JOURNAL OFVIROLOGY,
0022-538X/99/$04.00⫹0 Sept. 1999, p. 7641–7657 Vol. 73, No. 9
Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Localization of Mouse Hepatitis Virus Nonstructural Proteins
and RNA Synthesis Indicates a Role for Late
Endosomes in Viral Replication
YVONNEVAN DERMEER,1ERIC J. SNIJDER,1JESSIKA C. DOBBE,1SIBYLLE SCHLEICH,2 MARK R. DENISON,3,4,5WILLY J. M. SPAAN,1ANDJACOMINE KRIJNSE LOCKER2*
Department of Virology, Leiden University Medical Center, 2300 RC Leiden, The Netherlands1; EMBL, 69117
Heidelberg, Germany2; and Department of Pediatrics,3Department of Microbiology and Immunology,4
and the Elizabeth B. Lamb Center for Pediatric Research,5Vanderbilt University
Medical Center, Nashville, Tennessee 37232
Received 29 March 1999/Accepted 8 June 1999
The aim of the present study was to define the site of replication of the coronavirus mouse hepatitis virus (MHV). Antibodies directed against several proteins derived from the gene 1 polyprotein, including the 3C-like protease (3CLpro), the putative polymerase (POL), helicase, and a recently described protein (p22) derived from the C terminus of the open reading frame 1a protein (CT1a), were used to probe MHV-infected cells by indirect immunofluorescence (IF) and electron microscopy (EM). At early times of infection, all of these proteins showed a distinct punctate labeling by IF. Antibodies to the nucleocapsid protein also displayed a punctate labeling that largely colocalized with the replicase proteins. When infected cells were metabolically labeled with 5-bromouridine 5ⴕ-triphosphate (BrUTP), the site of viral RNA synthesis was shown by IF to colocalize with CT1a and the 3CLpro. As shown by EM, CT1a localized to LAMP-1 positive late endosomes/ lysosomes while POL accumulated predominantly in multilayered structures with the appearance of endocytic carrier vesicles. These latter structures were also labeled to some extent with both anti-CT1a and LAMP-1 antibodies and could be filled with fluid phase endocytic tracers. When EM was used to determine sites of BrUTP incorporation into viral RNA at early times of infection, the viral RNA localized to late endosomal membranes as well. These results demonstrate that MHV replication occurs on late endosomal membranes and that several nonstructural proteins derived from the gene 1 polyprotein may participate in the formation and function of the viral replication complexes.
The replication complex of viruses which multiply in the cytoplasm of the host cell has been described to be associated with either the cytoskeleton (35, 36, 39) or the cytosolic surface of intracellular membranes. The latter possibility appears to be preferred by positive-stranded RNA viruses (see, e.g., refer-ences 5, 9, 13, 18, 23, 33, 60, 64, 65, 68, 79, and 86). The replication of the poliovirus genome, for example, occurs on vesicular structures which are derived from intracellular mem-branes (8), possibly by a process which resembles autophagy (67). In the case of alphaviruses, there appears to be a rela-tionship between virus entry (via receptor-mediated endosis) and the formation of the replication complex on the cyto-solic surface of endocytic organelles (23). Recently, the RNA synthesis of the arterivirus equine arteritis virus (EAV) was also shown to occur on modified intracellular membranes, which are derived from the endoplasmic reticulum (ER) or intermediate compartment (IC) (60, 79).
The coronavirus mouse hepatitis virus (MHV) is a positive-stranded RNA virus with a genome of about 32 kb. The rep-lication strategy of coronaviruses is clearly related to that of arteriviruses (21, 72, 74), and the two families have recently been united in the new orderNidovirales(14). This name refers to one of the key features of the life cycle of corona- and arteriviruses, the discontinuous transcription of an extensive nested set of subgenomic mRNAs. The replicase genes of both families consist of two large open reading frames (ORFs),
ORF1a and ORF1b, of which the latter is expressed upon ribosomal frameshifting (12, 19). The ORF1a and ORF1ab translation products are large nonstructural polyprotein pre-cursors. Despite an enormous size difference, the organization of arteri- and coronaviruses is quite similar; e.g., their (puta-tive) RNA polymerase (POL) and helicase (HEL) activities are located in the ORF1b-encoded part of the protein (19, 26, 81). Two main functions of the ORF1a-encoded replicase part are proteolytic processing (see below) and membrane associ-ation (60, 81). Most nidovirus replicase subunits are thought to assemble into an RNA-protein complex which is presumably held together by protein-protein and/or protein-RNA interac-tions (66, 79, 81). Hydrophobic domains in the ORF1a protein have been proposed to mediate the membrane association of this complex, but how targeting to and recognition of mem-branes occur remains largely unknown.
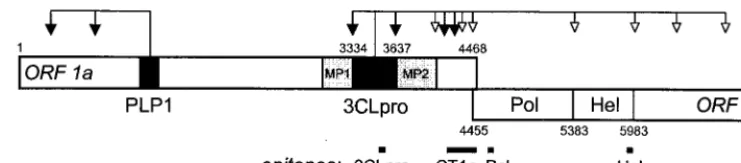
An apparently complete replicase processing map has been obtained for the arterivirus EAV (81, 82, 84). Our understand-ing of the proteolytic processunderstand-ing of the much larger coronavi-rus ORF1a and ORF1ab polyproteins is also rapidly increasing (Fig. 1). In the N-terminal half of the ORF1a protein of co-rona- and arteriviruses, multiple papain-like cysteine protease domains (PLPs) have been identified. In coronaviruses, the proteolytic activity of one of these (PLP-1) has been demon-strated and characterized in detail (3, 10, 22, 37, 40). The functionality of the second PLP (PLP-2) (49) remains to be proven. The most important nonstructural protease of both nidovirus families is located in the central region of the ORF1a polyprotein. These enzymes, the 3C-like cysteine protease (3CLpro) of coronaviruses (50, 52, 88) and the nsp4 serine * Corresponding author. Mailing address: EMBL, Meyerhofstrasse
1, 69117 Heidelberg, Germany. Phone: 49 6221 387508. Fax: 49 6221 387306 or 387512. E-mail: [email protected].
7641
on November 9, 2019 by guest
http://jvi.asm.org/
protease of arteriviruses (71, 82), belong to the chymotrypsin-like protease superfamily and are both flanked by hydrophobic sequences. The latter (called MP1 and MP2 in the case of MHV) have been proposed to function as membrane anchors for the coronavirus protease, since the activity of the 3CLpro is greatly enhanced in the presence of membranes (53, 62, 66, 78). The region between MP2 and the C terminus of the ORF1a protein of MHV contains two proven and three puta-tive 3CLpro cleavage sites. Recently, a 22-kDa cleavage prod-uct from this region of the MHV replicase was identified in infected cells and shown to be generated by the activity of 3CLpro (51).
Because of the striking similarities between the replication cycles of corona- and arteriviruses, it was tempting to speculate that coronavirus RNA synthesis might also occur on mem-branes of the ER or IC. Indeed, several studies that relied on indirect immunofluorescence (IF) microscopy as well as bio-chemical techniques suggested that the replication of corona-viruses is membrane bound (11, 38, 66, 68). In addition, it has recently been shown that the putative helicase of MHV local-izes to cytoplasmic complexes that are also the site of MHV RNA synthesis (20). However, the precise intracellular loca-tion of coronavirus replicaloca-tion complexes has until now re-mained an open question. In this study, we analyzed the sub-cellular localization of a number of MHV replicase subunits by indirect IF and electron microscopy (EM). Furthermore, we used 5-bromouridine 5⬘-triphosphate (BrUTP) for metabolic in situ labeling of newly synthesized viral RNA. Surprisingly, our combined data strongly suggest that MHV replication oc-curs on late endosomal and/or lysosomal membranes.
MATERIALS AND METHODS
Cells, viruses, and antibodies.Mouse L cells, Sac⫺cells, and DBT cells were grown in Dulbecco’s modified Eagle’s medium (Life Technologies) containing 10% fetal calf serum (FCS) and antibiotics. MHV-A59 and MHV temperature-sensitive (ts) mutantts379 were propagated in L cells as described previously (44, 75).
Antisera directed against four subunits of the MHV replicase were used in this study. New rabbit antisera were raised by using synthetic peptides as described by Snijder et al. (73). A serum directed against the 3CLpro-containing subunit p27 (hereafter referred to as anti-3CLpro serum) was raised by using a peptide representing residues 3517 to 3533 of the ORF1a polyprotein. Antiserum anti-CT1a (previously referred to as B4) was raised by using a bacterial fusion protein (containing residues 4032 to 4460 of the ORF1a protein) and has been described previously (51). Antisera recognizing the putative RNA polymerase (anti-POL) and helicase (anti-HEL) subunits were raised using peptides corresponding to residues 4517 to 4532 and 5965 to 5982 of the ORF1ab protein, respectively. Testing of antisera by indirect IF assays included the appropriate controls using mock-infected cells or cell lysates and the preimmunization serum. The anti-3CLpro serum detected anti-3CLpro in MHV-infected DBT cells with a sensitivity and specificity similar to those of the previously described SP9 and B3 sera (52, 53). The anti-HEL serum (96.8) has recently been described and has been used to define the expression, processing, and localization of the putative HEL in MHV-infected DBT cells (20). It detects a 67-kDa cleavage product of the ORF1ab polyprotein.
Antibodies to protein disulfide isomerase (PDI) mouse monoclonal antibody (MAb 1D3) were provided by S. Fuller (EMBL, Heidelberg, Germany [83]). Anti-mannosidase II rabbit antibodies were from K. Moremen (58), and the anti-SEC13 antibodies were from W. Hong (77). The anti-LAMP-1 and -2 rat MAbs (1D4B and ABL-93, respectively) were obtained from the Developmental Studies Hybridoma Bank (University of Iowa [15, 16]). Rat and mouse anti-BrdU MAbs were purchased from Harlan Sera-Lab (Loughborough, United Kingdom) and Boehringer Mannheim (Mannheim, Germany), respectively. For detection of the MHV M and N proteins, MAbs 5A5.2 and 5B188.2 were used (76). The fluorescent conjugates used were Cy3-conjugated donkey anti-rabbit immuno-globulin G (IgG), fluorescein isothiocyanate (FITC)-conjugated donkey anti-mouse IgG (both from Jackson ImmunoResearch Laboratories), and FITC-conjugated rabbit anti-rat IgG (DAKO, Glostrup, Denmark).
Indirect IF assays.L cells, Sac⫺cells, and DBT cells were grown on coverslips to 50% confluency, infected with MHV-A59 at a multiplicity of infection (MOI) of 1 PFU/cell, and fixed at 5 or 7 h postinfection (p.i.) with 3% paraformaldehyde in phosphate-buffered saline (PBS). In experiments wheretsmutants were used, cells were kept at 39.5°C during the infection and fixed at 4 or 6 h p.i. After being washed with 10 mM glycine in PBS, cells were permeabilized for 10 min with 0.1% Triton X-100 in PBS. The antibodies were diluted in PBS containing 5% FCS. After antibody incubations, cells were embedded in Mowiol 4-88 (Hoechst).
EM.Mouse L cells were grown in 10-cm2dishes, infected at an MOI of 10, and fixed at 5 or 7 h p.i. For routine fixation, cells were washed five to six times with ice-cold PBS containing 5 mM EDTA and EGTA. The cells were incubated for 5 to 10 min on ice in PBS-EDTA-EGTA and then gently squirted off the dish. Cells were collected by a 2-min centrifugation at 2,500⫻gin a microcentrifuge, and the pellet was overlaid with 4% paraformaldehyde–0.1% glutaraldehyde in 200 mM HEPES-KOH (pH 7.4). After fixing for 30 min at room temperature, the fixative was replaced by 8% paraformaldehyde in 200 mM HEPES-KOH. The fixed cells were prepared for cryosectioning and used for single- or double-labeling experiments as described previously (28, 70). To label endocytic or-ganelles with endocytic tracers, L cells were incubated for 2 h at 37°C with bovine serum albumin (BSA) coupled to 16-nm gold particles, washed with PBS, and chased overnight. Subsequently, the cells were infected with MHV as usual. To one dish of cells, 5-nm gold–BSA was added at 4.5 h p.i., followed by a 30-min incubation at 37°C. The cells were then rinsed three times with prewarmed medium and subsequently fixed by addition of an equal volume of 8% paraform-aldehyde–0.2% glutaraldehyde in 0.2 M PHEM buffer (120 mM PIPES, 50 mM HEPES, 4 mM MgCl2, and 20 mM EGTA [pH 6.9]) to the medium. Using the same fixation procedure, another dish of cells was fixed directly at 5 h p.i. After 2 h of fixing at room temperature, the cells were scraped from the dish and collected by centrifugation, and the cell pellet was overlaid with 8% paraformal-dehyde in 0.1 M PHEM buffer and kept overnight at 4°C. The cells were washed with PBS-glycine and resuspended in PBS with 2% low-melting-point agarose at 37°C. The cells were spun for 5 min in a microcentrifuge at 10,000⫻gand immediately put on ice. The agarose-embedded cells were cut in small blocks, infiltrated with 2.3 M sucrose, and frozen in liquid nitrogen.
BrUTP lipofection and immunolabeling.The metabolic labeling of viral RNA synthesis with BrUTP (Sigma) has been described recently (79). Briefly, MHV-infected L cells were given 10g of dactinomycin (Sigma)/ml 30 min prior to labeling. BrUTP (final concentration, 10 mM) was introduced into the cells by using cationic liposomes (Lipofectamine Plus; Life Technologies). Cells were labeled for 1 h at 4 or 6 h p.i. and then fixed and processed for either IF microscopy or EM as described above. BrUTP was localized by using either the rat MAb hybridoma supernatant at a dilution of 1:10 or the mouse MAb that was dissolved at a concentration of 50g/ml and used at a dilution of 1:4.
RESULTS
[image:2.612.116.487.71.152.2]IF microscopy reveals similar early labeling patterns for ORF1a- and ORF1b-encoded replicase subunits.To study the
FIG. 1. Coronavirus gene 1 expression and processing. The schematic of gene 1 shows the organization into ORF1a and ORF1b, with a number of important domains: papain-like proteinase 1 (PLP-1), 3C-like proteinase (3CLpro), hydrophobic domains (MP1 and MP2), RNA-dependent RNA polymerase (Pol), and helicase (Hel). Arrows indicate confirmed (black) or predicted (white) cleavages by PLP-1 and 3CLpro in MHV-A59. The black lines below the schematic indicate the positions of the protein sequences which were used to raise the antibodies used in this study. These were either synthetic peptides (3CLpro, Pol, and Hel) or expressed as part of a bacterial fusion protein (anti-CT1a).
7642 VAN DER MEER ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
subcellular localization of the MHV replicase by IF micros-copy, antisera raised against several regions of the ORF1ab polyprotein were tested on MHV-A59-infected L cells at 5 and 7 h p.i. After an initial screening, including the appropriate controls (see Materials and Methods), antisera recognizing a number of key replicase subunits were selected for further
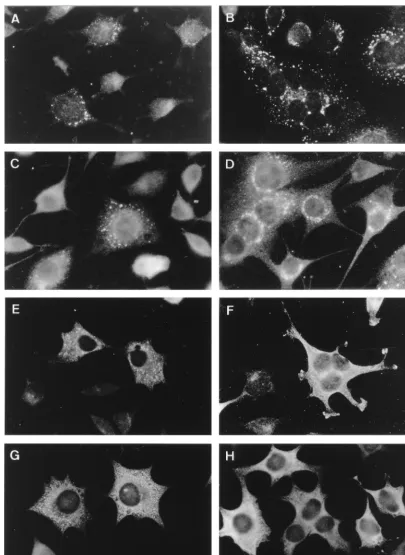
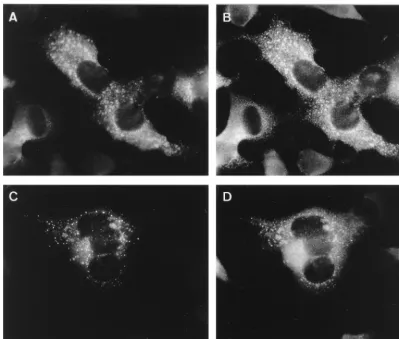
[image:3.612.100.505.70.625.2]studies. These were anti-peptide sera recognizing the main viral protease 3CLpro (anti-3CLpro), the putative RNA poly-merase (anti-POL), or the presumed helicase (anti-HEL) and an antiserum raised against a bacterial fusion protein which represented the C-terminal 429 residues of the ORF1a protein (anti-CT1a; Fig. 1). The recently described 22-kDa protein is FIG. 2. Indirect IF analysis of nonstructural proteins in MHV-infected cells fixed at 5 (panels A, C, E, and G) and 7 (panels B, D, F, and H) h p.i. Fixed cells were labeled with anti-3CLpro (96.6; A and B), anti-CT1a (C and D), anti-POL (778; E and F) and anti-HEL (96.8; G and H).
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7643
on November 9, 2019 by guest
http://jvi.asm.org/
the major cleavage product recognized by the latter antiserum (51). However, it should be stressed that, due to the relatively slow proteolytic processing of the coronavirus replicase, the antisera used in this study probably also react with polyprotein precursors and processing intermediates.
At 5 h p.i., MHV-infected L cells labeled with anti-3CLpro showed a perinuclear punctate pattern, mostly concentrated on one side of the nucleus (Fig. 2A). At 7 h p.i., more cells were positive and the labeled spots seemed to have increased in size (Fig. 2B). As described by Lu et al. (53), and in contrast to the anti-CT1a serum, the anti-3CLpro antibody recognizes mainly the mature 3CLpro protein (p27). In biochemical studies, this antibody recognized only small amounts of 3CLpro-containing precursor proteins, suggesting that the liberation of this pro-tease is a relatively rapid process. Interestingly, the anti-CT1a serum, which does recognize several precursor proteins (see above), showed an IF pattern at 5 h p.i. (Fig. 2C) and 7 h p.i. (Fig. 2D) that was very similar to the 3CLpro labeling.
The labeling obtained with the anti-POL serum at 5 h p.i. was mostly perinuclear and punctate, but at 7 h p.i. a less punctate labeling pattern predominated (Fig. 2E and F). A similar labeling was obtained with anti-HEL—a punctate pat-tern at 5 h p.i., but a more dispersed patpat-tern later in infection (Fig. 2G and H).
We also studied the labeling of the replicase proteins in two other cell lines, DBT and Sac⫺cells, but observed no major
differences compared to mouse L cells (data not shown). At later times (7 h), small syncytia in which the labeling for all four antibodies appeared to be less bright were observed (not shown). Since syncytium formation may lead to reorgani-zation and relocalireorgani-zation of cell compartments (48), we also studied the localization of the replicase using a ts mutant (ts379) that does not form syncytia at the restrictive tempera-ture (39.5°C [54]). The labeling of cells infected with wild-type
MHV or withts379 infected at the nonpermissive temperature was not substantially different, except that the lack of syncy-tium formation in thetsmutant resulted in a brighter labeling at 7 h p.i. (data not shown). In summary, at early times of infection, the labeling patterns for all four antibodies were very similar, suggesting colocalization of the replicase proteins.
The MHV nucleocapsid protein colocalizes with the repli-cation complex.Since all four antisera appeared to show sim-ilar labeling at early times of infection, we wanted to establish whether their respective labeling patterns colocalized. When testing a MAb to the MHV nucleocapsid (N) protein early in infection, punctate labeling which was very similar to the replicase labeling pattern was observed. Double-labeling experiments were therefore carried out at 5 h p.i., using the N-protein-specific mouse MAb (5B188.2) and the four anti-replicase rabbit sera described above.
Clear colocalization between the anti-3CLpro and anti-N could be observed (Fig. 3A and B, respectively), and similar results were obtained for the anti-POL and anti-N sera (Fig. 3C and D), as well as with the anti-HEL and anti-N antibodies (data not shown). Finally, the CT1a and N labeling also colo-calized to some extent, but although the two images largely overlapped, we also detected some spots that were either N positive and CT1a negative or vice versa (data not shown). In conclusion, these IF data suggest that the MHV N protein colocalizes with the viral replication complex during the early stages of infection (see also below). Furthermore, these dou-ble-labeling experiments suggest that 3CLpro, POL, HEL, and to some extent CT1a localize to the same structures at early times of infection.
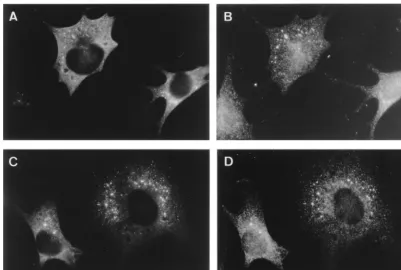
Partial colocalization of replicase subunits with a late en-dosomal marker protein.Subsequently, marker antibodies for various cellular compartments were used in double-labeling IF studies to characterize the intracellular location of the repli-FIG. 3. IF double labeling of the MHV N protein (A and C) with the replicase antisera 3CLpro (B) and POL (D) at 5 h p.i. The labeling for N and 3CLpro in panels A and B, respectively, largely overlap. Panels C and D show the double labeling for N and POL, which also clearly shows colocalization.
7644 VAN DER MEER ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.102.503.72.342.2]cation complex. We have recently shown that the replication of the arterivirus EAV occurs on modified ER membranes (60, 79), and therefore we initially focused on markers of the ER-IC-Golgi region.
At 5 h p.i., none of the four replicase antisera revealed colocalization with PDI, a marker for ER and IC (not shown). As a marker for the Golgi complex, a MAb specific for the MHV M protein, a triple-spanning membrane protein of the coronavirus envelope, was used. M is known to be retained in the Golgi complex (42, 45), and in IF the signals for anti-M and anti-mannosidase II (a marker for medial Golgi) colocalize (unpublished results). In double-labeling experiments, how-ever, the proteins recognized by the replicase antisera 3CLpro and CT1a did not colocalize with M in the Golgi complex (data not shown).
Markers of endocytic compartments were subsequently tested. Antibodies to the mouse lysosome-associated mem-brane proteins (LAMPs), which have been shown to localize to late endocytic structures as well as to lysosomes (e.g., see references 25 and 30), were used for this purpose. Double labeling with anti-LAMP-2 and anti-CT1a showed quite sub-stantial colocalization (Fig. 4A and B, respectively). The anti-LAMP-2 and anti-POL staining localized to the same region of the infected cell at 5 h p.i., but only a partial overlap of the two proteins was observed (Fig. 4C and D). Similar results were obtained with the anti-3CLpro and anti-HEL sera; they
local-ized to the same area as LAMP-2 with only partial colocaliza-tion (data not shown). While these data indicated that the protein recognized by anti-CT1a may be associated with late endosomes/lysosomes, the pattern obtained with the other three replicase antisera remained unclear. Thus, we extended our studies to the ultrastructural level by using EM.
The CT1a replicase antiserum labels late endocytic struc-tures.For the localization of MHV replicative proteins by EM, mouse L cells were fixed at 5 h p.i. (and sometimes 7 h p.i.; see text), and thawed cryosections were labeled with the various antibodies. Initial antibody screening revealed that by using this method, only the anti-CT1a and anti-POL antibodies gave specific labeling (data not shown).
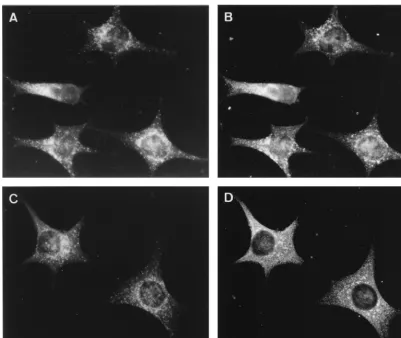
[image:5.612.102.503.71.408.2]Consistent with the IF data, the CT1a antibody labeled membranes that were clearly reminiscent of late endocytic structures, typically containing many internal vesicular profiles (Fig. 5). As shown in the IF studies, these labeled structures were often found in the perinuclear region of the cell, although some of them were found close to the plasma membrane (Fig. 6A). To determine the origin of the CT1a-positive membranes, antibodies to LAMP-1 were used in double-labeling studies. The labeling patterns of the two antisera largely overlapped, and the proteins clearly colocalized to vesicular membranes of late endocytic origin (Fig. 6A). Additional support was ob-tained by using internalized BSA-coupled 16-nm gold particles. This marker was internalized for 2 h by fluid phase endocytosis, FIG. 4. IF double labeling of LAMP-2 (A and C), a marker for late endosomes and lysosomes, and the MHV replicase proteins CT1a (B) and POL (D). There is substantial overlap between LAMP-2 (A) and CT1a (B). In panels C and D, the most brightly labeled LAMP-2 (C)-positive spots show colocalization with POL, while the POL serum also labels structures that are LAMP-2 negative.
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7645
on November 9, 2019 by guest
http://jvi.asm.org/
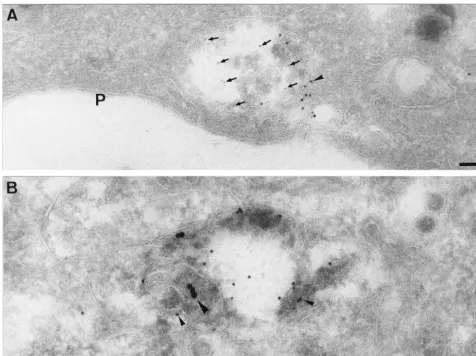
FIG. 5. Localization of CT1a as seen by EM. MHV-infected cells were fixed at 5 (B and C) and 7 (A) h p.i., and thawed cryosections were labeled with antibodies to CT1a (arrowheads). In panel A, note extensive labeling over structures with multivesicular appearance (stars). In panel B, the CT1a-positive membranes are attached to a vacuolar structure (star), the surrounding membranes of which are also weakly labeled (arrowheads). Panel C shows another profile of CT1a-positive (arrowheads) multivesicular membranes (stars). Note the membrane structure (large arrow) at the outside of the cell that is significantly labeled with CT1a (arrowhead). P, plasma membrane. Bars, 100 nm.
7646
on November 9, 2019 by guest
http://jvi.asm.org/
after which it was chased overnight into late endosomes/lyso-somes. Subsequently, the cells were infected with MHV, fixed at 5 h p.i., and prepared for cryosectioning. The CT1a labeling clearly localized to compartments in which the 16-nm gold had accumulated, confirming the late endocytic nature of these structures (Fig. 6B). Other organelles, such as the Golgi com-plex, the ER and IC (see below), early endosomes, and the plasma membrane, were essentially devoid of labeling by the CT1a serum (data not shown). In addition, even at this rela-tively early time of infection, in some cells typical MHV-bud-ding profiles in the IC which were essentially devoid of labeling for anti-CT1a were observed (not shown; see also below).
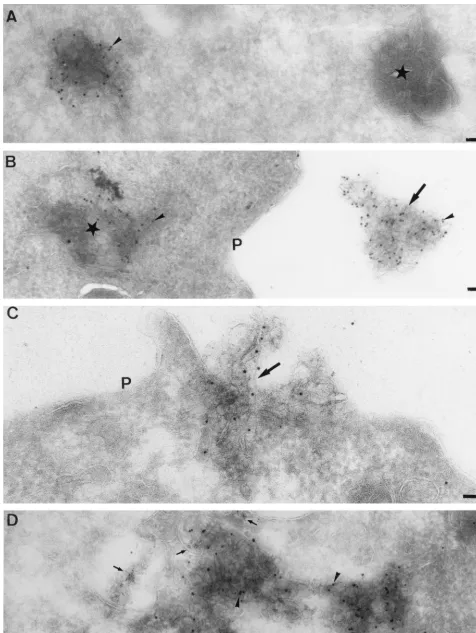
The localization of the RNA-dependent RNA polymerase by EM.When infected cells were labeled with antibodies to the putative MHV RNA polymerase domain (anti-POL), abun-dant labeling was found on structures that were morphologi-cally distinct from the CT1a-positive membranes. Strong anti-POL labeling appeared to be associated with structures of discrete size containing multiple membrane sheets, reminis-cent of endocytic carrier vesicles (ECVs) or multivesicular bodies (MVBs [32, 41, 56, 80]) (Fig. 7).
To determine the relationship between the CT1a- and POL-positive structures, the POL labeling was subsequently com-pared to CT1a and LAMP-1 in double-labeling studies. The
bulk of CT1a and POL accumulated clearly in morphologically different structures. Upon closer inspection, however, the CT1a-positive membranes did show low but significant labeling with the anti-POL serum (see also below) and, conversely, the POL-positive membranes did label to some extent for CT1a, representative pictures of which are shown in Fig. 8.
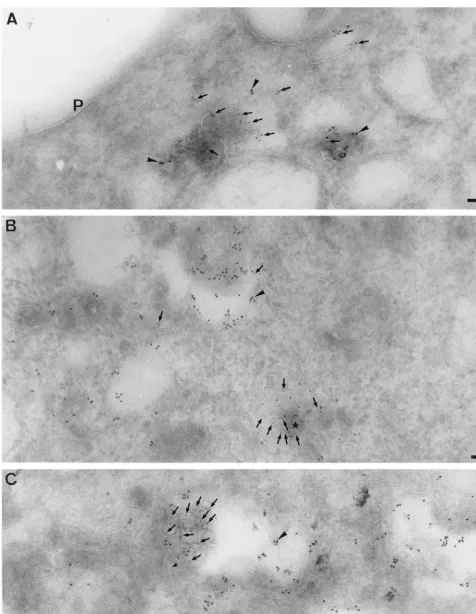
When POL was compared to LAMP-1, again the bulk of the labeling for both markers was clearly in distinct structures, LAMP-1 in typical late endosome/lysosome-like membranes and POL in the ECV-like structures. However, as for the CT1a serum, some POL labeling could be found on LAMP-1-posi-tive membranes (Fig. 9B), whereas the multilayered POL-pos-itive structures labeled for LAMP-1 to some extent, confirming their endocytic origin (Fig. 9A). The bulk of the POL-positive structures appeared as distinct entities, separated from the late endosomes/lysosomes. In some profiles, however, they seemed to share membrane continuities with or to be part of LAMP-1 positive late endosomes (Fig. 7D and Fig. 9).
Since these results strongly suggested that the POL-positive membranes were also of endocytic origin, L cells were subse-quently given two different sizes of BSA-gold to mark late as well as late and early endocytic organelles (see Materials and Methods). No profiles of POL-positive membranes filled with the BSA-gold that had been chased overnight were seen, sug-FIG. 6. Double labeling of MHV-infected cells with CT1a and LAMP-1 and with internalized 16-nm gold–BSA. Panel A shows cryosections of MHV-infected cells fixed at 5 h p.i. and double labeled with CT1a (small arrows, 5-nm gold) and LAMP-1 (arrowhead, 10-nm gold). In this particular profile, the CT1a-positive membranes are quite close to the plasma membrane (P). Panel B shows L cells that were filled with 16-nm gold–BSA (large arrowhead) that was chased overnight into late endosomes/lysosomes prior to MHV infection. Cells were fixed at 5 h p.i. and double labeled with antibodies to CT1a (arrowheads, 10-nm gold). Bars, 100 nm.
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7647
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.65.541.72.428.2]FIG. 7. Localization of the RNA-dependent RNA polymerase as seen by EM. Thawed cryosections of MHV-infected L cells were fixed at 5 h p.i. and labeled with anti-POL serum (arrowheads, 10-nm gold). Note in panel A two multilamellar structures, one that is strongly labeled for POL (arrowhead) and one that is not labeled (star). In panel B, a strongly labeled POL (10-nm gold, arrowhead)-positive structure can be seen inside (star), while another labeled membrane structure (large arrow) can be seen outside the cell. In panel C, the POL-positive membranes are apparently in the process of being secreted (large arrow) from the plasma membrane (P). In panel D, MHV-infected cells were filled with 5-nm gold–BSA 30 min prior to fixation at 5 h p.i. The POL-positive membranes (10-nm gold, arrowheads) are clearly filled with the 5-nm gold–BSA (small arrows). Bars, 100 nm.
7648
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 8. Double labeling of MHV-infected cells with POL and CT1a. MHV-infected cells were fixed at 5 h p.i., and cryosections were double labeled with antibodies to POL (5-nm gold, small arrows) and CT1a (10-nm gold, arrowheads). In panel A, several membrane structures show colocalization of both markers. In panels B and C, the typical multilamellar membrane structures that label heavily for POL are also labeled to some extent with CT1a. Panel D shows a profile labeled for both POL and CT1a (that also contains 16-nm gold–BSA) that seems to be in the process of exiting the cell. P, plasma membrane. Bars, 100 nm.
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7649
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 9. Double labeling of MHV-infected cells at 5 h p.i. with POL and LAMP-1. Panel A shows several structures that label both for POL (10-nm gold, arrowheads) and LAMP-1 (5-nm gold, small arrows). Panel B shows an area with extensive labeling for LAMP-1 (10-nm gold, arrowheads) that is also labeled to some extent with POL (5-nm gold, small arrows). Note especially the membrane structure in the bottom right (small star) where LAMP-1-positive membranes seem to surround membranes that label heavily for POL. In panel C, a similar area that is heavily labeled with LAMP-1 can be seen (10-nm gold, arrowhead). A strongly labeled POL (5-nm gold, small arrows) multilamellar structure seems to be surrounded by LAMP-1-positive membranes. Bars, 100 nm.
7650 VAN DER MEER ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
gesting that they may not be of late endocytic origin. They did, however, fill with the BSA-gold that was given to infected cells during the 30 min prior to fixation (but not with BSA-gold that was internalized for only 5 min [data not shown]) (Fig. 7D). These data are most consistent with the POL-positive struc-tures being intermediate between early and late endosomes/ lysosomes (see Discussion).
When comparing the POL labeling at 5 and 7 h p.i., the ECV-like structures clearly appeared to increase in size and number during the course of infection, suggesting that the viral infection was inducing their formation (data not shown). Most intriguingly, we observed that some of the membrane struc-tures labeled with the CT1a or POL serum (or both) appeared to be secreted, especially later in infection (7 h p.i.). Not only were such labeled membranes observed outside the cells (Fig. 5C and 7B), but profiles were also seen, suggesting that they were in the process of being secreted (Fig. 7C and 8D).
These data show that the MHV POL also localizes to mem-branes of endocytic origin. Consistent with the IF data, how-ever, the majority of the labeling was associated with structures reminiscent of ECVs and the protein only partially colocalized with CT1a or LAMP-1.
Subcellular localization of viral RNA synthesis.To establish the site of MHV RNA synthesis directly, we employed meta-bolic RNA labeling by using BrUTP in the presence of dacti-nomycin to block cellular mRNA transcription (60, 79). The
label was introduced into the cells by using cationic liposomes and was detected in IF assays by using antibodies to BrdU, which also recognize BrUTP-labeled RNA. When uninfected cells were treated in exactly the same way, no BrUTP-positive signal was detected by IF, indicating that the dactinomycin treatment efficiently prevented cellular RNA synthesis (not shown).
MHV-infected cells were BrUTP labeled from 5 to 6 h p.i. Cells were double labeled using an anti-BrdU MAb and either the anti-3CLpro or the anti-CT1a rabbit antiserum (Fig. 10). Clear overlap could be detected between the labeling patterns of the anti-3CLpro and anti-BrdU antibodies (Fig. 10A and B). The BrUTP labeling also colocalized partially with CT1a (Fig. 10C and D), but the latter antibody appeared to label addi-tional structures that were BrUTP negative. These IF results strongly suggested that MHV replicase subunits and MHV-specific RNA synthesis colocalize to a great extent in infected cells.
[image:11.612.102.502.72.411.2]Next, the site of viral RNA synthesis was studied by EM. Initially, infected cells were labeled with BrUTP and fixed at 6 h p.i., and cryosections were labeled with the anti-BrdU antibody. In apparent disagreement with the IF results, BrUTP labeling did not appear to be concentrated at any location in the cell. Labeling could be found in patches in the cytosol and on the cytosolic surface of the rough ER, as well as to some extent associated with endosomal membranes (see Fig. 12A; FIG. 10. Localization of BrUTP in MHV-infected cells as seen by IF. Double labeling of BrUTP (A and C) and the replicase antisera 3CLpro (B) and CT1a (D). MHV-infected cells were transfected with BrUTP at 5 h p.i. and fixed 1 h later. In panels A and B, the labeling for BrUTP and 3CLpro is shown. Panels C and D represent the double labeling for BrUTP and CT1a. In both double labelings, the bright spots clearly overlap.
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7651
on November 9, 2019 by guest
http://jvi.asm.org/
7652 VAN DER MEER ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
see below). We believe that the apparent discrepancy between the IF and EM data can be explained by the fact that the former technique preferentially highlights the brightest spots (where incorporated BrUTP is concentrated), whereas in thin-section EM all labeling can be seen, irrespective of concentra-tion.
Subsequently, the BrUTP labeling was performed at an early time point (5 h p.i.). Before infection and labeling, late endo-cytic compartments were filled with 16-nm BSA-gold. When labeled at this early time p.i., a fraction (less than 5%) of the cells now showed a pattern similar to that at 6 h p.i. (see Fig. 12A). The labeling for BrUTP in most cells, however, clearly differed from this pattern. The multivesicular structures that were filled with the BSA-gold were labeled by BrUTP to some extent (Fig. 11B). The bulk of the label, however, was prefer-entially associated with membranes of more tubular vesicular appearance that were clearly attached to the 16-nm BSA–gold-positive membranes (Fig. 11C and D). Since membranes of the IC also typically consist of tubular vesicular structures, trans-fected cells were also double labeled with antibodies to BrUTP and a marker protein of the IC (sec13 [69, 77]). However, no colocalization was detected, implying that the BrUTP-positive membranes were not of IC origin (data not shown). As as-sessed by double labeling, the BrUTP-positive membranes were also labeled with both CT1a (Fig. 11B and C) and POL (Fig. 11A and D). The typical ECV-like structures where the bulk of POL accumulated were generally not labeled with the BrdU antibody (data not shown).
Since the BrUTP-positive membranes appeared to label for CT1a as well as POL, we believe that they represent domains of late endosomes/lysosomes that may be the primary site of viral RNA synthesis. These combined data show that MHV replicative proteins, and probably the replication process itself, localize to membranes of endocytic origin at early times of infection.
The nucleocapsid protein also localizes to late endosomes early in infection. In view of the IF data obtained with the anti-N MAb (see above) and our conclusion that MHV repli-cation may occur on endosomal membranes, it was of interest to determine whether the N protein also localizes to this com-partment early in infection, before the onset of virus assembly. Indeed, the distinct, punctate labeling pattern observed for N by IF microscopy (Fig. 3A and C) was found by EM to repre-sent late endosomes/lysosomes, since it colocalized with struc-tures in which 16-nm BSA-gold accumulated during an over-night chase (Fig. 12B). Most importantly, the membranes to which the N protein localized structurally resembled those that labeled for BrUTP (see above). These had a tubular vesicular appearance and often extended away from the multivesicular part in which the BSA-gold had accumulated. However, be-cause of cross-reactivity between the two MAbs, we were un-able to prove this point by an N-BrUTP double labeling. These data show that at early times of infection the N protein also localizes to late endosomal membranes.
DISCUSSION
Membrane association of the MHV replicase.Corona- and arteriviruses have been united in the order of theNidovirales (14). Despite their common properties at the level of replica-tion and transcripreplica-tion, we have now revealed an important difference: the membranes used for formation of the viral replication complex. Arterivirus replication was recently found to be associated with ER or IC membranes, which are modified into vesicular structures with a double membrane (60, 79). Our combined data for MHV strongly suggest that coronavirus replication occurs on late endosomal membranes. This was shown by colocalization of a number of important replicase subunits, including the putative RNA POL and HEL, with established endosomal markers as well as by showing that newly synthesized viral RNA also localizes to endosomes. It should be kept in mind that, to a variable extent, the antisera used in this study recognize a mixture of mature replicase subunits and processing intermediates. For the two sera which are specific for the ORF1a protein (3CLpro and anti-CT1a), the labeling did not change in the course of infection, suggesting the membrane association of both precursors and cleavage products. Similar observations were previously made for the ORF1a-encoded proteins of the arterivirus EAV (79). In contrast, the staining for the MHV POL and HEL proteins became more dispersed during the course of infection (Fig. 2), suggesting the liberation of these subunits from the complex by proteolytic processing.
At first glance, some of the IF microscopy data were hard to reconcile with those obtained by EM. While both IF micros-copy and EM indicated that anti-CT1a labeled LAMP-positive membranes, the proteins recognized by 3CLpro, anti-HEL, and anti-POL did not seem to colocalize substantially with this late endosomal/lysosomal marker, as indicated by IF. We now assume that only the brightest spots (reflecting the sites with the highest concentrations of protein) can be easily discerned by IF microscopy. Thus, when it comes to, e.g., the anti-POL labeling, it can be expected that IF microscopy will reveal mainly the ECV-like structures that showed only weak LAMP labeling by EM. That the POL-positive structures were part of the endocytic pathway was demonstrated by our obser-vations that they labeled to some extent for LAMP-1 but not for ER or IC markers (data not shown), that they clearly shared membrane continuities with late endosomes, and that they could be filled with endocytic tracers. Furthermore, the BrUTP labeling experiments strongly suggested that viral RNA synthesis is also associated with late endosomes.
Our results apparently contradict recent data of Bi et al. (7) showing by IF in MHV-infected BHK cells the colocalization of two proteins derived from ORF1a with an established Golgi marker. In the present study, no Golgi labeling was detected with any of the antibodies tested either at the IF or the EM level. It should be kept in mind that organelles that have a strong tendency to localize to the perinuclear region (like the Golgi complex and late endosomes) can only in exceptional
FIG. 11. Localization of BrUTP as seen by EM. L cells were filled for 2 h with 16-nm gold–BSA that was subsequently chased overnight into late endosomes/ lysosomes. The cells were MHV infected, labeled with BrUTP at 4 h p.i., fixed 1 h later, and prepared for cryosectioning. In panels A and B, two rare profiles of BrUTP labeling can be seen. Panel A shows a POL (5-nm gold, small arrows)-positive structure that labels to some extent with anti-BrUTP (10-nm gold, arrowhead), and in panel B, the compartment where the internalized 16-nm gold–BSA (large arrowhead) accumulates is labeled with both CT1a (10-nm gold, arrowheads) and anti-BrUTP (5-nm gold, small arrows). Panels C and D show typical profiles of BrUTP labeling (marked by a large arrow in panel D). Tubular vesicular structures that are attached to the membranes in which the 16-nm gold–BSA accumulates (large arrowheads) are strongly labeled for anti-BrUTP (5-nm gold, small arrows in panel C; 10-nm gold arrowheads in panel D). CT1a in panel C (arrows, 10-nm gold) and POL (small arrows, 5-nm gold) in panel D also label these BrUTP-positive membranes to some extent. N, nucleus. Bars, 100 nm.
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7653
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 12. Cryosections of MHV-infected cells, labeled with BrUTP or anti-N. Panel A shows double labeling of anti-POL (5-nm gold, small arrows) and BrUTP (10-nm gold). This picture represents a rare section of BrUTP labeling at 5 h p.i. with a pattern typical of 7 h p.i. (see text). Patches of BrUTP labeling are seen apparently randomly distributed over the cell but are occasionally associated with the cytosolic side of the ER and with late endosomes (large arrowhead, 16-nm gold). Note also the colocalization of POL and BrUTP in some areas of the cell. Panels B and C show labeling for N (small arrows) at early stages (5 h p.i.) of infection. Note the clear association of N labeling with membranes that are attached to the 16-nm gold–BSA-containing (big arrow) late endosomes/lysosomes. P, plasma membrane; N, nucleus. Bars, 100 nm.
7654 VAN DER MEER ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
cases be unequivocally distinguished by light microscopy (29). Therefore, the unequivocal localization of the two antibodies described by Bi et al. to the Golgi complex awaits detailed characterization by EM.
The MHV N protein may play a role in RNA synthesis.The MHV N protein is an abundant structural component of the virion. From its sequence, N is predicted to be a cytosolic protein and labeling of infected cells with antibodies to N was expected to give a general cytosolic staining. Several reports, however, have also implicated the N protein in replication (4, 17, 47). Moreover, although N lacks a typical membrane-span-ning domain, the protein has been shown to be able to bind to membranes (1). These observations were confirmed and ex-tended by our finding that, at least at the onset of N protein synthesis and possibly throughout infection, the N protein co-localizes with the viral replication complex. Furthermore, these data are consistent with recent results showing coimmunopre-cipitation of N with an antibody directed to the viral putative helicase (20). These combined data thus suggest that MHV N has a role in MHV RNA synthesis or, alternatively, that en-capsidation by the N protein occurs at the site of RNA syn-thesis.
Is there a relation between virus entry and replication on endocytic membranes?Although our results for the coronavi-rus MHV differed from those obtained with arterivicoronavi-rus EAV (60, 79), they are consistent with the generally accepted idea that positive-stranded RNA viruses use cytoplasmic mem-branes for their replication. Other examples of viruses that appear to use endosomes for their replication include Semliki Forest virus and rubella virus, both members of theTogaviridae (23, 55). Since Semliki Forest virus enters the cell by receptor-mediated endocytosis, its replication occurs in close proximity to its site of disassembly. The logical question, therefore, is whether MHV might use a similar mechanism and also enter by endocytosis. The available literature on MHV entry, how-ever, is far from unequivocal. Detailed morphological entry studies have not yet been performed, and experiments have centered around two aspects: to determine the optimal pH of S-protein-mediated fusion and (in relation to this) to assess whether infectivity is affected by lysosomotropic drugs. Whereas it was clearly established that MHV-induced fusion is optimal at neutral pH (43, 85), the effect of lysosomotropic drugs on MHV entry is less clear (see, e.g., references 43, 46, and 57). It should be kept in mind that the insensitivity of viral infection to these drugs does not prove entry at the plasma membrane; it merely demonstrates that entry does not require a low pH. It has been shown that point mutations in the coiled-coil domain of the S protein render virus-induced fusion acid dependent (24). The acquisition of low-pH-dependent fusion could furthermore be correlated with the (increased) sensitivity of virus infection to lysosomotropic drugs (24, 59). Whether this indicates that some MHV strains enter by endo-cytosis while others enter by fusion at the plasma membrane (see reference 59), or whether MHV generally enters using both mechanisms (see reference 43), remains to be demon-strated.
The membrane compartments which carry MHV replicase subunits.Our observations imply that MHV replicase subunits and BrUTP labeling localize to several (sub)domains of the same compartment, with morphologically quite distinct ap-pearances. First, there were the CT1a-positive membranes that were mostly multivesicular and in which internalized BSA-gold accumulated. The bulk of the BrUTP labeling localized to more tubular vesicular membranes that were also labeled with anti-CT1a. Finally, there were the multilayered, ECV-like structures where POL-containing proteins accumulated in
large amounts. This compartment has all the hallmarks of the so-called prelysosomal compartment (PLC or late endosome) as described extensively for NRK cells (27, 30). This PLC has been shown to contain several morphologically distinct subdo-mains, and marker proteins (like the mannose-6-phosphate receptor and LAMPs) may preferentially accumulate in one of those domains (25, 30). The late endosomal compartment is generally enriched in lysosomal hydrolases as well as LAMPs (31).
The multilayered POL-positive structures were certainly most intriguing. Based on their characteristic morphology, they might be classified as ECVs or multivesicular bodies (MVBs). Functionally, ECVs have been proposed to transport cargo along microtubules from early to late endosomes (2). In our study, however, the images more frequently suggested mem-brane continuities with strongly LAMP-positive late endocytic structures, suggesting that the POL-positive membranes may not be typical ECVs. However, consistent with the idea that ECVs may be intermediates between early and late endo-somes, the POL-positive structures were not filled with BSA-gold that had been chased overnight into late endosomes/ lysosomes but could be filled following a 30-min incubation with this marker. The same treatment also labeled typical early endosomal profiles, which were clearly distinct in our sections (data not shown). One dispute concerning the structure of ECVs or MVBs is whether the internal membranes are infold-ings of one continuous membrane and thus exposed to the cytosol or whether these membranes represent distinct inter-nalized vesicles (see, e.g., references 6, 34, 56, and 87 and references therein). In favor of the former model, we observed that the POL-containing proteins (which are likely to be cyto-solic proteins) were present over the entire structure and not only on the outer cytoplasmic surface. This suggests that the internal membranes are also functionally exposed to the cy-tosol.
A very intriguing observation was that CT1a- and POL-positive membranes appeared to be secreted. Interestingly, similar observations have been made for major histocompati-bility complex (MHC) class II-positive structures. Intracellu-larly, MHC class II molecules localize to membranes that, like the MHV POL- and CT1a-positive structures, contain markers for late endosomes (61). A subset of the MHC class II mole-cules localize to vesicles that have all the hallmarks of ECVs or MVBs (41), and recent evidence has shown that some of this subpopulation of MHC class II-containing vesicles is secreted (63). It has been speculated that this might be a way of trans-porting MHC class II molecule-peptide complexes between cells (63). In the case of the CT1a- and POL-containing mem-branes, we can only speculate about the role of their secretion. It might be a way to discard an excess of protein(s) involved in replication, or it might be a general cellular response to a viral insult. Alternatively, it could be a way to transport certain (viral) molecules from cell to cell. More information about the protein content of these structures will certainly help to eluci-date this question.
Targeting of the replicase complex.Our observations raise the interesting question of how the coronavirus replicative proteins are targeted to the correct membranes. In the case of the arterivirus EAV, the scenario could be the conventional one: one or several replicase subunits containing hydrophobic domains may be translated and inserted into the rough ER and would then be transported (either alone or in association with other proteins) to (sub)domains of the ER, where replication takes place (79). In the case of MHV, the same scenario could be followed initially, but then one or several membrane-bound proteins need to be targeted to endosomes for replication to
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7655
on November 9, 2019 by guest
http://jvi.asm.org/
occur. Obviously, the MP1 and MP2 regions in the ORF1a gene are strong candidates to play a role in the membrane anchoring of the complex. Inconsistent with this idea is the fact that early in infection no ER or Golgi localization (the oblig-atory route to reach endosomes) with any of the replicase antibodies was seen, either by IF microscopy or by EM. The alternative possibility is that the proteins are synthesized and inserted directly into endosomal membranes, by means of a mechanism without (known) precedent. Thus, unravelling the molecular details of such a process could have implications for hitherto unknown mechanisms of membrane insertion of cel-lular proteins, bypassing the ER translocation machinery.
ACKNOWLEDGMENTS
We thank Gareth Griffiths, Fred Wassenaar, Sasha Gorbalenya, and Xiaotao Lu for excellent technical assistance and/or for critical reading of the manuscript.
M.R.D. is supported by Public Health Service grant AI-26603.
REFERENCES
1.Anderson, R., and F. Wong.1993. Membrane and phospholipid binding by murine coronaviral nucleocapsid protein. Virology194:224–232.
2.Aniento, F., N. Emans, G. Griffiths, and J. Gruenberg.1993. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol.123:1373–1387.
3.Baker, S. C., K. Yokomori, S. Dong, R. Carlisle, A. E. Gorbalenya, E. V. Koonin, and M. M. Lai.1993. Identification of the catalytic sites of a papain-like cysteine proteinase of murine coronavirus. J. Virol.67:6056–6063. 4.Baric, R. S., G. W. Nelson, J. O. Fleming, R. J. Deans, J. G. Keck, N. Casteel,
and S. A. Stohlman.1988. Interaction between coronavirus nucleocapsid protein and viral RNAs: implications for viral transcription. J. Virol.62: 4280–4287.
5.Barton, D. J., S. G. Sawicki, and D. L. Sawicki.1991. Solubilization and immunoprecipitation of alphavirus replication complexes. J. Virol.65:1496– 1506.
6.Berg, T., T. Gjøen, and O. Bakke.1995. Physiological functions of endosomal proteolysis. Biochem. J.307:313–326.
7.Bi, W., J. D. Pinon, S. Hughes, P. J. Bonilla, K. V. Holmes, S. R. Weiss, and J. L. Leibowitz.1998. Localization of mouse hepatitis virus open reading frame 1A derived proteins. J. Neurovirol.4:594–605.
8.Bienz, K., D. Egger, and L. Pasamontes.1987. Association of poliovirus proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic im-munocytochemistry and autoradiography. Virology160:220–226.
9.Bienz, K., D. Egger, T. Pfister, and M. Troxler.1992. Structural and func-tional characterization of the poliovirus replication complex. J. Virol.66: 2740–2747.
10. Bonilla, P. J., S. A. Hughes, and S. R. Weiss.1997. Characterization of a second cleavage site and demonstration of activity intransby the papain-like proteinase of the murine coronavirus mouse hepatitis virus strain A59. J. Vi-rol.71:900–909.
11. Brayton, P. R., M. M. C. Lai, C. D. Patton, and S. A. Stohlman.1982. Characterization of two RNA polymerase activities induced by mouse hep-atitis virus. J. Virol.42:847–853.
12. Brierly, I., M. E. Boursnell, M. M. Binns, B. Bilimoria, V. C. Blok, T. D. K. Brown, and S. C. Inglis.1987. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J.6:3779– 3785.
13. Caliguiri, L. A., and I. Tamm.1969. Membranous structures associated with translation and transcription of poliovirus RNA. Science166:885–886. 14. Cavanagh, D.1997. Nidovirales: a new order comprising Coronaviridae and
Arteriviridae. Arch. Virol.142:629–633.
15. Chen, J. W., T. L. Murphy, M. C. Willingham, I. Pastan, and J. T. August. 1985. Identification of two lysosomal membrane glycoproteins. J. Cell Biol. 101:85–95.
16. Chen, J. W., W. Pan, M. P. D’Souza, and J. T. August.1985. Lysosome-associated membrane proteins: characterization of LAMP-1 of macrophage P388 and mouse embryo 3T3 cultured cells. Arch. Biochem. Biophys.239: 574–585.
17. Compton, S. R., D. B. Rogers, K. V. Holmes, D. Fertsch, J. Remenick, and J. J. McGowan.1987. In vitro replication of mouse hepatitis virus strain A59. J. Virol.61:1814–1820.
18. Dales, S.1965. Effects of streptovitacin A on the initial events in the repli-cation of vaccinia and reovirus. Proc. Natl. Acad. Sci. USA54:462–468. 19. den Boon, J. A., E. J. Snijder, E. D. Chirnside, A. A. F. de Vries, M. C.
Horzinek, and W. J. M. Spaan.1991. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J. Virol.65:2910–2920.
20. Denison, M. R., W. J. M. Spaan, Y. van der Meer, C. A. Gibson, A. C. Sims, E. Prentice, and X. T. Lu.1999. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. J. Virol.73: 6862–6871.
21. De Vries, A. A. F., M. C. Horzinek, P. J. M. Rottier, and R. J. de Groot.1997. The genome organization of the Nidovirales: similarities and differences between arteri-, toro-, and coronaviruses. Semin. Virol.8:33–47. 22. Dong, S., and S. C. Baker.1994. Determinants of the p28 cleavage site
recognized by the first papain-like cysteine proteinase of murine coronavirus. Virology204:541–549.
23. Froshauer, S., J. Kartenbeck, and A. Helenius.1988. Alphavirus RNA rep-licase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol.107:2075–2086.
24. Gallagher, T. M., C. Escarmis, and M. J. Buchmeier.1991. Alterations of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. J. Virol.65:1916–1928.
25. Geuze, H. J., W. Stoorvogel, G. J. Strous, J. W. Slot, J. Zijderhand-Bleeke-molen, and I. Mellman.1988. Sorting of mannose 6-phosphate receptors and lysosomal membrane proteins in endocytic vesicles. J. Cell Biol.107:2491– 2501.
26. Gorbalenya, A. E., E. V. Koonin, A. P. Donchenko, and V. M. Blinov.1989. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res.17:4847–4861.
27. Griffiths, G., R. Matteoni, R. Back, and B. Hoflack.1990. Characterization of the cation-independent mannose 6-phosphate receptor-enriched prelyso-somal compartment in NRK cells. J. Cell Sci.95:441–461.
28. Griffiths, G.1993. Fine structure immunocytochemistry. Springer-Verlag, Heidelberg, Germany.
29. Griffiths, G., R. G. Parton, J. Lucocq, B. van Deurs, D. Brown, J. W. Slot, and H. J. Geuze.1993. The immunofluorescence era of membrane traffic. Trends Cell Biol.3:214–219.
30. Griffiths, G., B. Hoflack, K. Simons, I. Mellman, and S. Kornfeld.1988. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell52:329– 341.
31. Griffiths, G.1996. On vesicles and membrane compartments. Protoplasma 195:37–58.
32. Griffiths, G., R. Back, and M. Marsh.1989. A quantitative analysis of the endocytic pathway in BHK cells. J. Cell Biol.109:2703–2720.
33. Grimley, P. M., J. G. Levin, I. K. Berezesky, and R. M. Friedman.1972. Specific membranous structures associated with the replication of group A arboviruses. J. Virol.10:492–503.
34. Gruenberg, J., G. Griffiths, and K. E. Howell.1989. Characterization of the early endosome putative endocytic carrier vesicles: insights from cell-free assays. J. Cell Biol.108:1301–1316.
35. Gupta, S., B. P. De, J. A. Drazba, and A. K. Banerjee.1998. Involvement of actin microfilaments in the replication of human parainfluenza virus type 3. J. Virol.72:2655–2662.
36. Hamaguchi, M., K. Nishikawa, T. Toyoda, T. Yoshida, T. Hanaichi, and Y. Nagai.1985. Transcriptive complex of Newcastle disease virus. Virology 147:295–308.
37. Herold, J., A. E. Gorbalenya, V. Thiel, B. Schelle, and S. G. Siddell.1998. Proteolytic processing at the amino terminus of human coronavirus 229E gene 1-encoded polyproteins: identification of a papain-like proteinase and its substrate. J. Virol.72:910–918.
38. Heusipp, G., C. Groetzinger, J. Herold, S. G. Siddell, and J. Ziebuhr.1997. Identification and subcellular localization of a 41 kDa, polyprotein 1ab processing product in human coronavirus 229E-infected cells. J. Gen. Virol. 78:2789–2794.
39. Hill, V. M., S. A. Harmon, and D. F. Summers.1986. Stimulation of vesicular stomatitis virus in vitro RNA synthesis by microtubule-associated proteins. Proc. Natl. Acad. Sci. USA83:5410–5413.
40. Hughes, S. A., P. J. Bonilla, and S. R. Weiss.1995. Identification of the murine coronavirus p28 cleavage site. J. Virol.69:809–813.
41. Kleijmeer, M. J., S. Morkowski, J. M. Griffith, A. Y. Rudensky, and H. J. Geuze.1997. Major histocompatibility complex class II compartments in human and mouse B lymphoblasts represent conventional endocytic com-partments. J. Cell Biol.139:639–649.
42. Klumperman, J., J. Krijnse Locker, A. Meijer, M. C. Horzinek, H. J. Geuze, and P. J. M. Rottier.1994. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol.68:6523–6534. 43. Kooi, C., M. Cervin, and R. Anderson.1991. Differentiation of
acid-pH-dependent and -nonacid-pH-dependent entry pathways for mouse hepatitis virus. Virology180:108–119.
44. Koolen, M. J., A. D. Osterhaus, G. van Steenis, M. C. Horzinek, and B. A. M. van der Zeijst.1983. Temperature-sensitive mutants of mouse hepatitis virus strain A59: isolation, characterization, and neuropathogenic properties. Vi-rology125:393–402.
45. Krijnse Locker, J., D.-J. Opstelten, M. Ericsson, M. C. Horzinek, and P. J. M. Rottier.1995. Oligomerization of a trans-Golgi/trans-Golgi network
7656 VAN DER MEER ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
retained protein occurs in the Golgi complex and may be part of its reten-tion. J. Biol. Chem.270:8815–8821.
46.Krzystyniak, K., and J. M. Dupuy.1984. Entry of mouse hepatitis virus 3 into cells. J. Gen. Virol.65:227–231.
47. Lai, M. M. C., and D. Cavanagh.1997. The molecular biology of coronavi-ruses. Adv. Virus Res.48:1–100.
48. Lavi, E., Q. Wang, S. R. Weiss, and N. K. Gonatas.1996. Syncytia formation induced by coronavirus infection is associated with fragmentation and re-arrangement of the Golgi apparatus. Virology221:325–334.
49. Lee, H. J., C. K. Shieh, A. E. Gorbalenya, E. V. Koonin, N. La Monica, J. Tuler, A. Bagdzhadhyan, and M. M. Lai.1991. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology180:567–582.
50. Liu, D. X., and T. D. Brown.1995. Characterisation and mutational analysis of an ORF 1a-encoding proteinase domain responsible for proteolytic pro-cessing of the infectious bronchitis virus 1a/1b polyprotein. Virology209: 420–427.
51. Lu, X. T., A. C. Sims, and M. R. Denison.1998. Mouse hepatitis virus 3C-like protease cleaves a 22-kilodalton protein from the open reading frame 1a polyprotein in virus-infected cells and in vitro. J. Virol.72:2265–2271. 52. Lu, Y., X. Lu, and M. R. Denison.1995. Identification and characterization
of a serine-like proteinase of the murine coronavirus MHV-A59. J. Virol. 69:3554–3559.
53. Lu, Y., X. Lu, and M. R. Denison.1996. Intracellular and in vitro-translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology222:375–382.
54. Luytjes, W., H. Gerritsma, E. C. W. Bos, and W. J. M. Spaan.1997. Char-acterization of two temperature-sensitive mutants of coronavirus mouse hep-atitis virus strain A59 with maturation defects in the spike protein. J. Virol. 71:949–955.
55. Magliano, D., J. A. Marshall, D. S. Bowden, N. Vardaxis, J. Meanger, and J.-Y. Lee.1998. Rubella virus replication complexes are virus-modified lyso-somes. Virology240:57–63.
56. McDowall, A., K. Roemisch, J. Gruenberg, and G. Griffiths. 1989. The structure of organelles of the endocytic pathways in hydrated cryo-sections of cultured cells. Eur. J. Cell Biol.49:281–294.
57. Mizzen, L., A. Hilton, S. Cheley, and R. Anderson.1985. Attenuation of murine coronavirus infection by ammonium chloride. Virology142:378–388. 58. Moremen, K. W., and O. Touster.1985. Biosynthesis and modification of Golgi mannosidase II in HeLa and 3T3 cells. J. Biol. Chem.260:6654–6662. 59. Nash, T. C., and M. J. Buchmeier.1997. Entry of mouse hepatitis virus into
cells by endosomal and nonendosomal pathways. Virology233:1–8. 60. Pedersen, K. W., Y. van der Meer, N. Roos, and E. J. Snijder.1999.
ORF1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double membrane vesicles which carry the viral replication complex. J. Virol.73:2016–2026.
61. Peters, P. J., G. Raposo, J. J. Neefjes, V. Oorschot, R. L. Leijendekker, H. J. Geuze, and H. L. Ploegh.1995. Major histocompatibility complex class II compartments in human B lymphoblastoid cells are distinct from early en-dosomes. J. Exp. Med.182:325–334.
62. Pinon, J. D., R. R. Mayreddy, J. D. Turner, F. S. Khan, P. J. Bonilla, and S. R. Weiss.1997. Efficient autoproteolytic processing of the MHV-A59 3C-like proteinase from the hydrophobic domains requires membranes. Vi-rology230:309–322.
63. Raposo, G., H. W. Nijman, W. Stoorvogel, R. Leijendekker, C. V. Harding, C. J. M. Melief, and H. J. Geuze.1996. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med.183:1161–1172.
64. Restrepo-Hartwig, M. A., and P. Ahlquist.1996. Brome mosaic virus heli-case- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol.70:8908–8916.
65. Schaad, M. C., P. E. Jensen, and J. C. Carrington.1997. Formation of plant RNA virus replication complexes on membranes: role of an endoplasmic reticulum-targeted viral protein. EMBO J.16:4049–4059.
66. Schiller, J. J., A. Kanjanahaluethai, and S. C. Baker.1998. Processing of the coronavirus MHV-JHM polymerase polyprotein: identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a. Virology242: 288–302.
67. Schlegel, A. S., T. H. Giddings, M. S. Ladinsky, and K. Kirkegaard.1996.
Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol.70:6576–6588.
68. Sethna, P. B., and D. A. Brian.1997. Coronavirus genomic and subgenomic minus-strand RNAs copartition in membrane-protected replication com-plexes. J. Virol.71:7744–7749.
69. Shaywitz, D. A., L. Orci, M. Ravazzola, A. Swaroop, and C. A. Kaiser.1995. Human SEC13Rp functions in yeast and is located on transport vesicles budding from the endoplasmic reticulum. J. Cell Biol.128:769–777. 70. Slot, J. W., H. J. Geuze, S. Gigengack, G. E. Lienhard, and D. E. James.
1991. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol.113:123–135.
71. Snijder, E. J., A. L. M. Wassenaar, L. C. van Dinten, W. J. M. Spaan, and A. E. Gorbalenya.1996. The arterivirus nsp4 protease is the prototype of a novel group of chymotrypsin-like enzymes, the 3C-like serine proteases. J. Biol. Chem.271:4864–4871.
72. Snijder, E. J., and J. J. M. Meulenberg.1998. The molecular biology of arteriviruses. J. Gen. Virol.79:961–979.
73. Snijder, E. J., A. L. M. Wassenaar, and W. J. M. Spaan.1994. Proteolytic processing of the replicase ORF1a protein of equine arteritis virus. J. Virol. 68:5755–5764.
74. Snijder, E. J., and W. J. M. Spaan.1995. The coronaviruslike superfamily.In S. G. Siddell (ed.), The Coronaviridae. Plenum Press, New York, N.Y. 75. Spaan, W. J. M., P. J. M. Rottier, M. C. Horzinek, and B. A. M. van der
Zeijst.1981. Isolation and identification of virus-specific mRNAs in cells infected with mouse hepatitis virus (MHV-A59). Virology108:424–434. 76. Talbot, P. J., R. L. Knobler, and M. J. Buchmeier.1984. Western and dot
immunoblotting analysis of viral antigens and antibodies: application to murine hepatitis virus. J. Immunol. Methods73:177–188.
77. Tang, B. L., F. Peter, J. Krijnse-Locker, S. H. Low, G. Griffiths, and W. Hong.1997. The mammalian homologue of yeast sec13p is enriched in the intermediate compartment and is essential for protein transport from the endoplasmic reticulum to the Golgi complex. Mol. Cell. Biol.17:256–266. 78. Tibbles, K. W., I. Brierley, D. Cavanagh, and T. D. Brown.1996.
Character-ization in vitro of an autocatalytic processing activity associated with the predicted 3C-like proteinase domain of the coronavirus avian infectious bronchitis virus. J. Virol.70:1923–1930.
79. van der Meer, Y., H. G. van Tol, J. Krijnse Locker, and E. J. Snijder.1998. ORF1a-encoded replicase subunits are involved in membrane association of the arterivirus replication complex. J. Virol.72:6689–6698.
80. van Deurs, B., O. W. Petersen, S. Olsnes, and K. Sandvig.1989. The ways of endocytosis. Int. Rev. Cytol.117:131–177.
81. van Dinten, L. C., A. L. M. Wassenaar, A. E. Gorbalenya, W. J. M. Spaan, and E. J. Snijder.1996. Processing of equine arteritis virus replicase ORF1b protein: identification of cleavage products containing the putative viral polymerase and helicase domains. J. Virol.70:6625–6633.
82. van Dinten, L. C., S. Rensen, A. E. Gorbalenya, and E. J. Snijder.1999. Proteolytic processing of the open reading frame 1b-encoded part of arteri-virus replicase is mediated by nsp4 serine protease and is essential for arteri-virus replication. J. Virol.73:2027–2037.
83. Vaux, D., J. Tooze, and S. Fuller.1990. Identification by idiotypic anti-bodies of an intracellular membrane protein that recognizes a mammalian endoplasmic reticulum retention signal. Nature345:495–502.
84. Wassenaar, A. L. M., W. J. M. Spaan, A. E. Gorbalenya, and E. J. Snijder. 1997. Alternative proteolytic processing of the arterivirus replicase ORF1a polyprotein: evidence that NSP2 acts as a cofactor for the NSP4 serine protease. J. Virol.71:9313–9322.
85. Weismiller, D. G., L. S. Sturman, M. J. Buchmeier, J. O. Fleming, and K. V. Holmes.1990. Monoclonal antibodies to the peplomer glycoprotein of coro-navirus mouse hepatitis virus identify two subunits and detect a conforma-tional change in the subunit released under mild alkaline conditions. J. Virol. 64:3051–3055.
86. Westaway, E. G., J. M. Mackenzie, M. T. Kenney, M. K. Jones, and A. A. Khromykh.1997. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol.71:6650–6661.
87. Williams, M. C.1987. Vesicles within vesicles: what role do multivesicular bodies play in alveolar type II cells? Am. Rev. Respir. Dis.135:744–746. 88. Ziebuhr, J., J. Herold, and S. G. Siddell.1995. Characterization of a human
coronavirus (strain 229E) 3C-like proteinase activity. J. Virol.69:4331–4338.
VOL. 73, 1999 LOCALIZATION OF MHV REPLICATION COMPLEX 7657