Copyright © 1998, American Society for Microbiology
A Lymph Node-Derived Cytopathic Simian Immunodeficiency
Virus Mne Variant Replicates in Nonstimulated
Peripheral Blood Mononuclear Cells
JASON T. KIMATA, AFSANEH MOZAFFARIAN,ANDJULIE OVERBAUGH*
Department of Microbiology, University of Washington, Seattle, Washington 98195
Received 15 November 1996/Accepted 6 October 1997
Lymph nodes (LNs) are sites of active human immunodeficiency virus type 1 (HIV-1) and simian immuno-deficiency virus (SIV) replication and disease at both early and late stages of infection. Consequently, variant viruses that replicate efficiently and subsequently cause immune dysfunction may be harbored in this tissue. To determine whether LN-associated SIVs have an increased capacity to replicate and induce cytopathology, a molecular clone of SIV was isolated directly from DNA extracted from unpassaged LN tissue of a pig-tailed
macaque (Macaca nemestrina) infected with SIVMne. The animal had declining CD41T-lymphocyte counts at
the time of the LN biopsy. In human CD41T-cell lines, the LN-derived virus, SIVMne027, replicated with
relatively slow kinetics and was minimally cytopathic and non-syncytium inducing compared to other SIVMne clones. However, in phytohemagglutinin-stimulated pig-tailed macaque peripheral blood mononuclear cells
(PBMCs), SIVMne027 replicated efficiently and was highly cytopathic for the CD41T-cell population.
Inter-estingly, unlike other SIVMne clones, SIVMne027 also replicated to a high level in nonstimulated macaque PBMCs. High-level replication depended on the presence of both the T-cell and monocyte/macrophage popu-lations and could be enhanced by interleukin-2 (IL-2). Finally, the primary determinant governing the ability of SIVMne027 to replicate in nonstimulated and IL-2-stimulated PBMCs mapped to gag-pol-vif. Together, these data demonstrate that LNs may harbor non-syncytium-inducing, cytopathic viruses that replicate efficiently and are highly responsive to the effects of cytokines such as IL-2.
In both human immunodeficiency virus type 1 (HIV-1)- and simian immunodeficiency virus (SIV)-infected individuals, high viral load and generalized immune activation herald CD41T-cell decline and progression to AIDS (22, 36, 42, 49,
54). This correlation most likely reflects the fact that these lentiviruses require cellular activation signals to productively infect CD41T lymphocytes (7, 46, 57, 64). It also raises the
possibility that chronic immune stimulation plays a central role in HIV and SIV pathogenesis. Support for this hypothesis comes from studies of HIV-1 that have demonstrated transient increases in plasma viremia and proviral burden in infected individuals immunized against hepatitis B or tetanus toxoid, or having an intercurrent secondary infection (11, 13, 55), and from recent studies showing that continuous rounds of virus infection and replication drive rapid CD41T-cell turnover (28,
62). On the other hand, the persistent replication of HIV and SIV that occurs throughout the course of infection could be the primary driving force underlying the maintenance of a chronic immune activation state (22). In fact, a unique strain of SIVsmm, SIVPBj14, causes massive T-cell activation both in vivo and in vitro, demonstrating that a lentivirus is capable of inducing an immune activation state (52). Together, these data not only demonstrate the importance of virus-host interactions in the regulation of HIV and SIV replication but also suggest that a determinant of virulence may be defined by a virus’s ability to induce and utilize immune activation signals for ef-ficient replication.
The requirement for cellular activation signals in the initia-tion and maintenance of HIV-1 and SIV replicainitia-tion is well
established in vitro (7, 45, 46, 57, 64), but the mechanism by which the virus interacts with cellular signalling pathways to enhance viral replication remains poorly understood. In in-fected T cells, activation signals (e.g., via T-cell receptor/CD3 and CD28 and cytokine receptors) release the arrest in viral replication that occurs at postentry steps prior to integration of the provirus, including reverse transcription and nuclear trans-location of the viral preintegration complex (7, 45, 46, 57, 64). Furthermore, in productively infected cells, cellular activation is important for upregulating viral transcription from the long terminal repeat (LTR) (2, 27, 29). Additionally, there is evi-dence that HIV and SIV proteins such as envelope (Env) gp120 and Nef can promote or suppress viral replication through the induction of cytokines or modulation of T-cell activation, respectively (6, 20, 26, 37). In the context of these data, virulence may potentially be influenced by genetic vari-ation in viral determinants that affect infectivity, postentry steps in the virus life cycle, and transcription.
Genetic variation has been shown to influence the pheno-type of HIV-1 (34). Indeed cytopathic variants of HIV-1 that emerge in the peripheral blood (PB) during the course of infection may play an active role in determining the rate of disease progression. These viruses are associated with an in-crease in viral load, accelerated CD41T-cell decline, and onset
of AIDS; they are frequently able to infect T-cell lines, and they may be rapidly replicating, highly cytopathic, and syncy-tium inducing (T-tropic, rapid-high/SI) in tissue culture (1, 12, 14, 15, 23, 59, 60). By contrast, viruses isolated early after infection, when CD41 T-cell counts are stable or slowly
de-clining, are commonly macrophage-tropic, slowly replicating, minimally cytopathic, and non-syncytium inducing (M-tropic, slow-low/NSI) (1, 12, 14, 15, 23, 59, 60). We have observed a similar shift in the phenotype of viruses isolated from the PB of SIVMne-infected macaques at early and late stages of infec-* Corresponding author. Mailing address: Department of
Microbi-ology, Box 357242, University of Washington, Seattle, WA 98195-7242. Phone: (206) 543-3146. Fax: (206) 543-8297. E-mail: overbaug@u .washington.edu.
245
on November 9, 2019 by guest
http://jvi.asm.org/
tion (50). Together, these data have led to the hypothesis that T-tropic, rapid-high/SI viruses are more pathogenic than M-tropic, slow-low/NSI viruses. However, immune dysfunction is evident early in infection (53), when the virus population tends to be M-tropic, slow-low/NSI, suggesting that these viruses may also have an impact on disease progression.
Immune activation in HIV-1- and SIV-infected individuals is prominent in secondary lymphoid tissues such as lymph nodes (LNs), which serve as major reservoirs for the viruses (10, 21, 22, 44). In LNs, manifestations of disease are evident at early and late stages of infection (10, 22, 44), perhaps due to viral replication associated with chronic immune stimulation within this tissue. Moreover, it has been shown, at least for SIV, that the predominant variants harbored in lymphoid tissue are ge-netically distinguishable from those found in PB (8). While there is a high level of viral replication and evidence for disease in LNs of HIV-1- or SIV-infected individuals, primary full-length LN-derived molecular variants of HIV-1 or SIV have not been directly cloned (i.e., without prior selection in cul-ture) and characterized. Thus, in this study we describe the direct cloning and in vitro characterization of a LN-derived molecular clone of SIVMne.
MATERIALS AND METHODS
Isolation of a SIVMne molecular clone from LN tissue.DNA was extracted from mesenteric LN tissue of a pig-tailed macaque (Macaca nemestrina, animal T78027) infected with an uncloned isolate of SIVMne (5). This isolate repro-ducibly causes an AIDS-like syndrome in pig-tailed and rhesus (M. mulatta) macaques in 1 to 3 years (5). The mesenteric LN tissue was biopsied at 16 months postinfection when the animal had declining CD41T-cell counts and early signs of AIDS. The DNA was digested with EcoRI, layered onto a 10 to 40% contin-uous sucrose gradient in STE buffer (1 M NaCl, 20 mM Tris [pH 8], 5 mM EDTA [pH 8]), and fractionated by centrifugation at 26,000 rpm for 20 h at 15°C in a Beckman (Fullerton, Calif.) SW41.1 rotor. Fractions containing DNA fragments 10 to 20 kb in size were concentrated by ethanol precipitation, ligated into the EcoRI sites of thelDash II arms, and packaged by using the Gigapack II XL system as specified by the manufacturer (Stratagene, La Jolla, Calif.). Approxi-mately 107plaques were screened for SIV, using32P-labeled gag and env DNA probes derived from a pathogenic clone of SIVMne (SIVMneCl8 [4, 38, 43]). Plaques that scored positive for both probes were isolated and purified. One phage clone encoded a full-length provirus which we designated SIVMne027. The provirus was excised from thelDash II vector with EcoRI and ligated into the EcoRI site of pUC18. The full DNA sequence of SIVMne027 was deter-mined by manual sequencing using Sequenase version 2.0 (United States Bio-chemical, Cleveland, Ohio) as well as an ABI automated sequencer.
Construction of recombinant viruses.To construct a chimeric virus containing the 59half of SIVMne027 (R-U5-gag-pol-59vif) and 39half of SIVMneCl8 (39 vif-vpx-vpr-rev-tat-env-nef-U3-R) or the reciprocal recombinant virus, BstBI-SalI fragments from the plasmid proviral clones of SIVMne027 and SIVMneCl8 (pMneCl8) were exchanged. BstBI cleaves both proviruses in vif (position 5343, which is 536 bp into the vif gene), and SalI cuts in the polylinker region of both proviral plasmid clones at a site downstream of the cellular sequences flanking the 39LTR of each provirus. The chimeric virus that consisted of the 59half of SIVMne027 and 39half of SIVMneCl8 was designated SIVMne027/Cl8, and the reciprocal virus was designated SIVMneCl8/027.
Generation of virus stocks.Ten million CEMx174 cells were transfected with 10mg of each plasmid proviral construct by the DEAE-dextran method and cultured for 10 days in RPMI complete medium (RPMI 1640 supplemented with 10% heat-inactivated [56°C for 30 min] fetal bovine serum, 2 mML-glutamine,
penicillin [100 U/ml], streptomycin [100mg/ml], and amphotericin B [250 ng/ ml]). Conditioned supernatants were clarified by centrifugation at 1,500 rpm in a Beckman clinical centrifuge, filtered through 0.22-mm-pore-size filters (Milli-pore, Bedford, Mass.), and stored in 1-ml aliquots at270°C. The tissue culture infectious dose (TCID) of each virus supernatant stock per milliliter was deter-mined by the sMAGI assay (9). Briefly, sMAGI indicator cells were infected with 1 to 50ml of viral supernatant and stained forb-galactosidase expression 3 days postinfection (p.i.) as previously described (9).
Infection of human CD41cell lines.The human CD41T-cell lines Jurkat, CEM, MT4, and Molt4 clone 8, and a CD41T-B-cell hybrid cell line, CEMx174, were maintained in RPMI complete medium. To examine viral tropism for these cell lines, triplicate cultures of 53105cells from each line were infected with 1,000 TCID of each virus derived from the molecular clones of SIVMne and propagated in 2 ml of RPMI complete medium. Every 3 days, the total cell number in each culture was adjusted to 53105, and fresh medium was added. If there were cytopathic effects in a culture that reduced the total cell number
below 53105, then no cells were removed, but the culture medium was replaced with 2 ml of fresh medium. At 6, 12, and 18 days p.i., 1 ml of supernatant from each culture was removed and stored at270°C. Viral replication was assessed by examining the supernatants for cell-free SIV p27gagby antigen enzyme-linked
immunosorbent assay (ELISA) specific for the SIV p27gagcapsid protein
(Im-munotech, Westbrook, Maine). Cultures were scored positive for viral replica-tion if the cell-free supernatants taken at 12 and 18 days p.i. were positive for SIV p27gag(.50 pg/ml).
Replication and cytopathicity of SIVMne variants in CEMx174 cells. To compare the replication rates and cytopathicity of the different viruses in the CEMx174 cell line, duplicate cultures of 83105CEMx174 cells were infected with 200 TCID of each virus for 4 h in RPMI complete medium. Following the incubation, the cells were pelleted by centrifugation, washed twice with phos-phate-buffered saline (PBS) to remove residual free virions, and resuspended in 2 ml of fresh RPMI complete medium. Every 2 to 3 days, both the viable and total cell numbers were determined by trypan blue dye exclusion, and the total syncytia per culture were counted by visual inspection. Only syncytia larger than 5 cell diameters were scored. Viral replication was monitored by assaying dilu-tions of the culture supernatants for cell-free SIV p27gagby antigen ELISA. All
p27gagvalues were obtained in the linear range of the assay. The cell number in
each culture was adjusted to 83105, and fresh RPMI complete medium was added to a final volume of 2 ml. If the cell number was below 83105, then no cells were removed, but the culture medium was replaced with 2 ml of fresh RPMI complete medium.
Isolation and infection of macaque PBMCs, monocytes/macrophages, and T cells.Macaque PB mononuclear cells (PBMCs) were isolated from whole blood of SIV- and simian type D retrovirus-negative pig-tailed macaques (M. nemest-rina) by Ficoll-Hypaque centrifugation as previously described (50). To examine viral replication in nonstimulated PBMCs (i.e., in the absence of exogenous mitogens) or interleukin-2 (IL-2)-stimulated PBMCs, 3 3106PBMCs were infected with 6,000 TCID of each virus in duplicate in 1 ml of RPMI complete medium. Following a 24-h incubation, the cells were pelleted by centrifugation, washed twice with PBS, and resuspended in RPMI complete medium. Nonstimu-lated PBMC cultures were grown continuously in the absence of exogenous IL-2, while IL-2-stimulated PBMC cultures were grown continuously in the presence of exogenous IL-2 (20 U/ml) for the duration of the experiment. Every 3 to 4 days, supernatants were harvested, stored at270°C, and used to monitor viral replication by antigen ELISA for p27gag.
To examine viral replication in PBMCs prestimulated with phytohemaggluti-nin (PHA), PBMCs were cultured with 10mg of PHA-P (Difco Laboratories, Detroit, Mich.) per ml in RPMI complete medium for 3 days. The cells were pelleted by centrifugation and washed with RPMI complete medium to remove the PHA, and duplicate cultures of 23106cells were infected with 2,000 TCID of each virus in 1 ml of RPMI complete medium plus 20 U of recombinant human IL-2 (Boehringer Mannheim, Indianapolis, Ind.) per ml. The following day, the cells were pelleted, washed with PBS twice to remove residual cell-free virions, and resuspended in RPMI complete medium supplemented with 20 U of IL-2 per ml. Every 3 days, culture supernatant was removed and replaced with fresh RPMI complete plus IL-2 (20 U/ml). The supernatants were stored at 270°C and used to monitor virus replication by assaying for cell-free SIV p27gag
antigen by ELISA.
To analyze the cytopathicity of the viruses for CD41T cells, 43106PBMCs were infected with each virus at a multiplicity of infection (MOI) ranging from 0.001 to 0.1. Over a 14-day period, the percentage of CD41T cells in each of the cultures was monitored by fluorescence-activated cell sorting (FACS) analysis using a Becton Dickinson FACScan. For the analysis, 30,000 viable cells were counted. Forward and side scatter light characteristics were used to exclude dead cells from the analysis. Anti-human CD4 and anti-human CD8 monoclonal antibodies (Becton Dickinson, San Jose, Calif.) that cross-react with macaque CD4 and CD8, respectively, were used for enumeration in addition to an anti-macaque CD3 monoclonal antibody obtained from Biosource International (Camarillo, Calif.).
Monocytes/macrophages were isolated from macaque PBMCs by adherence to plastic tissue culture flasks (Corning Glass Works, Corning, N.Y.), cultured for 5 days in macrophage medium (RPMI 1640 supplemented with 10% heat-inactivated [56°C, 30 min] human AB serum, 5% heat-heat-inactivated fetal bovine serum, 10% GCT conditioned medium [obtained from the AIDS Research Reagent and Reference Program], 2 mM glutamine, penicillin [100 U/ml], strep-tomycin [100 mg/ml], and amphotericin B [250 ng/ml]), and infected as previ-ously described (50). Duplicate cultures were infected with each virus at an MOI ranging from 0.001 to 0.1. Viral replication was monitored by assaying superna-tants taken at 3- to 4-day intervals p.i. for the presence of SIV p27gagby ELISA.
Enriched populations of primary macaque T cells were isolated from PBMCs by a modification of the method used by Polacino et al. (46). Macaque PBMCs were first cultured for 2 h in RPMI complete medium, using Corning T75 flasks to remove the adherent population of cells. The nonadherent cell population was concentrated by centrifugation at 1,200 rpm for 5 min, resuspended in serum-free RPMI 1640 medium, loaded onto a 30%/40%/60% Percoll step gradient, and spun at 2,900 rpm in a Beckman clinical centrifuge for 20 min at 4°C. Cells at the 40%/60% interphase were recovered, washed twice with PBS, and resus-pended in RPMI complete medium. By FACS analysis, this cell population contained mainly T cells (.98% CD31). The T cells (1.53106/culture) were
on November 9, 2019 by guest
http://jvi.asm.org/
infected in 1 ml of RPMI complete medium with each virus at an MOI of 0.001. After 24 h, the cells were pelleted by centrifugation, washed twice with PBS, and resuspended in 2 ml of RPMI complete medium. Every 3 days, 1 ml of condi-tioned supernatant from each culture was removed and stored at270°C to monitor viral replication by ELISA for SIV p27gag.
Nucleotide sequence accession number.The complete nucleotide sequence of SIVMne027 was entered into GenBank under accession no. U79412.
RESULTS
Isolation of an infectious molecular clone of SIVMne from
LN tissue.To study the properties of viruses found in LNs, we
first isolated a proviral clone directly from unpassaged LN tissue of a pig-tailed macaque (animal T78027) that had been infected with an uncloned, pathogenic isolate of SIVMne (5). Macaque T78027 had been infected with SIVMne for 16 months and had declining CD41T-cell counts and early signs
of AIDS at the time of the LN biopsy. Using recombinant lambda phage cloning, we obtained a single full-length proviral variant of SIVMne, designated SIVMne027, from this animal’s LN DNA sample. CEMx174 cells transfected with the proviral clone of SIVMne027 expressed virus that was infectious for macaque and human cells (Table 1). The host cell range of SIVMne027 was similar to that of SIVMneCl8, a pathogenic virus that was cloned from the SIVMne isolate that was inoc-ulated into macaque T78027 (4, 38, 43). SIVMne027 replicated in pig-tailed macaque PBMCs and to low levels in macaque monocytes/macrophages. It also replicated in the human CD41cell lines CEMx174 and MT4. However, the host range
of SIVMne027 was different from those of uncloned mixtures of late variant viruses previously isolated from PBMCs of other pig-tailed macaques with AIDS. These uncloned mixtures of late variants had an expanded host range for CD41human cell
lines that included Molt4 clone 8, but they replicated poorly or not at all in monocyte/macrophage cultures (Table 1 and ref-erence 50).
SIVMne027 is rapidly replicating and highly cytopathic but
non-syncytium inducing. We previously demonstrated that
CEMx174 cells are highly sensitive to the cytopathic effects of SIV and can be used as an indicator for the identification of rapid-high/SI variants of SIVMne (50). To determine the bio-logical characteristics of SIVMne027, we compared its repli-cation kinetics, cytopathicity, and syncytium-inducing ability in CEMx174 cells with those of SIVMneCl8, a slow-low/NSI virus (50), and SIVMne170, a rapid-high/SI virus molecularly de-rived from an uncloned mixture of late variant viruses isolated by cocultivation from PBMCs (31). The phenotype of SIVM neCl8 is typical of viruses present early in infection, while the phenotype of SIVMne170 is representative of variants found
late in infection (50). When CEMx174 cells were infected with each virus at a low MOI (0.00025), SIVMne027 replicated with delayed kinetics compared to SIVMneCl8 and SIVMne170, reaching a peak SIV p27gagantigen later than SIVMneCl8 and
SIVMne170, respectively (Fig. 1A). Furthermore, SIVMne027 was minimally cytopathic for CEMx174 cells, a phenotype sim-ilar to that of SIVMneC18 (Fig. 1B and C); specifically, it reduced the viable cell number fivefold and percentage of cells that were viable to 40%. In contrast, the rapid-high/SI virus, SIVMne170, reduced the viable cell number approximately 100-fold and the percentage of viable cells to less than 10%. Increasing the MOI to 0.025 or greater did not enhance the cytopathicity of SIVMne027 (data not shown). Finally, like SIVMneC18, SIVMne027 induced few syncytia compared to SIVMne170 (Fig. 1D). Syncytia were not observed in any other infected cell lines, including MT4 (data not shown). By these criteria, SIVMne027 is a slow-low/NSI variant of SIVMne.
Differences in the replication kinetics and cytopathicity of SIV and HIV-1 have been elucidated in cultures of PBMCs prestimulated with PHA (1, 12, 14–16, 23, 58–60, 63). Thus, to further characterize SIVMne027, we examined its phenotype in PHA-stimulated macaque PBMCs. SIVMne027 replicated to a 50-fold-higher level than SIVMneCl8 (Fig. 2A), and it replicated with the most rapid kinetics of any SIVMne variant examined to date (data not shown). Furthermore, the CD41
T-cell (CD31CD41) population in infected PBMC cultures
was decreased to a greater extent by SIVMne027 (37% at day 6 and 90% at day 13 p.i.) than SIVMneCl8 (11% at day 11 and 31% at day 14) infection (Fig. 2B). The decrease in CD41cells
in the infected cultures was not simply due to down regulation of CD4 expression on the surface of cells. It was more likely caused by a depletion of the CD31 CD41 cell population
because almost all of the CD31 CD42 cell population
ex-pressed CD8 (data not shown). These observations were con-firmed in assays using PBMCs isolated from a second macaque (Fig. 2C and D). In PBMCs from this animal, SIVMneCl8 replicated to a fivefold-higher level than in the PBMCs from the first donor. However, SIVMneCl8’s maximum p27gaglevel
was still 15-fold lower than that achieved by SIVMne027, and its highest level of CD41T-cell killing was also significantly
less than that for SIVMne027. Similar results were obtained with different MOIs over a 100-fold range and in multiple independent experiments using PBMCs isolated from two other pig-tailed macaques (data not shown). In all experi-ments, SIVMne027 was consistently more cytopathic than SIVMneCl8. However, the extent of cell killing was different for each experiment and was dependent on the donor PBMCs as well as the MOI. The decrease in the CD41T-cell
popula-tion at 2 weeks p.i. ranged from 10 to 50% for SIVMneCl8-infected PBMCs and from 65 to 92% for SIVMne027-SIVMneCl8-infected PBMCs. Last, syncytia were not observed in any of the infected PBMC cultures (data not shown). Together, these data dem-onstrate that SIVMne027 is a non-syncytium-inducing virus in both macaque PBMCs and a CD41cell line, but it is rapidly
replicating in macaque PBMCs and is highly cytopathic for the CD41 T-cell population. Additionally, they suggest that an
increase in the replication kinetics and cytopathicity of SIV can evolve independent of the syncytium-inducing property.
SIVMne027 replicates in nonstimulated PBMCs. A few
studies have investigated the ability of HIV-1 and SIV to rep-licate in leukocyte cultures or PBMCs that had not been stim-ulated with potent mitogens such as PHA or concanavalin A (17, 20, 25, 32, 33, 47, 48, 61). To determine the efficiency of replication of SIVMne027 in PBMC cultures that had not been prestimulated with exogenous mitogens, we compared the rep-lication kinetics of SIVMne027 and SIVMneCl8 in pig-tailed TABLE 1. Replication and tropism of SIVMne027a
Virus
Viral replication
PBMC mMf CEMx174 Jurkat CEM MT4 Molt4clone 8
SIVMneCl8 1 1 1 2 2 1 2
SIVMne027 1 1 1 2 2 1 2
SIVMne variants 1 1/2 1 NT NT NT 1
aThe cultures were scored positive (1) for viral replication if, during the
3-week culture period, the cell-free supernatants were positive for SIV p27gagby
antigen ELISA. A minus sign indicates that SIV p27gagwas not detected at any
time point during the culture period. NT, not tested. PBMC and monocyte-derived macrophages (mMf) were prepared from SIV- and simian type D retrovirus-negative pig-tailed macaques. The replication and tropism of the un-cloned SIVMne variant mixture (M87004 170 week [PBMC]) were previously reported (50) but are shown for comparison.
on November 9, 2019 by guest
http://jvi.asm.org/
macaque PBMCs in the presence or absence of exogenous IL-2. Interestingly, SIVMne027 replicated to significant levels in the nonstimulated PBMC cultures, whereas SIVMneCl8 replicated poorly (Fig. 3A and C). The addition of IL-2 at 24 h p.i. enhanced the production of p27gag in the
SIVMne027-infected cultures 5- to 20-fold. In contrast, while the produc-tion of p27gag in the SIVMneCl8-infected cultures was also
increased by IL-2, the highest level of p27gagwas still 100- to
200-fold less than that attained by SIVMne027 (Fig. 3B and D). We observed these characteristics in seven independent experiments using PBMCs from three different pig-tailed ma-caques (data not shown). Furthermore, the PB-derived SIVMne variant clone, SIVMne170, behaved like SIVMneCl8 in nonstimulated PBMC cultures, demonstrating that among variants of the SIVMne strain, the ability to replicate in non-stimulated PBMCs was unique to SIVMne027 (data not shown). These data demonstrate that SIVMne027 may have an increased capacity to replicate in PBMCs, and they also show that this virus is highly responsive to signals induced by IL-2. Additionally, the ability of SIVMne027 to replicate in non-stimulated PBMCs appeared to occur without the induction of cellular proliferation because we could not detect a differ-ence in cell number between infected and uninfected control PBMCs by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide)] colorimetric assay. Furthermore, like infection with other variants of SIVMne, infection with
SIVMne027 did not increase expression of the T-cell activation marker CD69 or CD25. These data suggest that virus produc-tion may not involve CD31lymphocytes. However, selection
for CD31cells from SIVMne027-infected PBMC cultures by
immunomagnetic bead separation using an anti-macaque CD3 monoclonal antibody and subsequent analysis of the CD31
cells by antigen ELISA for p27gagor PCR for proviral DNA
demonstrated that the T-cell population was productively in-fected (data not shown).
Comparison of the replication kinetics of SIVMne027 and
other SIV clones.To further characterize SIVMne027, we
com-pared its ability to replicate in nonstimulated PBMCs with those of a molecular clone of the immunostimulatory viral isolate, SIVPBj14 (clone SIVPBj1.9 [16]), and a highly mac-rophage-tropic clone, SIVmac1A11 (3), as well as SIVMneCl8. SIVMne027, SIVPBj1.9, and SIVmac1A11 replicated to sub-stantial levels in nonstimulated PBMCs compared to SIVM neCl8 as measured by SIV p27gagantigen production (Fig. 4A).
The peak SIV p27gagantigen level achieved in the
SIVMne027-and SIVmac1A11-infected cultures was approximately 10,000 pg/ml, while the SIVPBj14-infected cultures produced a sev-enfold-higher maximal level of antigen (72,000 pg/ml). In con-trast, the SIVMneCl8-infected cultures reached a maximum SIV p27gaglevel at 300 pg/ml.
To further characterize the phenotype SIVMne027, we ex-amined whether the virus could replicate in cultures enriched FIG. 1. Replication and cytopathicity of SIVMne027 in CEMx174 cells. Eight hundred thousand CEMx174 cells were infected with 200 TCID of virus. At 2-day intervals p.i., supernatants were harvested to monitor viral replication by antigen ELISA for SIV p27gag. The viable cell number was determined by trypan blue dye
exclusion, and syncytia were scored as described in Materials and Methods. (A) SIV p27gaglevels versus days p.i.; (B) extrapolated viable cell number versus days p.i.;
(C) percentage of the total number of cells that are viable versus days p.i.; (D) total number of syncytia versus days p.i. The value shown for each time point is the average of duplicate cultures. Similar data were obtained in three independent experiments.
on November 9, 2019 by guest
http://jvi.asm.org/
for macaque resting T lymphocytes or monocyte-derived mac-rophages. Virus replication was low in resting T-cell cultures infected with SIVMne027, similar to the cultures infected with SIVmac1A11, while SIVMneCl8 replication was undetectable (Fig. 4B). By contrast, SIVPBj1.9 efficiently replicated in rest-ing T-cell cultures in the absence of the monocyte/macrophage population. In monocyte-derived macrophage cultures, SIVMne027 replicated to a low level, three- to fourfold lower than the level achieved by SIVMneCl8 (Fig. 4C). On the other hand, both SIVmac1A11 and SIVPBj1.9 replicated efficiently and to high levels in these monocyte-derived macrophage cul-tures, as previously demonstrated by Banapour et al. (3) and Fletcher et al. (24), respectively. Together, these data demon-strate that SIVMne027’s requirements for replication in non-stimulated PBMC cultures are different from those of SIVPBj, which replicates efficiently in both resting T cells and mono-cytes/macrophages, and SIVmac1A11, which efficiently repli-cates in monocytes/macrophages. SIVMne027, instead, re-quires both the T-cell and monocyte/macrophage populations for high-level replication in nonstimulated macaque PBMCs, suggesting that T-cell–macrophage interactions are important for stimulating SIVMne027 replication.
Genetic comparison of SIVMne027 with other SIVs. We
determined the sequence of the entire SIVMne027 genome and compared the predicted amino acid sequences of the gene products with the homologous regions of SIVMneCl8, SIVPBj14 (16), SIVmac1A11 (35), and a prototype
AIDS-inducing SIV clone, SIVmac239 (30, 35) (Table 2). SIVMne027 encodes a complete open reading frame for each gene. SIVMne027 had the strongest sequence similarity to SIVMneCl8; the percentage of amino acid differences ranged from a low value of 0.7% in reverse transcriptase (RT) and integrase (IN) to a high value of 4.0 to 5.3% in Vpr, Tat, Rev, and Nef. In contrast, SIVMne027 differed overall from SIVPBj14 by 12.7%. SIVMne027 was most similar to SIVPBj14 in IN (3.8% different) and most different in Env, Vif, Tat, Rev, and Nef (17.3 to 24.6%). Our sequence analysis also revealed that SIVMne027 did not have an additional Src homology 2 (SH2) binding motif (YXXL) in Nef or a dupli-cation of the NF-kB binding site in the U3 LTR region (data not shown), two of the known determinants of SIVPBj14 that contribute to its ability to replicate in nonstimulated PBMCs (16–19). Additionally, SIVMne027 differed from SIVmac239 and SIVmac1A11 by a greater percentage than SIVMneCl8 in each viral protein, except Vpx, which was identical. However, both SIVmac239 and SIVmac1A11 had fewer amino acid dif-ferences with SIVMne027 than did SIVPBj14. Finally, SIVMne027 was also found to be genetically distinguishable from other clones of SIV and HIV-1 (data not shown).
Replication of SIVMne027 in nonstimulated PBMCs
re-quires a determinant located within the 5* half of the viral
genome.To identify the determinant in SIVMne027 that
con-fers the ability to replicate in nonstimulated PBMC cultures, we constructed reciprocal recombinant proviruses that ex-FIG. 2. Replication and cytopathicity of SIVMne027 in PHA-stimulated macaque PBMCs. (A and C) SIV p27gaglevels versus days p.i. Two million PHA-stimulated
PBMCs were infected with 2,000 TCID of virus in duplicate. Virus production was monitored by antigen ELISA for SIV p27gag. The average antigen value for the
duplicate cultures at each time point is shown, and the viruses used for infection are indicated. (B and D) Decrease in CD41T cells versus days p.i. Four million PHA-stimulated PBMCs were infected with 40,000 TCID of virus. The decrease in CD41T cells at the indicated time point (6 and 13 days p.i.) was monitored by FACS analysis as described in Materials and Methods. The values in panels B and D represent the decrease in the percentage of CD31CD41cells in the virus-infected cultures relative to the uninfected culture at each time point.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.126.477.70.379.2]change the 59and 39regions of SIVMne027 and the parental virus, SIVMneCl8 (Fig. 5A), and compared the abilities of viruses derived from these constructs to replicate in nonstim-ulated and IL-2-stimnonstim-ulated PBMCs. When nonstimnonstim-ulated PBMCs were infected with these viruses, similar patterns of replication were observed in the cultures infected with the wild-type SIVMne027 and the chimeric virus, SIVMne027/Cl8, which encodes the R-U5-gag-pol-59vif region of SIVMne027
and 39vif-vpx-vpr-tat-rev-env-nef-U3-R region from SIVMneCl8
(Fig. 5B). In contrast, the reciprocal clone, SIVMneCl8/027, re-sembled SIVMneCl8 in that it did not show an appreciable level of virus replication as measured by p27gag expression.
Furthermore, the SIVMne027/Cl8 chimeric virus was respon-sive to the addition of IL-2. However, its peak production of p27gag was approximately threefold lower than that of
SIVMne027 (Fig. 5C). The SIVMneCl8/027 chimera, like SIVMneCl8, produced only a low level amount of p27gagunder
these conditions. These results indicate that the 39 region of SIVMne027, including nef and the U3-R region of the LTR, does not contain the primary determinants for replication in nonstimulated or IL-2-stimulated PMBCs. Instead, the pri-mary determinant(s) that confers SIVMne027’s ability to rep-licate in nonstimulated and IL-2-stimulated PBMCs lies in a region of the 59half of the virus, which includes U5-gag-pol and the 59end of vif.
The U5, Gag, Pol, and amino-terminal Vif sequences from SIVMne027 and SIVMneCl8 were further analyzed to
deter-mine more specifically which 59 region of SIVMne027 may contain the determinant conferring the ability to replicate in nonstimulated and IL-2-stimulated PBMCs. The analysis re-vealed that the SIVMne027 and SIVMneCl8 U5 sequences and the untranslated region upstream of the gag translational initiation codon were highly conserved. There were four nu-cleotide differences, all were located in U5; none was in the primer binding site (data not shown). Pol was also highly con-served (99.1% identical) (Fig. 6 and Table 2). Only one of the amino acid differences (position 573 in RT, Lys to Arg) oc-curred at a position that is conserved among the SIVs, includ-ing SIVMneCl8 (Fig. 6 and reference 39). On the other hand, the Gag sequence of SIVMne027 differed from that of SIVMne-Cl8 by 2.5% and contained conservative and nonconservative amino acid differences at nine positions (amino acids 48, 63, 132, 182, 276, 286, 287, 362, and 492) that are conserved among other SIVs, including SIVMneCl8 (Fig. 7 and reference 39). Finally, while there were four amino acid differences (Glu-64, Tyr-85, Tyr-104, and Asn-143) between SIVMne027 and SIVMneCl8 in the amino-terminal region of Vif, a comparison with the Vif protein of other SIVs revealed that none of the mutations were unique to SIVMne027 (data not shown).
To examine whether the determinants in SIVMne027 that enhanced replication were also responsible for its cytopathic-ity, we also examined the cytopathic properties of the recom-binant viruses in PHA-stimulated PBMCs (Fig. 8). Both chi-meric viruses, SIVMne027/Cl8 and SIVMneCl8/027, were less FIG. 3. Replication of SIVMne027 in nonstimulated and IL-2-stimulated pig-tailed macaque PBMCs. Three million nonstimulated PBMCs were infected with 6,000 TCID of virus and cultured in the presence or absence of exogenous IL-2. Virus production was monitored by antigen ELISA for SIV p27gag. Each antigen value is
the average of duplicate cultures. (A and C) Virus production from nonstimulated PBMCs. (B and D) Virus production from IL-2-stimulated PBMCs. IL-2 was added 24 h p.i. and was maintained at 20 U/ml for the duration of the experiment. Panels A and B and panels C and D represent independent experiments using PBMCs prepared from different pig-tailed macaques.
on November 9, 2019 by guest
http://jvi.asm.org/
cytopathic than the parent virus SIVMne027, which reduced the CD41T-cell population by 35 and 63% after 7 and 14 days
of infection, respectively, in this experiment. SIVMne027/Cl8 depleted 27% of the CD41T cells by 7 days postinfection; by
day 14, this value had increased to only 31%. The reciprocal virus SIVMneCl8/027 killed 9% of the CD41T-cell population
after 7 days of infection and 35% by 14 days p.i. Although both chimeric viruses were less cytopathic than SIVMne027, they were more cytopathic than the parent virus, SIVMneCl8, which reduced the CD41 T-cell population by 10 and 20%
after 7 and 14 days of infection, respectively. Thus, determi-nants in both halves of SIVMne027 may contribute to its cy-topathicity.
DISCUSSION
The predominant SIV variants in lymphoid tissue have been shown to be genetically different from those found in the pe-ripheral blood of infected macaques (8). To begin to evaluate the contribution of LN-derived lentivirus variants to viral pathogenesis, we molecularly cloned a variant of SIV directly from DNA prepared from unpassaged LN tissue of a pig-tailed macaque inoculated with SIVMne. Importantly, this is the first infectious HIV-1 or SIV molecular clone isolated directly from unpassaged LN tissue. The virus, SIVMne027, displayed a distinct phenotype in culture compared to other SIVMne clones. SIVMne027 was macrophage-tropic, replicated poorly in T-cell lines, and was non-syncytium inducing. However, it replicated efficiently in PHA-stimulated PBMCs and was highly cytopathic for the CD41 T-cell population.
Interest-ingly, SIVMne027 also replicated efficiently in nonstimulated PBMCs in the absence of potent mitogens (e.g., PHA or con-canavalin A), and it was responsive to IL-2 under these culture conditions. These data demonstrate that in SIV- and, by im-plication, HIV-1-infected individuals, LNs may harbor cyto-pathic variant viruses with an increased ability to utilize, and perhaps deregulate, cellular activation signals that are typically required for viral replication.
[image:7.612.51.289.63.591.2]We previously demonstrated that SIVMne, like HIV-1, evolves from a slow-low/NSI virus to a rapid-high/SI virus pop-ulation during the course of an infection (50). Interestingly, the primary determinant that conferred the rapid replication ki-netics and increased cytopathic effects of a rapid-high/SI SIVMne variant molecular clone mapped to gag, but not the syncytium induction determinant, which mapped to the enve-lope surface protein coding region (31, 51), suggesting that rapidly replicating, highly cytopathic SIVMne variants may FIG. 4. Comparison of the replication kinetics of SIVMne027 and of other
strains of SIV in nonstimulated macaque PBMCs and macaque T-cell-enriched cultures. (A) Virus production from nonstimulated macaque PBMCs. The in-fection and analysis were performed as described in the legend to Fig. 3. (B) Virus production from primary macaque T-cell cultures. Cultures of T cells were infected with each virus at an MOI of 0.001. (C) Virus production from macaque monocyte-derived macrophage cultures. Monocyte-derived macrophage cultures were infected with each virus at an MOI of 0.001. For panels B and C, virus production was monitored by SIV p27gagantigen ELISA, and cultures were
maintained as described in Materials and Methods.
TABLE 2. Percentages of amino acid differences between SIVMne027 and other SIVs
Viral protein % Amino acid difference
SIVMneCl8 SIVmac239 SIVmac1A11 SIVPBj14
Gag 2.5 6.8 6.5 8.4
Pol
Protease 2.8 2.8 6.6 12.0
RT 0.7 2.1 2.9 5.5
IN 0.7 2.7 3.1 3.8
Env 2.9a 8.5 8.8a 19.2
Vif 2.3 6.5 7.0 17.3
Vpx 1.0 0 0 9.8
Vpr 4.0 6.9 11.0a 8.9
Tat 4.6 11.5 14.5 24.6
Rev 4.7 13.1 14.0 22.4
Nef 5.3 13.3a 14.8 22.1
aThe protein is predicted to be truncated due to a premature stop codon in
the sequence of the gene. The premature stop codons were considered single mutations, and the predicted amino acid sequences encoded downstream from these stop codons were included in the calculations.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.309.549.87.234.2]evolve independent of the syncytium-inducing phenotype. The data presented here demonstrate that a SIVMne variant, SIVMne027, can be rapidly replicating and highly cytopathic in the absence of an overt syncytium-inducing phenotype. The experimental inoculation of macaques with SIVMne027 will allow us to directly examine whether the increased in vitro virulence of SIVMne027 is predictive of increased in vivo pathogenicity. Furthermore, comparative studies of SIVMne-Cl8 and SIVMne027 may allow us to further define the viral genetic determinants and in vitro biological characteristics of virulence. In regard to the latter, it is noteworthy that the chimeric viruses, SIVMne027/Cl8 and SIVMneCl8/027, were less cytopathic for CD41T cells in PHA-stimulated PBMCs
and replicated less efficiently than the parent virus SIVMne027 in IL-2-stimulated PBMCs, demonstrating that determinants
in both halves of the virus may contribute to its overall viru-lence.
Unlike other SIVMne clones, SIVMne027 replicated effi-ciently in nonstimulated and IL-2-stimulated PBMCs. SIVMne-027’s ability to replicate in nonstimulated PBMCs initially ap-peared to be similar to that of the immunostimulatory virus, SIVPBj14. However, SIVMne027 was distinguishable from SIVPBj14 by several criteria. First, in contrast to the molecular clone of SIVPBj14, SIVPBj1.9, SIVMne027 did not replicate well in either macaque resting T-cell or monocyte-derived macrophage-enriched cultures; instead, it required the pres-ence of both cell populations for efficient replication. Second, SIVMne027 did not contain either a duplication of the NF-kB site in the U3 region of the LTR or an additional YXXL motif in the amino-terminal region of Nef, two mutations that have been shown to be critical for the phenotype of SIVPBj14 (16– 20). We further verified that mutations in these regions of SIVMne027 were not the primary determinants conferring the ability to replicate in nonstimulated PBMCs with recombinant viruses. Third, SIVMne027 was slowly replicating and mini-mally cytopathic in CEMx174 cells, whereas SIVPBj14 has been shown to be highly cytopathic for this cell line (16). Fourth, while SIVPBj14 is able to induce T-cell proliferation (25), we could not detect an increase in lymphocyte prolifera-tion in SIVMne027-infected PBMCs by the MTT assay in preliminary experiments. We also could not detect an up reg-ulation of the T-cell activation markers CD69 and CD25 on CD41T cells in SIVMne027-infected cultures. Nevertheless,
we did find infected CD31 lymphocytes, demonstrating the
importance of T cells in SIVMne027 replication in nonstimu-lated PBMC cultures. These data suggest that SIVMne027 may either induce immune activation signals that are sufficient for viral replication but not cellular proliferation or bypass the requirement for T-cell activation. However, IL-2 enhanced SIVMne027 production from infected PBMCs. This observa-tion indirectly suggests that the SIVMne027-infected cells may be activated, because resting T cells are refractory to the effects of IL-2 (56). Our failure to detect T-cell activation in SIVMne027 infected-cultures by other methods, such as FACS, may be explained by a high rate of turnover of produc-tively infected CD31CD41T cells. This interpretation is
con-sistent with our data demonstrating that SIVMne027 is highly cytopathic for the CD31CD41T cells. Together, these data
demonstrate that SIVMne027 is genetically and phenotypically distinct from SIVPBj14.
Cell-cell interactions play an important role in regulating lentivirus replication. Several recent studies have demon-strated that efficient replication of both SIV and HIV-1 re-quires contact between mononuclear phagocytes or antigen-presenting cells, such as macrophages or dendritic cells, and T cells (32, 33, 47, 48, 61). Furthermore, introduction of an additional SH2 binding domain into the amino-terminal re-gion of Nef of SIVmac239 results in a virus (SIVmac239YE) that is capable of replicating in nonstimulated PBMCs in a macrophage-dependent manner (20). We demonstrate here that SIVMne027, like the SIVmac239YE mutant (20) but unlike SIVPBj1.9, depends on monocytes/macrophages for efficient replication in nonstimulated PBMCs. SIVMne027 also appeared to resemble the macrophage-tropic virus, SIVmac1A11, in its ability to replicate in nonstimulated PB-MCs because SIVmac1A11 required the monocyte/macro-phage population. However, SIVMne027 differed from SIV-mac1A11 because it replicated poorly in cultures enriched for monocyte-derived macrophages, whereas SIVmac1A11 repli-cated to a high level. Indeed, replication of SIVmac1A11 in nonstimulated PBMCs could be explained entirely by macro-FIG. 5. Construction and analysis of SIVMne027 and SIVMneCl8 chimeric
viruses. (A) Schematic diagram of the chimeric viruses. B, the BstBI restriction site used for generation of the chimeric viruses. (B) Virus production from nonstimulated PBMCs. (C) Virus production from IL-2-stimulated PBMCs. For panels B and C, infection of PBMCs and the analysis of virus production were carried out as described in the legend to Fig. 3 and Materials and Methods.
on November 9, 2019 by guest
http://jvi.asm.org/
phage infection. By contrast, the level of SIVMne027 replica-tion in monocyte-derived macaque macrophages is too low to account for the high-level replication in nonstimulated ma-caque PBMCs. Therefore, it seems unlikely that replication of SIVMne027 in the monocyte/macrophage population alone can fully explain its ability to replicate to high levels in non-stimulated PBMCs. In support of this interpretation, we dem-onstrated here and have previously shown that both SIVM-neCl8 and SIVMne170 replicated with kinetics similar to those of SIVMne027 in monocyte-derived macrophage cultures (31, 50, 51), but neither virus replicated to appreciable levels in either nonstimulated or IL-2-stimulated PBMCs, further sug-gesting that macrophage infection is insufficient for viral
rep-lication in nonstimulated or IL-2-stimulated PBMCs. A model consistent with our data is that SIVMne027 has a greater capacity to utilize activation signals resulting from mononu-clear phagocyte–T-cell interactions for replication than the other molecular variants of SIVMne. Although the significance of the SIVMne027 phenotype is unclear, the selection for vi-ruses during infection that are highly responsive to cellular activation signals seems plausible, especially because persistent SIV and HIV-1 replication correlates with immune activation and disease progression (22, 36, 42, 49, 54), productive infec-tion of CD41T cells requires activation signals (7, 46, 57, 64),
[image:9.612.83.505.64.543.2]and vaccinations or secondary infections elevate viral loads in HIV-1-infected individuals (11, 13, 55).
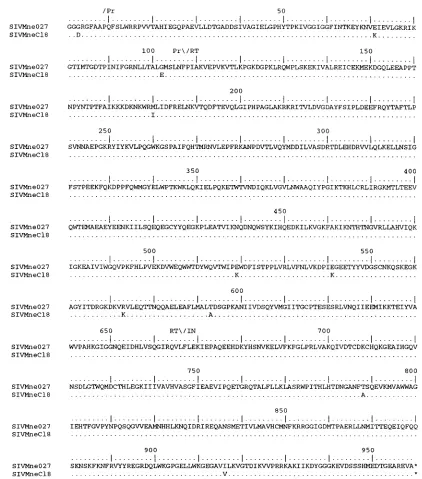
FIG. 6. Comparison of the predicted amino acid sequences of Pol from SIVMne027 and SIVMneCl8. The predicted amino acid sequence of SIVMne027 Pol serves as the reference. The regions encoding protease (Pr), RT, and IN are shown above the sequences. Similarities and differences between SIVMne027 Pol and the other SIV Pol proteins are shown with the same notation as described in the legend to Fig. 5.
on November 9, 2019 by guest
http://jvi.asm.org/
The primary determinant(s) conferring the ability of SIVMne027 to replicate in nonstimulated and IL-2-stimulated PBMCs lies within gag-pol-59vif. While we have not
[image:10.612.86.508.64.449.2]identi-fied the specific mutation(s) that determine the ability of SIVMne027 to replicate in nonstimulated and IL-2-stimulated PBMCs in a functional assay, a U5, Pol, or Vif determinant seems unlikely because these regions are highly conserved be-tween SIVMne027 and SIVMneCl8. On the other hand, the Gag polyprotein of SIVMne027 encodes nine unique muta-tions that distinguish it from SIVMneCl8 and other SIVs, mak-ing it a more likely candidate for the determinant that confers SIVMne027’s ability to replicate in nonstimulated and IL-2-stimulated PBMCs. Interestingly, Novembre et al. showed, us-ing recombinant viruses between SIVPBj clones and the closely related clones SIVsmmH4 and SIVsmmH9, that muta-tions in gag and the central region of the proviral genome which encodes for regulatory genes affect the phenotype of SIVPBj14 (40, 41). Our data further underscore the influence that mutations selected in genes located in the 59 half of the viral genome exert on viral replication. Identification of the specific mutations and the mechanism of action will be impor-tant for understanding how variation in gag, pol, or vif affects FIG. 7. Comparison of the predicted amino acid sequences of Gag from SIVMne027 and SIVMneCl8. The regions coding for MA, CA, and NC are indicated above the sequences. Amino acid similarities and differences are noted as described in the legend to Fig. 5.
FIG. 8. Cytopathicity of the chimeric viruses for PHA-stimulated PBMCs. Four million PBMCs prestimulated with PHA for 3 days were infected with 40,000 TCID and cultured in RPMI complete medium and IL-2 (20 U/ml). Cytopathicity of the viruses for CD41T cells was monitored by FACS as de-scribed in Materials and Methods.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.53.289.522.681.2]viral replication in vitro and the impact that this may have on virus replication and pathology in vivo.
ACKNOWLEDGMENTS
We thank Jen Rohn and Mary Poss for critical reviews of the manu-script. The CEMx174 cell line and GCT conditioned medium were obtained from the AIDS Research Reagent and Reference Program. Virus stocks of SIVmac1A11 were kindly provided by Marta Marthas. This work was supported by NIH grant RO1 AI34251. J.T.K. was supported in part by NIH training grants T32 CA09229 and T32 AI07140 and NRSA individual postdoctoral fellowship F32 AI09337. J.O. is a scholar of the Leukemia Society.
REFERENCES
1. Åsjo¨, B., J. Albert, A. Karlsson, L. Morfeldt-Manson, B. Biberfeld, K.
Lid-man, and E. M. Fenyo¨.1986. Replicative capacity of human immunodefi-ciency virus from patients with varying severity of HIV infection. Lancet
ii:660–662.
2. Åsjo¨, B., D. Cefai, P. Debre, Y. Dudoit, and B. Autran. 1993. A novel mode of human immunodeficiency virus type 1 (HIV-1) activation: ligation of CD28 alone induces HIV-1 replication in naturally infected lymphocytes. J. Virol. 67:4395–4398.
3. Banapour, B., M. L. Marthas, R. J. Munn, and P. A. Luciw. 1991. In vitro macrophage tropism of pathogenic and nonpathogenic molecular clones of simian immunodeficiency virus (SIVMAC). Virology 183:12–19.
4. Benveniste, R. E., R. W. Hill, L. J. Eron, U. M. Csaikl, W. B. Knott, L. E.
Henderson, R. C. Sowder, K. Nagashima, and M. A. Gonda.1990. Charac-terization of clones of HIV-1 infected HuT 78 cells defective in gag gene processing and of SIV clones producing large amounts of envelope glyco-protein. J. Med. Primatol. 19:351–366.
5. Benveniste, R. E., W. R. Morton, E. A. Clark, C.-C. Tsai, H. D. Ochs, J. M.
Ward, L. Kuller, W. B. Knott, R. W. Hill, M. J. Gale, and M. E. Thouless.
1988. Inoculation of baboons and macaques with simian immunodeficiency virus/mne, a primate lentivirus closely related to human immunodeficiency virus type 2. J. Virol. 62:2091–2101.
6. Borghi, P., L. Fantuzzi, B. Varano, S. Gessani, P. Puddu, L. Conti, M.
Rosaria Capobianchi, F. Ameglio, and F. Belardelli. 1995. Induction of interleukin-10 by human immunodeficiency virus type 1 and its gp120 protein in human monocytes/macrophages. J. Virol. 69:1284–1287.
7. Bukrinsky, M. I., T. L. Stanwick, and M. Stevenson. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV1 infection. Science 254: 423–427.
8. Campbell, B. J., and V. M. Hirsch. 1994. Extensive envelope heterogeneity of simian immunodeficiency virus in tissues from infected macaques. J. Virol.
68:3129–3137.
9. Chackerian, B., N. L. Haigwood, and J. Overbaugh. 1995. Characterization of a CD4-expressing macaque cell line that can detect virus after a single replication cycle and can be infected by diverse simian immunodeficiency virus isolates. Virology 213:386–394.
10. Chakrabarti, L., M.-C. Cumont, L. Montagnier, and B. Hurtrel. 1994. Vari-able course of primary simian immunodeficiency virus infection in lymph nodes: relation to disease progression. J. Virol. 68:6634–6642.
11. Cheeseman, S. H., R. E. Davaro, and R. T. Ellison III. 1996. Hepatitis B vaccination and plasma HIV-1 RNA. N. Engl. J. Med. 334:1272. 12. Cheng-Mayer, C., D. Seto, M. Tateno, and J. A. Levy. 1988. Biologic features
of HIV-1 that correlate with virulence in the host. Science 240:80–82. 13. Claydon, E. J., J. Bennett, D. Gor, and S. M. Forster. 1991. Transient
elevation of serum HIV antigen levels associated with intercurrent infection. AIDS 5:113–114.
14. Connor, R. I., and D. D. Ho. 1994. Human immunodeficiency virus type 1 variants with increased replicative capacity develop during the asymptomatic stage before disease progression. J. Virol. 68:4400–4408.
15. Connor, R. I., H. Mohri, Y. Cao, and D. D. Ho. 1993. Increased viral burden and cytopathicity correlate temporally with CD41T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected indi-viduals. J. Virol. 67:1772–1777.
16. Dewhurst, S., J. E. Embretson, D. C. Anderson, J. I. Mullins, and P. N. Fultz. 1990. Sequence analysis and acute pathogenicity of molecularly cloned SIVSMM-PBj14. Nature 345:636–640.
17. Dittmar, M. T., K. Cichutek, P. N. Fultz, and R. Kurth. 1995. The U3 promoter region of the acutely lethal simian immunodeficiency virus clone smmPBj1.9 confers related biological activity on the apathogenic clone agm3mc. Proc. Natl. Acad. Sci. USA 92:1362–1366.
18. Dollard, S. C., S. Gummuluru, S. Tsang, P. N. Fultz, and S. Dewhurst. 1994. Enhanced responsiveness to nuclear factorkB contributes to the unique phenotype of simian immunodeficiency virus variant SIVsmmPBj14. J. Virol.
68:7800–7809.
19. Du, Z., P. O. Ilyinskii, V. G. Sasseville, M. Newstein, A. A. Lackner, and R. C.
Desrosiers.1996. Requirements for lymphocyte activation by unusual strains
of simian immunodeficiency virus. J. Virol. 70:4157–4161.
20. Du, Z., S. M. Lang, V. G. Sasseville, A. A. Lackner, P. O. Ilyinskii, M. D.
Daniel, J. U. Jung, and R. C. Desrosiers.1995. Identification of a nef allele that causes lymphocyte activation and acute disease in macaque monkeys. Cell 82:665–674.
21. Embretson, J., M. Zupancic, J. L. Ribas, A. Burke, P. Racz, K. Tenner-Racz,
and A. T. Haase.1993. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 362: 359–362.
22. Fauci, A. S. 1993. Multifactorial nature of human immunodeficiency virus disease: implications for therapy. Science 262:1011–1018.
23. Fenyo¨, E. M., L. Morfeldt-Manson, F. Chiodi, B. Lind, A. von Gegerfelt, J.
Albert, E. Olausson, and B. Åsjo¨.1988. Distinct replicative and cytopathic characteristics of human immunodeficiency virus isolates. J. Virol. 62:4414– 4419.
24. Fletcher, T. M., III, B. Brichacek, N. Sharova, M. A. Newman, G. Stivahtis,
P. M. Sharp, M. Emerman, B. H. Hahn, and M. Stevenson.1996. Nuclear import and cell cycle arrest functions of the HIV-1 Vpr protein are encoded by two separate genes in HIV-2/SIVsm. EMBO J. 15:6155–6165. 25. Fultz, P. N. 1991. Replication of an acutely lethal simian immunodeficiency
virus activates and induces proliferation of lymphocytes. J. Virol. 65:4902– 4909.
26. Gessani, S., P. Puddu, B. Varano, P. Borghi, L. Conti, L. Fantuzzi, and F.
Belardelli.1994. Induction of beta interferon by human immunodeficiency virus type 1 and its gp120 protein in human monocytes-macrophages: role of beta interferon in restriction of virus replication. J. Virol. 68:1983–1986. 27. Hannibal, M. C., D. M. Markovitz, N. Clark, and G. J. Nabel. 1993.
Differ-ential activation of human immunodeficiency virus type 1 and 2 transcription by specific T-cell activation signals. J. Virol. 67:5035–5040.
28. Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M.
Markowitz.1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126.
29. Jeang, K. T., and A. Gatignol. 1994. Comparison of regulatory features among primate lentiviruses. Current Top. Microbiol. Immunol. 188:123–144. 30. Kestler, H., T. Kodama, D. Ringler, M. Marthas, N. Pedersen, A. Lackner,
D. Regier, P. Sehgal, M. Daniel, N. King, and R. Desrosiers.1990. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science 248:1109–1112.
31. Kimata, J. T., and J. Overbaugh. 1997. The cytopathicity of a simian immu-nodeficiency virus Mne variant is determined by mutations in Gag and Env. J. Virol. 71:7629–7639.
32. Kinter, A. L., G. Poli, L. Fox, E. Hardy, and A. S. Fauci. 1995. HIV repli-cation in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. J. Immunol.
154:2448–2459.
33. Knight, S. C. 1996. Bone-marrow-derived dendritic cells and the pathogen-esis of AIDS. AIDS 10:807–817.
34. Levy, J. A. 1993. Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev. 57:183–289.
35. Luciw, P. A., K. E. S. Shaw, R. E. Unger, V. Planelles, M. W. Stout, J. E.
Lackner, E. Pratt-Lowe, N. J. Leung, B. Banapour, and M. L. Marthas.1992. Genetic and biological comparisons of pathogenic and nonpathogenic mo-lecular clones of simian immunodeficiency virus (SIVMAC). AIDS Res. Hum. Retroviruses 8:395–402.
36. Mellors, J. W., C. R. Rinaldo, Jr., P. Gupta, R. M. White, J. A. Todd, and
L. A. Kingsley.1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272:1167–1170.
37. Merrill, J. E., Y. Koyanagi, and I. S. Y. Chen. 1989. Interleukin-1 and tumor necrosis factor a can be induced from mononuclear phagocytes by human immunodeficiency virus type 1 binding to the CD4 receptor. J. Virol. 63: 4404–4408.
38. Morton, W. R., R. E. Benveniste, E. A. Clark, C.-C. Tsai, M. J. Gale, M. E.
Thouless, J. Overbaugh, and M. G. Katze.1989. Transmission of the simian immunodeficiency virus SIVmne in macaques and baboons. J. Med. Prima-tol. 18:237–245.
39. Myers, G., B. H. Hahn, J. W. Mellors, L. E. Henderson, B. Korber, K. T.
Jeang, F. E. McCutchan, and G. N. Pavlakis (ed.).1995. Human retroviruses and AIDS. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, N.Mex.
40. Novembre, F. J., P. R. Johnson, M. G. Lewis, D. C. Anderson, S. Klump,
H. M. McClure, and V. M. Hirsch.1993. Multiple viral determinants con-tribute to pathogenicity of the acutely lethal simian immunodeficiency virus SIVsmmPBj variant. J. Virol. 67:2466–2474.
41. Novembre, F. J., M. M. Saucier, V. M. Hirsch, P. R. Johnson, and H. M.
McClure.1994. Viral genetic determinants in SIVsmmPBj pathogenesis. J. Med. Primatol. 23:136–145.
42. O’Brien, W. A., P. M. Hartigan, D. Martin, J. Esinhart, A. Hill, S. Benoit, M.
Rubin, M. S. Simberkoff, J. D. Hamilton, and V. A. C. S. G. O. AIDS.1996. Changes in plasma HIV-1 RNA and CD41lymphocyte counts and the risk of progression to AIDS. N. Engl. J. Med. 334:426–431.
43. Overbaugh, J., L. M. Rudensey, M. D. Papenhausen, R. E. Benveniste, and
W. R. Morton.1991. Variation in simian immunodeficiency virus env is
on November 9, 2019 by guest
http://jvi.asm.org/
confined to V1 and V4 during progression to simian AIDS. J. Virol. 65: 7025–7031.
44. Pantaleo, G., C. Graziosi, J. F. Demarest, L. Butini, M. Montroni, C. H. Fox,
J. M. Orenstein, D. P. Kotler, and A. S. Fauci.1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of dis-ease. Nature 362:355–358.
45. Polacino, P. S., H. A. Liang, and E. A. Clark. 1995. Formation of simian immunodeficiency virus long terminal repeat circles in resting T cells re-quires both T cell receptor- and IL-2-dependent activation. J. Exp. Med.
182:617–621.
46. Polacino, P. S., H. A. Liang, E. J. Firpo, and E. A. Clark. 1993. T-cell activation influences initial DNA synthesis of simian immunodeficiency virus in resting T lymphocytes from macaques. J. Virol. 67:7008–7016. 47. Pope, M., M. G. H. Betjes, N. Romani, H. Hirmand, P. U. Cameron, L.
Hoffman, S. Gezelter, G. Schuler, and R. M. Steinman.1994. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell 78:389–398.
48. Pope, M., D. Elmore, D. Ho, and P. Marx. 1997. Dendritic cell-T cell mix-tures, isolated from the skin and mucosae of macaques, support the repli-cation of SIV. AIDS Res. Hum. Retroviruses 13:819–827.
49. Popov, J., T. McGraw, B. Hofmann, B. Vowels, A. Shum, P. Nishanian, and
J. L. Fahey.1992. Acute lymphoid changes and ongoing immune activation in SIV infection. J. Acquired Immune Defic. Syndr. 5:391–399.
50. Rudensey, L. M., J. T. Kimata, R. E. Benveniste, and J. Overbaugh. 1995. Progression to AIDS in macaques is associated with changes in the replica-tion, tropism, and cytopathic properties of the simian immunodeficiency virus variant population. Virology 207:528–542.
51. Rudensey, L. M., J. T. Kimata, E. M. Long, B. Chackerian, and J.
Over-baugh.1998. Changes in the extracellular envelope glycoprotein of variants that evolve during the course of simian immunodeficiency virus SIVMne infection affect neutralizing antibody recognition, syncytium formation, and macrophage tropism but not replication, cytopathicity, or CCR-5 coreceptor recognition. J. Virol. 72:209–217.
52. Schwiebert, R., and P. N. Fultz. 1994. Immune activation and viral burden in acute disease induced by simian immunodeficiency virus SIVsmmPBj14: correlation between in vitro and in vivo events. J. Virol. 68:5538–5547. 53. Shearer, G. M., and M. Clerici. 1991. Early T-helper cell defects in HIV
infection. AIDS 5:245–253.
54. Sheppard, H. W., and M. S. Ascher. 1992. The natural history and patho-genesis of HIV infection. Annu. Rev. Microbiol. 46:533–564.
55. Stanley, S. K., M. A. Ostrowski, J. S. Justement, K. Gantt, S. Hedayati, M.
Mannix, K. Roche, D. J. Schwartzentruber, C. H. Fox, and A. S. Fauci.1996. Effect of immunization with a common recall antigen on viral expression in patients infected with human immunodeficiency virus type 1. N. Engl. J. Med. 334:1222–1230.
56. Stern, J. B., and K. A. Smith. 1986. Interleukin-2 induction of T-cell G1 progression and c-myb expression. Science 233:203–206.
57. Stevenson, M., T. L. Stanwick, M. P. Dempsey, and C. A. Lamonica. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 9:1551–1560.
58. Tao, B., and P. N. Fultz. 1995. Molecular and biological analyses of quasi-species during evolution of a virulent simian immunodeficiency virus, SIVsmmPBj14. J. Virol. 69:2031–2037.
59. Tersmette, M., R. E. Y. De Goede, B. J. M. Al, I. N. Winkel, R. A. Gruters,
H. T. Cuypers, H. G. Huisman, and F. Miedema.1988. Differential syncyti-um-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immuno-deficiency syndrome (AIDS) and AIDS-related complex. J. Virol. 62:2026– 2032.
60. Tersmette, M., R. A. Gruters, F. De Wolf, R. E. Y. De Goede, J. M. A. Lange,
P. T. A. Schellekens, J. Goudsmit, H. G. Huisman, and F. Miedema.1989. Evidence for a role of virulent human immunodeficiency virus (HIV) vari-ants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J. Virol. 63:2118–2125.
61. Tsunetsugu-Yokota, Y., K. Akagawa, H. Kimoto, K. Suzuki, M. Iwasaki, S.
Yasuda, G. Hausser, C. Hultgren, A. Meyerhans, and T. Takemori.1995. Monocyte-derived cultured dendritic cells are susceptible to human immu-nodeficiency virus infection and transmit virus to resting T cells in the process of nominal antigen presentation. J. Virol. 69:4544–4547. 62. Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch,
J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, M. S. Saag, and G. M. Shaw.1995. Viral dynamics in human immunodeficiency virus type 1 infec-tion. Nature 373:117–122.
63. Yu, X., M. F. McLane, L. Ratner, W. O’Brien, R. Collman, M. Essex, and
T.-H. Lee.1994. Killing of primary CD41T cells by non-syncytium-inducing macrophage-tropic human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 91:10237–10241.
64. Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Y.
Chen.1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals labile, latent viral structure. Cell 61:213–222.