCUCUCUA GA--C CGAUAGA G--UC GAGAGAAAAGUGGCUC
3 bp 100 CCCACAAAUCUCUCUA GA--- CGAUAGA U---C GAGAGAAAAGUGGCUC
16 nt 39 CCCACAAAUCUCU--- --- CGAUAGA --- ---AGAAAAGUGGCUC
26 nt 6 CCCACAAA--- --- CGAUAGA --- ---AGUGGCUC
36 nt ,0.1 CCC--- --- CGAUAGA --- ---CUC
Loop and adjacent stem deletion
13 nt 45 CCCACAAAUCUCUCUA --- --- --- GAGAGAAAAGUGGCUC
43 nt ,0.1 CCC--- --- --- --- ---CUC
Deletion of 13 nt from HDV and insertion of foreign sequences
57 nt of WHV epsilonf 20 1 CCCACAAAUCUCUCUAGA 57 nt of WHV epsilon UCGAGAGAAAAGUGGCUC
55 nt of DHBV epsilon 1 CCCACAAAUCUCUCUAGA 55 nt of DHBV epsilon UCGAGAGAAAAGUGGCUC
a–cSee Table 1, footnotes a to c.
dThe RNA sequences correspond to positions 769 to 817. Nucleotides different from the wild-type sequence are indicated in lowercase.
eTo create random mutants of the loop sequence, we used oligonucleotide primer R (Fig. 2), which was degenerate in this loop region. Six clones of unique sequence were used in transfection.
fSee the text.
FIG. 3. Time course of accumulation of antigenomic RNA in Huh7 cells transfected with mutated cDNA constructs. Panels A and B represent top and bottom mutants, respectively. The vertical axes correspond to the accumulation of HDV antigenomic RNA per unit mass of cellular RNA, expressed in arbitrary units. The mutants used are indicated at the bottom, with the nomenclatures based on Tables 1 and 2. WT, wild type.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.318.555.479.676.2]DISCUSSION
One of the objectives in carrying out this mutagenesis study was to investigate essential cis-acting elements at the bottom end of the HDV rodlike genome. We found that the bottom region was highly tolerant of base changes in the loop and of insertions as well as deletions in the adjacent stem region (Table 2). If we removed larger regions extending closer but not even into the ribozyme domains, as predicted from studies in vitro (34) and in vivo (18, 30), we did observe inhibition of accumulation. We speculate that this may be because the de-letions create novel RNA foldings which interfere with the correct folding of the essential cis-acting ribozyme domains. Consistent with this interpretation was the effect of a change from CU to UC (at nt 815 to 816) in the unpaired region
immediately adjacent to the predicted antigenomic ribozyme domain; this led to a 10-fold reduction in RNA accumulation ability (45). We next tested the simultaneous removal from the bottom of the rod of 13 nt and the replacement with more than 50 nt of foreign sequences. In one of two cases, the residual ability of the modified genome to replicate and accumulate RNA was 20% of that of wild-type HDV, which we estimate to be about 60,000 copies per cell (10). Also we confirmed that the modification was maintained during genome replication (Table 2). Thus, in this case, our data are supportive of the principle of using the HDV genome as a vector for such a short non-HDV sequence. At the same time, we found that after insertion of a very similar sequence genome, accumulation was only 1%, thus providing a warning that even for short inser-tions, success is not predictable.
The other objective of this study was to evaluate possible cis-acting sequences at the top of the rodlike structure (Fig. 1A). As mentioned in the introduction, this end of the rod is actually adjacent to, rather than overlapping, the region which Beard et al. have interpreted as a promoter for RNA-directed RNA synthesis (1). We found that variations of the terminal loop sequence, or even modest increases in its size, had no effect on HDV RNA accumulation (Table 1). This could mean that the loop region is not critical for RNA replication and accumulation. However, our studies do not exclude the inter-pretation that this region, independent of its sequence or size, is essential. It is maybe worth noting that early studies of viroid replication demonstrated by electron microscopy the binding of RNA polymerases to the terminal loop regions (14). In contrast to the loop region, we observed that even small changes in the adjacent stem region had major effects on the ability to accumulate HDV RNA (Table 1).
We undertook more extensive studies of certain top mutants with which we had detected an effect. By providing dAg-S in trans, from a separate plasmid, the ability of one top mutant to replicate and accumulate genomes was raised from 1% to $20% (Fig. 4 and 5). Nevertheless, the time course of accu-mulation was still demonstrably slower than for the wild type, suggesting more than one consequence for such a small muta-tion. With another top mutant, one that had a 2-bp insertion in the stem, the increase was from 16% (Table 1) to essentially 100% (45). Our interpretation of the rescuability of such mu-tants is that they suffered from a decrease in the ability to accumulate stable mRNA species needed to act as templates for the translation ofdAg. For example, the mutants may have a reduced ability to initiate RNA species which can go on to be stabilized by the addition of a 59-cap structure and a 39-poly(A) tail.
The mutations that we made near the top were designed to have a minimal effect on the rodlike structure. Nevertheless, when an RNA folding program was applied, changes were predicted in the local folding. For example, the 3-nt external bulge region on the genome, which includes nt 1631, the pu-tative mRNA initiation site (Fig. 1A), was reduced to 2 nt for one mutant (top-flip) and was replaced by 100% double-stranded RNA for another (top-1bp). Such nonpaired regions could be critical sites for the initiation of RNA synthesis. In-triguingly, the 3-nt bulge is very similar to the external bulge on the hepadnavirus pregenomic RNA, at which reverse tran-scription is initiated (41).
ACKNOWLEDGMENTS
J.M.T. was supported by grants AI-26522, AI-31927, and CA-06927 from the National Institutes for Health and by an appropriation from the Commonwealth of Pennsylvania. H.J.N. was supported in part by
FIG. 4. Time course of accumulation of antigenomic RNA in transfected COS7 cells stably expressing dAg-S. Panels A and B show results for cells transfected with cDNAs and in vitro-transcribed RNAs, respectively. The mu-tants used are indicated at the bottom.
FIG. 5. Time course of accumulation of antigenomic RNA in transfected COS7 cells stably expressing dAg-S. Panels A and B show results for cells transfected with cDNAs and in vitro-transcribed RNAs, respectively. The mu-tants used are indicated at the bottom. The (m) designation indicates an addi-tional mutation, a 2-nt deletion at the unique EcoRI site. This was introduced to ensure that the replicating genomes made no HDV-related protein. WT, wild type.
on November 9, 2019 by guest
http://jvi.asm.org/
an award from the Stipendienprogramm Infektionsforschung, BMBF, Bonn, Germany.
Constructive comments on the manuscript were given by William Mason, Glenn Rall, Kate Dingle, and Vadim Bichko. We thank Matt Bockol for assistance with construction of some of the mutants, Anita Cywinski and the DNA Sequencing Facility, and Tony Yeung and DNA Synthesis Facility.
REFERENCES
1. Beard, M. R., T. B. Macnaughton, and E. J. Gowans. 1996. Identification and characterization of a hepatitis delta virus RNA transcriptional promoter. J. Virol. 70:4986–4995.
2. Bichko, V., H. Netter, T.-T. Wu, and J. Taylor. 1994. Pathogenesis associated with replication of hepatitis delta virus. Infect. Agents Dis. 3:94–97. 3. Bichko, V., H. J. Netter, and J. Taylor. 1994. Introduction of hepatitis delta
virus into animal cell lines via cationic liposomes. J. Virol. 68:5247–5252. 4. Bichko, V. V., Y. E. Khudyakov, and J. M. Taylor. 1996. A novel form of
hepatitis delta antigen. J. Virol. 70:3248–3251.
5. Bichko, V. V., and J. M. Taylor. 1996. Redistribution of the delta antigens in cells replicating the genome of hepatitis delta virus. J. Virol. 70:8064–8070. 6. Casey, J. L., K. F. Bergmann, T. L. Brown, and J. L. Gerin. 1992. Structural requirements for RNA editing in hepatitis delta virus: evidence for a uridine-to-cytidine editing mechanism. Proc. Natl. Acad. Sci. USA 89:7149–7153. 7. Casey, J. L., T. L. Brown, E. J. Colan, F. S. Wignall, and J. L. Gerin. 1993.
A genotype of hepatitis D virus that occurs in northern South America. Proc. Natl. Acad. Sci. USA 90:9016–9020.
8. Chao, M. 1991. Ph.D. thesis. University of Pennsylvania, Philadelphia, Pa. 9. Chao, M., S.-Y. Hsieh, and J. Taylor. 1990. Role of two forms of the hepatitis
delta virus antigen: evidence for a mechanism of self-limiting genome rep-lication. J. Virol. 64:5066–5069.
10. Chen, P.-J., G. Kalpana, J. Goldberg, W. Mason, B. Werner, J. Gerin, and
J. Taylor.1986. Structure and replication of the genome of hepatitisdvirus. Proc. Natl. Acad. Sci. USA 83:8774–8778.
11. Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium-thiocyanate-phenol-chloroform extraction. Anal. Bio-chem. 162:156–159.
12. Fu, T.-B., and J. Taylor. 1993. The RNAs of hepatitis delta virus are copied by RNA polymerase II in nuclear homogenates. J. Virol. 67:6965–6972. 13. Gluzman, Y. 1981. SV40-transformed simian cells support the replication of
early SV40 mutants. Cell 23:175–182.
14. Goodman, T. C., L. Nagel, W. Rappold, G. Klotz, and D. Riesner. 1984. Viroid replication: equilibrium association constant and comparative activity measurements for the viroid-polymerase interaction. Nucleic Acids Res.
12:6231–6246.
15. Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467.
16. Hsieh, S.-Y., M. Chao, L. Coates, and J. Taylor. 1990. Hepatitis delta virus genome replication: a polyadenylated mRNA for delta antigen. J. Virol.
64:3192–3198.
17. Hsieh, S.-Y., and J. Taylor. 1992. Delta virus as a vector for the delivery of biologically active RNAs: possibility of a ribozyme specific for chronic hep-atitis B virus infections. Adv. Exp. Med. Biol. 312:125–128.
18. Jeng, K.-S., A. Daniel, and M. M. C. Lai. 1996. A pseudoknot structure is active in vivo and required for hepatitis delta virus RNA replication. J. Virol.
70:2403–2410.
19. Junker-Niepmann, M., R. Bartenschlager, and H. Schaller. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsida-tion and sufficient for packaging of foreign RNA. EMBO J. 9:3389–3396. 20. Kuo, M. Y.-P., M. Chao, and J. Taylor. 1989. Initiation of replication of the
human hepatitis delta virus genome from cloned DNA: role of delta antigen. J. Virol. 63:1945–1950.
21. Kuo, M. Y.-P., J. Goldberg, L. Coates, W. Mason, J. Gerin, and J. Taylor. 1988. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: sequence, structure, and applications. J. Virol. 62:1855– 1861.
22. Kuo, M. Y. P., L. Sharmeen, G. Dinter-Gottlieb, and J. Taylor. 1988. Char-acterization of self-cleaving RNA sequences on the genome and antigenome
of human hepatitis delta virus. J. Virol. 62:4439–4444.
23. Lai, M. M. C. 1995. The molecular biology of hepatitis delta virus. Annu. Rev. Biochem. 64:259–286.
24. Lazinski, D. W., and J. M. Taylor. 1993. Relating structure to function in the hepatitis delta virus antigen. J. Virol. 67:2672–2680.
25. Lazinski, D. W., and J. M. Taylor. 1994. Expression of hepatitis delta virus RNA deletions: cis and trans requirements for self-cleavage, ligation, and RNA packaging. J. Virol. 68:2879–2888.
26. Luo, G., M. Chao, S.-Y. Hsieh, C. Sureau, K. Nishikura, and J. Taylor. 1990. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 64:1021–1027.
27. Macnaughton, T. B., M. R. Beard, M. Chao, E. J. Gowans, and M. M. C. Lai. 1993. Endogenous promoters can direct the transcription of hepatitis delta virus RNA from a recircularized cDNA template. Virology 196:629–636. 28. Macnaughton, T. B., E. J. Gowans, S. P. McNamara, and C. J. Burrell. 1991.
Hepatitis delta antigen is necessary for access of hepatitis delta virus RNA to the cell transcriptional machinery but is not part of the transcriptional complex. Virology 184:387–390.
29. Macnaughton, T. B., and M. M. C. Lai. 1993. Identification of promoters of hepatitis delta virus RNA transcription on hepatitis delta virus cDNA, p. 13–20. In S. J. Hadziyannis, J. M. Taylor, and F. Bonino (ed.), Hepatitis delta virus: molecular biology, pathogenesis, and clinical aspects. Wiley-Liss, New York, N.Y.
30. Macnaughton, T. B., Y.-J. Wang, and M. M. C. Lai. 1993. Replication of hepatitis delta virus RNA: effect of mutations of the autocatalytic cleavage sites. J. Virol. 67:2228–2234.
31. Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chem-ically defined medium. Cancer Res. 42:3858–3863.
32. Netter, H. J., S. Y. Hsieh, D. Lazinski, and J. Taylor. 1993. Modified HDV as a vector for the delivery of biologically-active RNAs, p. 371–375. In S. Hadziyannis, J. Taylor, and F. Bonino (ed.), Hepatitis delta virus: molecular biology, pathogenesis, and clinical aspects. Wiley-Liss, Inc., New York, N.Y. 33. Netter, H. J., T.-T. Wu, M. Bockol, A. Cywinski, W.-S. Ryu, B. C. Tennant,
and J. M. Taylor.1995. Nucleotide sequence stability of the genome of hepatitis delta virus. J. Virol. 69:1687–1692.
34. Perrotta, A. T., and M. D. Been. 1991. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature 350:436–436. 35. Qu, F., C. Heinrich, P. Loss, G. Steger, P. Tien, and D. Riesner. 1993. Multiple pathways of reversion in viroids for conservation of structural elements. EMBO J. 12:2129–2139.
36. Sharmeen, L., M. Y. Kuo, G. Dinter-Gottlieb, and J. Taylor. 1988. The antigenomic RNA of human hepatitis delta virus can undergo self-clavage. J. Virol. 62:2674–2679.
37. Tanner, N. K. 1995. The catalytic RNAs from hepatitis delta virus: structure, function and applications, p. 11–32. In G. Dinter-Gottlieb (ed.), The unique hepatitis delta virus. R. G. Landes Co., Austin, Tex.
38. Tavis, J. E. 1996. The replication strategy of the hepadnaviruses. Viral Hepatitis Rev. 2:205–218.
39. Taylor, J. 1990. Hepatitis delta virus: cis and trans functions needed for replication. Cell 61:371–373.
40. Taylor, J. M., H. J. Netter, and T.-T. Wu. 1995. Potential for the use of modified hepatitis delta virus in the therapy of chronic hepatitis B virus infections. Viral Hepatitis Rev. 1:47–52.
41. Wang, G.-H., and C. Seeger. 1993. Novel mechanism for reverse transcrip-tion in hepatitis B viruses. J. Virol. 67:6507–6512.
42. Wang, K.-S., Q.-L. Choo, A. J. Weiner, J.-H. Ou, C. Najarian, R. M. Thayer,
G. T. Mullenbach, K. J. Denniston, J. L. Gerin, and M. Houghton.1986. Structure, sequence and expression of the hepatitis delta viral genome. Nature 323:508–513.
43. Weiner, A. J., Q.-L. Choo, K.-S. Wang, S. Govindarajan, A. G. Redeker, J. L.
Gerin, and M. Houghton.1988. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta anti-gen polypeptides p24 and p27. J. Virol. 62:594–599.
44. Wu, T.-T., V. V. Bichko, W.-S. Ryu, S. M. Lemon, and J. M. Taylor. 1995. Hepatitis delta virus mutant: effect on RNA editing. J. Virol. 69:7226–7231. 45. Wu, T.-T., H. J. Netter, D. W. Lazinski, and J. M. Taylor. 1996. Unpublished
observations.