Associated Transcript, and Programmed Death 1 Contribute to Higher
Levels of Herpes Simplex Virus 1 Latency
Kevin R. Mott, Sariah J. Allen, Mandana Zandian, Homayon Ghiasi
Center for Neurobiology and Vaccine Development, Ophthalmology Research, Department of Surgery, Cedars-Sinai Burns & Allen Research Institute, Los Angeles, California, USA
ABSTRACT
The latency-associated transcript (LAT) of herpes simplex virus 1 (HSV-1), CD8
␣
ⴙdendritic cells (DCs), and programmed death
1 (PD-1) have all been implicated in the HSV-1 latency-reactivation cycle. It is not known, however, whether an interaction
be-tween LAT and CD8
␣
ⴙDCs regulates latency and T-cell exhaustion. To address this question, we used LAT-expressing
[LAT(
ⴙ
)] and LAT-negative [LAT(
ⴚ
)] viruses. Depletion of DCs in mice ocularly infected with LAT(
ⴙ
) virus resulted in a
re-duction in the number of T cells expressing PD-1 in the trigeminal ganglia (TG), whereas depletion of DCs in mice similarly
in-fected with LAT(
ⴚ
) virus did not alter PD-1 expression. CD8
␣
ⴙDCs, but not CD4
ⴙDCs, infected with LAT(
ⴙ
) virus had higher
levels of ICP0, ICP4, thymidine kinase (TK), and PD-1 ligand 1 (PD-L1) transcripts than those infected with LAT(
ⴚ
) virus.
Co-culture of infected bone marrow (BM)-derived DCs from wild-type (WT) mice, but not infected DCs from CD8
␣
ⴚ/ⴚmice, with
WT naive T cells contributed to an increase in PD-1 expression. Transfer of bone marrow from WT mice but not CD8
␣
ⴚ/ⴚmice
to recipient Rag1
ⴚ/ⴚmice increased the number of latent viral genomes in reconstituted mice infected with the LAT(
ⴙ
) virus.
Collectively, these data indicated that a reduction in latency correlated with a decline in the levels of CD8
␣
ⴙDCs and PD-1
ex-pression. In summary, our results demonstrate an interaction among LAT, PD-1, and CD11c CD8
␣
ⴙcells that regulates latency
in the TG of HSV-1-infected mice.
IMPORTANCE
Very little is known regarding the interrelationship of LAT, PD-1, and CD8
␣
ⴙDCs and how such interactions might contribute
to relative numbers of latent viral genomes. We show here that (i) in both
in vivo
and
in vitro
studies, deficiency of CD8
␣
ⴙDCs
significantly reduced T-cell exhaustion in the presence of LAT(
ⴙ
) virus but not LAT(
ⴚ
) virus; (ii) HSV-1 infectivity was
signifi-cantly lower in LAT(
ⴚ
)-infected DCs than in their LAT(
ⴙ
)-infected counterparts; and (iii) adoptive transfer of bone marrow
(BM) from WT but not CD8
␣
ⴚ/ⴚmice to recipient Rag1
ⴚ/ⴚmice restored latency to the level in WT mice following infection
with LAT(
ⴙ
) virus. These studies point to a key role for CD8
␣
ⴙDCs in T-cell exhaustion in the presence of LAT, which leads to
larger numbers of latent viral genomes. Thus, altering this negative function of CD8
␣
ⴙDCs can potentially be used to generate a
more effective vaccine against HSV infection.
A
characteristic feature of infection with herpes simplex virus 1
(HSV-1) is the ability of the virus to establish latency in
sen-sory neurons of an infected host (
1–4
). Individuals who have
ac-quired a latent infection are subject to episodic recurrences and
serve as permanent carriers who are intermittently infectious (
5–
7
). The recurrences are caused by reactivation of the virus, which
results in its transit back to the original site of infection (
8
,
9
).
More than 80 HSV-1 genes are expressed in neurons during lytic
infection. This expression of HSV-1 genes is drastically curtailed
during latency. Indeed, the latency-associated transcript (LAT) is
the only gene product consistently detected in abundance during
latency in infected mice, rabbits, and humans (
1
,
2
,
4
,
10
,
11
).
Using LAT-expressing [LAT(
⫹
)] and LAT-negative [LAT(
⫺
)]
viruses, we recently demonstrated that the presence of LAT leads
to the generation of dysfunctional T-cell responses in the
trigem-inal ganglia (TG) of latently infected mice (
12
). Both LAT
expres-sion and enhanced latency correlated with increased mRNA levels
of CD8 and the inhibitory receptor programmed death 1 (PD-1)
in the TG. These results suggested that TG that are latently
in-fected with LAT(
⫹
) virus contain both more CD8
⫹T cells and
more CD8
⫹T cells expressing the exhaustion marker, PD-1, than
TG that are latently infected with LAT(
⫺
) virus. This was
con-firmed by flow cytometry analyses of expression of CD3, CD8,
PD-1, HSV-1 gC, the gB
498 –505-specific CD8
⫹T-cell pentamer,
interleukin-2 (IL-2), gamma interferon (IFN-
␥
), and tumor
ne-crosis factor alpha (TNF-
␣
). The functional significance of PD-1
and its ligands in HSV-1 latency was indicated by the significantly
lower levels of HSV-1 latency in mice that were deficient in PD-1
or PD-1 ligand 1 (PD-L1) than in wild-type (WT) mice. The levels
of HSV-1 latency were unaffected in PD-L2-deficent mice. We
have also shown that latency is enhanced by immunization of
infected WT mice with FMS-like tyrosine kinase 3 ligand (Flt3L)
DNA, which increases the number of dendritic cells (DCs) (
13
,
Received27 February 2014 Accepted23 March 2014 Published ahead of print26 March 2014
Editor:R. M. Sandri-Goldin
Address correspondence to Homayon Ghiasi, [email protected]. Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00590-14
on November 7, 2019 by guest
http://jvi.asm.org/
14
). Conversely, depletion of DCs was associated with reduced
latency. Latency was also significantly lower in infected Flt3L
⫺/⫺and CD8
⫺/⫺mice than in infected WT mice. Interestingly,
how-ever, although immunization of Flt3L
⫺/⫺mice with Flt3L DNA
enhanced latency, immunization of CD8
⫺/⫺mice with Flt3L
DNA did not. Transfer experiments using DCs expanded
ex vivo
in the presence of Flt3L or granulocyte-macrophage
colony-stim-ulating factor (GM-CSF) suggested that enhanced latency was
as-sociated with the presence of lymphoid-related (CD11c
⫹CD8
␣
⫹)
DCs, whereas reduced latency was associated with
myeloid-re-lated (CD11c
⫹CD8
␣
⫺) DCs. Modulation of DC numbers by
Flt3L DNA immunization or by depletion did not, however, alter
acute virus replication in the eye and TG or eye disease in ocularly
infected mice (
13
,
14
). It has been shown that CD8
⫹T cells
infil-trate TG by 3 days post-ocular infection, and it has been
postu-lated that they act to inhibit HSV-1 reactivation (
15
). During
la-tency, a subset of CD8
ⴙT cells remain in direct contact with
infected neurons (
16
). These cells can block HSV-1 reactivation
from latency in
ex vivo
cultures of TG from latently infected mice
(
15
,
16
). These results suggest a role for CD8
ⴙT cells in HSV-1
latency, while our published studies indicate an involvement of
DCs in the efficiency with which HSV-1 establishes latency (
13
,
14
). Using a combination of knockout mouse and adoptive
trans-fer approaches, we have now demonstrated clearly that CD8
␣
ⴙDCs, rather than CD8
ⴙT cells, are responsible for enhanced
HSV-1 latency (
17
).
Collectively, our published studies show that (i) mice latently
infected with WT HSV-1 have higher levels of LAT RNA, CD8
mRNA, and PD-1 mRNA in the TG than those in mice depleted of
DCs (
13
,
14
,
18
); (ii) HSV-1 latency is significantly lower in
PD-1
⫺/⫺and PD-L1
⫺/⫺mice than in WT mice (
12
); and (iii) the
presence of CD11c
⫹CD8
␣
⫹cells directly increases the number of
HSV-1 latent viral genomes in mouse TG (
13
,
14
). However, very
little is known regarding the interrelationship of LAT, PD-1, and
CD8
␣
⫹DCs and how such interactions might contribute to
la-tency. We therefore undertook studies to test the hypothesis that
CD11c
⫹CD8
␣
⫹DCs in the presence of LAT harbor more
non-productive virus, which contributes to an increase in T-cell
ex-haustion and thus enhances latency. We report here that a
defi-ciency of CD8
␣
⫹DCs significantly reduced T-cell exhaustion in
LAT(
⫹
)-infected mice but not LAT(
⫺
)-infected mice.
Addition-ally,
in vitro
assays revealed that incubation of HSV-1-infected
WT DCs, but not similarly infected CD8
␣
⫺DCs, with T cells
isolated from naive WT mice promoted T-cell exhaustion.
Fur-thermore, adoptive transfer of bone marrow (BM) from WT but
not CD8
␣
⫺/⫺mice to recipient Rag1
⫺/⫺mice restored latency to
the level in WT mice following infection with LAT(
⫹
) virus.
These studies point to a key role for CD8
␣
⫹DCs in T-cell
exhaus-tion in the presence of LAT, which leads to higher levels of virus.
MATERIALS AND METHODS
Viruses and mice.Plaque-purified virulent HSV-1 strains McKrae [WT; LAT(⫹)] and dLAT2903 [LAT(⫺)] were grown in rabbit skin (RS) cell monolayers in minimal essential medium (MEM) containing 5% fetal calf serum (FCS), as we described previously (19,20). dLAT2903 was derived from strain McKrae, but with both copies of the LAT promoter (one in each viral long repeat) and the first 1,667 nucleotides (nt) of the LAT transcript deleted (20).
WT C57BL/6, C57BL/6-CD8␣⫺/⫺, C57BL/6-
2m⫺/⫺,
C57BL/6-DTR, C57BL/6-PD-L1⫺/⫺, and C57BL/6-Rag1⫺/⫺mice were used. All animal procedures adhered to the Association for Research in Vision and
Ophthalmology (ARVO) statement on the use of animals in ophthalmic and vision research and were conducted according to institutional animal care and use guidelines.
Ocular infection.Mice were infected ocularly with 2⫻105PFU per
eye of LAT(⫹) or LAT(⫺) virus. Each virus was suspended in 2l of tissue culture medium and administered as an eye drop without prior corneal scarification.
DC culture.Six-week-old mice were used as a source of bone marrow (BM) for the generation of mouse DCs in culture. BM cells were isolated by flushing femurs with phosphate-buffered saline (PBS). Pelleted cells were briefly resuspended in red blood cell lysing buffer (Sigma-Aldrich, St. Louis, MO) to lyse red blood cells and stabilized by adding complete medium (RPMI 1640, 10% fetal bovine serum, 100 U/ml penicillin, 100
g/ml streptomycin, 2 mML-glutamine). The cells were centrifuged and resuspended in complete medium supplemented with GM-CSF (100 ng/ ml; Peprotech, NJ) to enhance replication of DCs (21). Afterwards, cells were plated in non-tissue-culture plastic petri dishes (cells from 1 bone per 10-cm dish) for 5 days at 37°C with CO2. After 5 days, the medium was
removed and the adherent cells were recovered by incubating them for 5 min at 37°C with Versene (Invitrogen, San Diego, CA). Cells were washed, counted, and plated onto tissue culture dishes for use the following day.
Infection of DCs.Monolayers of DCs from the different strains of mice described above were infected with 1 PFU/cell of LAT(⫹) or LAT(⫺) virus. One hour after infection at 37°C, virus was removed and the infected cells were washed three times with fresh medium and then incubated in growth medium for 24 h. Infected DCs were used for the following studies. (i) Studies were performed to measure differences in the susceptibility of WT,2m⫺/⫺, CD8␣⫺/⫺, and PD-L1⫺/⫺DCs to
in-fection with LAT(⫹) or LAT(⫺) virus. Infected DC monolayers were harvested, and the presence of gB transcript was determined by quantita-tive real-time PCR (qRT-PCR). (ii) To measure the differences in the susceptibility of CD8␣⫹DCs versus CD4⫹DCs, infected DCs were har-vested and were separated into CD4⫹DCs and CD8␣⫹DCs as described below. The presence of ICP0, ICP4, thymidine kinase (TK), and PD-L1 transcripts in each cell population were determined by qRT-PCR as de-scribed below. (iii) To measure the activation state of DCs following HSV-1 infection, DCs isolated from WT mice were mock infected or infected with 1 PFU/cell of HSV-1 strain McKrae for 24 or 48 h. Mock-infected and Mock-infected DCs were stained, and flow cytometric analyses were performed as described below. (iv) Finally, to determine if DCs contrib-uted to T-cell exhaustion, mock-infected or HSV-1-infected DCs from WT or CD8␣⫺/⫺mice were cocultured with different ratios of naive T cells from WT mice for 24, 48, 72, or 96 h. The monolayers were harvested at various times postcocultivation and stained for flow cytometric analy-ses as described below.
Isolation of CD4ⴙand CD8␣ⴙDCs.Cultured DCs were grown as described above, harvested on day 5, and infected with 1 PFU/cell of LAT(⫹) or LAT(⫺) HSV-1 for 24 h. At 24 h postinfection (p.i.), isolation of CD8␣⫹and CD4⫹DCs was performed using a two-step magnetic cell sorting kit (Miltenyi Biotec, Auburn, CA) per the manufacturer’s proto-col. Briefly, harvested cells were directly labeled with CD8 microbeads, and CD8␣⫹DCs were isolated from the enriched DC fraction by positive selection. The magnetically labeled CD8␣⫹DCs were retained on a col-umn and eluted after removal of the colcol-umn from the magnetic field. Subsequently, the remaining enriched DCs that passed through the col-umn were directly labeled with CD4 microbeads, and CD4⫹DCs were isolated by the same method of positive selection in a magnetic field.
Adoptive transfer of BM to recipient mice.BM from WT C57BL/6 or CD8␣⫺/⫺mice was isolated, and each recipient Rag1⫺/⫺mouse received BM from one donor mouse intravenously (i.v.). At 2 weeks posttransfer, the Rag1⫺/⫺recipient mice were ocularly infected with LAT(⫹) virus. TG from infected mice were isolated for detection of LAT RNA and gB DNA on day 30 p.i.
Depletion of DCs.C57BL/6-DTR mice were depleted of their DCs by use of 100 ng of diphtheria toxin (DT) in 100l of PBS via the
intraper-Mott et al.
on November 7, 2019 by guest
http://jvi.asm.org/
itoneal (i.p.) route 24 h prior to ocular infection and at days 2, 5, 10, and 14 p.i. As a negative control, a subset of mice was similarly injected with PBS alone and is therefore referred to as mock depleted. Efficiency of DC depletion in corneas and spleens was monitored by flow cytometric anal-ysis before ocular infection and at day 5 p.i. After the first depletion, more than 90% of DCs had been depleted.
Confocal microscopy and image analysis.DCs isolated from differ-ent strains of mice were grown on Lab-Tex chamber slides (Sigma-Al-drich) as we described previously (22). Briefly, cells were fixed by incu-bating slides in methanol for 10 min followed by acetone for 5 min at
⫺20°C. Afterwards, slides were rinsed three times for 5 min each at am-bient temperature in PBS containing 0.05% (vol/vol) Tween 20 (PBS-T). Slides were then blocked for 30 min at ambient temperature in PBS-T containing 1% (wt/vol) bovine serum albumin (BSA). Rat anti-CD8␣ (clone 53-6.7; eBioscience, San Diego, CA), rat anti-CD4 (clone Gk1.5; eBioscience), rat anti-CD8(clone YTS156.7.7; BioLegend), and hamster anti-CD11c (clone HL3; BD Biosciences) were used for immunohisto-chemistry (IHC). Immunostaining was done using a CD11c-CD4, CD11c-CD8␣, or CD11c-CD8antibody combination and staining for 1 h at 25°C. After three rinses for 5 min each in PBS, slides were incubated for 1 h at 25°C with secondary antibodies labeled with anti-hamster– fluorescein isothiocyanate (FITC) or anti-rat– tetramethyl rhodamine isocyanate (TRITC) (Invitrogen). Slides were washed three times with PBS, air dried, and mounted with Prolong Gold DAPI (4= ,6-diamidino-2-phenylindole) mounting medium (Invitrogen). Images were captured at 1,024 by 1,024 pixels (original magnification⫽ ⫻20) in independent fluorescence channels, using a Nikon C1 Eclipse inverted confocal micro-scope.
Flow cytometric analysis.Infected or mock-infected cells were har-vested at various times p.i. and stained with CD3, CD4, anti-CD8␣, anti-PD-1, and anti-CD11c, obtained from BD Pharmingen (San Diego, CA) and BioLegend (San Diego, CA), and then analyzed by flow cytometry as we previously described (23).
DNA extraction and PCR analysis of HSV-1 genomic DNA.DNA was isolated from homogenized individual TG by using a commercially available DNeasy Blood & Tissue kit (Qiagen, Stanford, CA) according to the manufacturer’s instructions. PCR analyses were done using gB-spe-cific primers (forward, 5=-AACGCGACGCACATCAAG-3=; reverse, 5=-C TGGTACGCGATCAGAAAGC-3=; and probe, 5=-6-carboxyfluorescein [FAM]-CAGCCGCAGTACTACC-3=). The amplicon length for this primer set was 72 bp. Relative copy numbers for gB DNA were calculated using standard curves generated from the plasmid pAc-gB1 (24). In all experiments, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization of transcripts.
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.RNA was extracted from latent TG or infected DCs as we described previously (22, 25,26). Following RNA extraction, 1g of total RNA was reverse tran-scribed using random hexamer primers and murine leukemia virus (MuLV) reverse transcriptase from a High Capacity cDNA reverse tran-scription kit (Applied Biosystems, Foster City, CA), in accordance with the manufacturer’s recommendations. The levels of various transcripts were evaluated using commercially available TaqMan gene expression assays (Applied Biosystems) with optimized primer and probe concentra-tions. Primer-probe sets consisted of two unlabeled PCR primers and a FAM-labeled TaqMan MGB probe formulated into a single mixture. Ad-ditionally, all cellular amplicons included an intron-exon junction to eliminate signals from genomic DNA contamination. The HSV-1 ICP0, ICP4, TK, gB, LAT, and PD-L1 primer and probe sequences were as fol-lows: (i) for ICP0, the forward primer sequence was 5=-CGGACACGGA ACTGTTCGA-3=, the reverse primer sequence was 5=-CGCCCCCGCAA CTG-3=, and the probe sequence was 5=-FAM-CCCCATCCACGCCCT G-3= (amplicon length ⫽111 bp); (ii) for ICP4, the forward primer sequence was 5=-GCGTCGTCGAGGTCGT-3=, the reverse primer se-quence was 5=-CGCGGAGACGGAGGAG-3=, and the probe sequence was 5=-FAM-CACGACCCCGACCACC-3=(amplicon length⫽69 bp);
(iii) for TK, the forward primer sequence was 5=-CAGTAGCGTGGGCA TTTTCTG-3=, the reverse primer sequence was 5=-CCTCGCCGGCAAC AAAA-3=, and the probe sequence was 5=-FAM-CTCCAGGCGGACTT C-3=(amplicon length⫽59 bp); (iv) for gB, the forward primer sequence was 5=-AACGCGACGCACATCAAG-3=, the reverse primer sequence was 5=-CTGGTACGCGATCAGAAAGC-3=, and the probe sequence was 5=-F AM-CAGCCGCAGTACTACC-3=(amplicon length ⫽72 bp); (v) for LAT, the forward primer sequence was 5=-GGGTGGGCTCGTGTTACA G-3=, the reverse primer sequence was 5=-GGACGGGTAAGTAACAGAG TCTCTA-3=, and the probe sequence was 5=-FAM-ACACCAGCCCGTT CTTT-3=(amplicon length⫽81 bp, corresponding to LAT nt 119553 to 119634); and (vi) for PD-L1, ABI Mm00452054_m1 assay primers were used (amplicon length⫽77 bp). As an internal control, a set of GAPDH primers from Applied Biosystems (m999999.15_G1 assay primers) (am-plicon length⫽107 bp) was used.
qRT-PCR was performed using an ABI Prism 7900HT sequence de-tection system (Applied Biosystems, Foster City, CA) in 384-well plates as we described previously (25,26). Real-time PCR was performed in tripli-cate for each tissue sample. Threshold cycle (CT) values, which represent
the PCR cycles at which there was a noticeable increase in the reporter fluorescence above baseline, were determined using SDS 2.2 software. The GAPDH transcript was used for normalization purposes.
Statistical analysis.Student’sttest and the chi-square test were per-formed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant when thePvalue was
⬍0.05.
RESULTS
DCs play an indispensable role in regulating T-cell exhaustion
in mice latently infected with HSV-1.
We have shown that
deple-tion of DCs reduces the number of HSV-1 latent genomes in
ocu-larly infected mice (
13
,
14
) and that a decrease in latency correlates
with a decrease in T-cell exhaustion in the TG of latently infected
mice (
12
). However, very little is known regarding the role of DCs
in T-cell exhaustion. As a proof-of-principal experiment, we
de-pleted DCs in diphtheria toxin receptor (DTR) transgenic mice by
treatment with DT 1 day before ocular infection with LAT(
⫹
) or
LAT(
⫺
) HSV-1 and at various times p.i. As controls, DTR mice
were mock depleted of DCs and infected ocularly, DTR mice were
both mock depleted and mock infected (and designated “naive”),
and WT C57BL/6 mice were infected with the viruses. Since 30
days p.i. is accepted as a point in time by which latency has been
established (
27
), the TG were isolated from individual mice at day
30 p.i., and single-cell suspensions of the TG from each mouse
were stained for CD3, CD8, and PD-1. The percentages of CD3
⫹CD8
⫹PD-1
⫹T cells in the TG of DC-depleted, mock-depleted,
and WT mice infected with LAT(
⫹
) virus, as well as naive mice,
are shown in
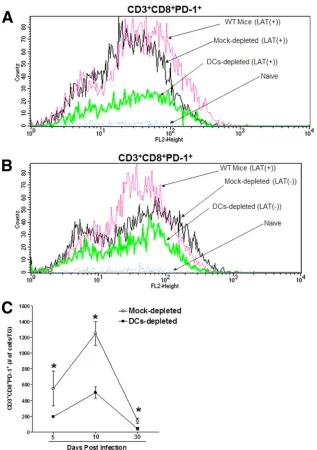
Fig. 1A
. As would be expected, there were few CD3
⫹CD8
⫹PD-1
⫹T cells in the TG from naive mice, and the largest
numbers of CD3
⫹CD8
⫹PD-1
⫹cells were found in the TG of the
WT and mock-depleted DTR control mice. The numbers of
CD3
⫹CD8
⫹PD-1
⫹cells in the TG of the DC-depleted mice were
significantly lower than those in the two virus-infected controls
(
Fig. 1A
). Moreover, the numbers of CD3
⫹CD8
⫹PD-1
⫹cells in
the TG of DC-depleted mice that were infected with LAT(
⫺
) virus
did not differ significantly from the numbers of CD3
⫹CD8
⫹PD-1
⫹cells in the TG of mock-depleted controls (
Fig. 1B
). These
results are consistent with a previously reported study of the
num-ber of CD8-positive T cells in TG of mice infected with LAT(
⫺
)
virus versus LAT(
⫹
) virus (
12
).
We next analyzed the kinetics of CD3/CD8/PD-1 expression
during both lytic (days 5 and 10) and latent (day 30) infection in
on November 7, 2019 by guest
http://jvi.asm.org/
the TG of DC-depleted or mock-depleted DTR mice infected with
LAT(
⫹
) virus (
Fig. 1C
). During both the lytic and latent phases of
HSV-1 infection, the numbers of CD3
⫹CD8
⫹PD-1
⫹T cells were
significantly lower in the TG of DC-depleted mice than in the TG of
the mock-depleted mice. In both groups, the numbers of CD3
⫹CD8
⫹PD-1
⫹cells increased through day 10 p.i. and declined by day
30 p.i. Similar to the results of this study, it was previously shown that
by 3 days p.i., CD8
⫹T cells begin infiltrating the TG, peaking at 14
days (clear of virus by day 11 p.i.), declining significantly after
clear-ance of lytic infection, and persisting as latent virus (
12
,
16
,
27–29
). In
addition, in line with our results, it was previously shown that the
level of PD-1 expression during the early stage of human hepatitis C
virus (HCV) infection is significantly higher for subjects who
prog-ress to chronic HCV infection than for those who clear infection (
30
).
Furthermore, during acute HCV infection, the loss of PD-1
expres-sion by CD8
⫹T cells was shown to correlate with viral clearance (
31
).
These data support our overall hypothesis that the presence of LAT
and DCs affects the level of T-cell exhaustion during both the lytic and
latent cycles of HSV-1 infection.
Expression of CD4 and CD8
␣
transcripts in BM-derived DCs
isolated from different strains of mice before HSV-1 infection.
Recently, we reported that the absence of CD8
␣
⫹DCs correlated
FIG 1Role of DCs in CD8 T-cell exhaustionin vivo. DTR mice were depleted of their DCs by use of diphtheria toxin or were mock depleted with PBS, as described in Materials and Methods. Mice were ocularly infected with LAT(⫹) or LAT(⫺) virus, and individual mouse TG were collected on days 5, 10, and 30 p.i. Controls included infected WT mice and noninfected naive DTR mice. On days 5, 10, and 30 p.i., TG from each mouse were isolated and digested with collagenase (400 IU/TG). The cell suspension was filtered through a 45-mm cell strainer, stained with PAC Blue–anti-CD8, FITC–anti-PD-1, and allophyco-cyanin (APC)–anti-CD3, and analyzed by flow cytometry. (A) Representative histograms of CD3⫹CD8⫹PD1⫹T cells on day 30 p.i. in mice infected with LAT(⫹) virus. (B) Representative histograms of CD3⫹CD8⫹PD1⫹T cells on day 30 p.i. in mice infected with LAT(⫺) virus. (C) Mean numbers of CD3⫹CD8⫹ PD1⫹T cells from DC-depleted and mock-depleted mice infected with LAT(⫹) virus on days 5, 10, and 30 p.i. Data for three mice per treatment are shown.Mott et al.
on November 7, 2019 by guest
http://jvi.asm.org/
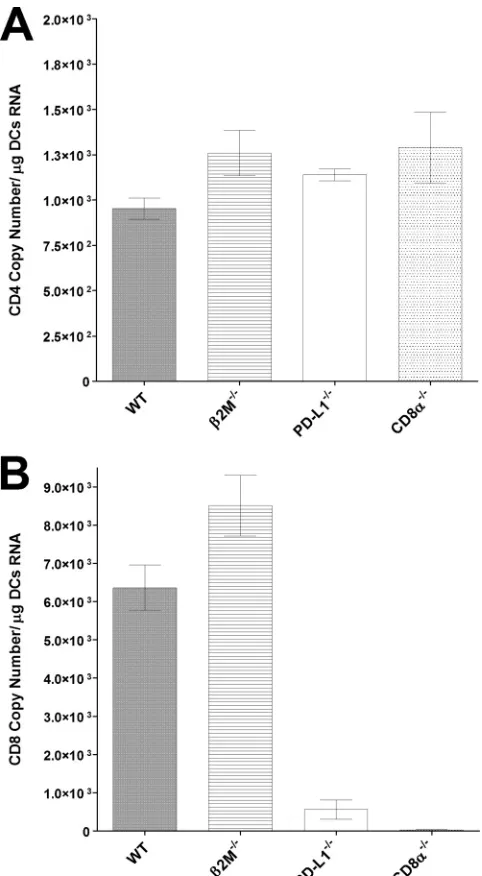
[image:4.585.134.451.63.513.2]with reduced latency (
17
). To determine if lower levels of latency
were associated with a reduction in CD8
␣
⫹transcripts, we
quan-tified CD4 and CD8
␣
transcripts in BM-derived DCs isolated
from WT,

2m
⫺/⫺, PD-L1
⫺/⫺, and CD8
␣
⫺/⫺naive mice. The
CD4 copy numbers were similar in the DCs from all four strains of
mice (
Fig. 2A
) (
P
⬎
0.05). Although the copy numbers of CD8
␣
transcripts in DCs isolated from WT mice were similar to the copy
numbers found in

2m
⫺/⫺mice, the copy numbers of the CD8
␣
transcripts were significantly lower in the DCs isolated from the
PD-L1
⫺/⫺mice (
Fig. 2B
) (
P
⬍
0.0001). As expected, no detectable
levels of CD8
␣
transcript were detected in DCs isolated from
CD8
␣
⫺/⫺mice (
Fig. 2B
). Thus, lower levels of CD8
␣
transcripts
in DCs may be associated with reduced latency
in vivo
, as we
ob-served for CD8
␣
⫺/⫺(
17
) and PD-L1
⫺/⫺(
12
) mice.
HSV-1 infectivity is reduced in DCs isolated from CD8
␣
ⴚ/ⴚmice.
As we have shown previously, CD8
␣
⫺/⫺mice that are
ocu-larly infected with HSV-1 harbor fewer viruses than CD8
␣
⫹/⫹mice that are similarly infected (
17
). Therefore, we sought to
de-termine whether CD8
␣
⫹DCs (from WT,

2m
⫺/⫺, or PD-L1
⫺/⫺mice) express more HSV-1 transcripts than CD8
␣
⫺DCs (from
CD8
␣
⫺/⫺mice) when infection is carried out
in vitro
. DCs were
isolated from naive WT,

2m⫺/⫺, PD-L1
⫺/⫺, and CD8
␣
⫺/⫺mice.
The DCs were then infected with 1 PFU/cell of LAT(
⫹
) virus. At
24 h p.i., RNAs were isolated, and the copy numbers of the gB
transcript were measured using qRT-PCR (
Fig. 3
, left side). The
levels of gB transcripts in DCs isolated from WT,

2m⫺/⫺, and
PD-L1
⫺/⫺mice and infected with LAT(
⫹
) virus were similar to
each other. In contrast, the levels of gB transcripts in the infected
CD8
␣
⫺/⫺DCs were at least 10-fold lower (
Fig. 3
) [
P
⬍
0.0001 for
LAT(
⫹
) virus].
We showed previously that HSV-1 latency after infection with
LAT(
⫺
) virus is significantly less than that after infection with
LAT(
⫹
) virus (
12
), although the replication of these viruses is
similar both
in vitro
and
in vivo
(
20
). To determine if the levels of
gB transcripts were similar in DCs infected with LAT(
⫺
) virus and
those infected with LAT(
⫹
) virus, we infected DCs from WT,

2m
⫺/⫺, PD-L1
⫺/⫺, or CD8
␣
⫺/⫺mice with 1 PFU/cell of LAT(
⫺
)
virus (
Fig. 3
, right side). Similar to the results obtained for
infec-tion of the mice with LAT(
⫹
) virus, the levels of gB transcripts
were significantly lower in the LAT(
⫺
)-infected CD8
␣
⫺/⫺DCs
than the LAT(
⫺
)-infected WT or

2m
⫺/⫺DCs (
Fig. 3
) [
P
⬍
0.05
for LAT(
⫺
) virus]. However, the LAT(
⫺
)-infected PD-L1
⫺/⫺ FIG 2Expression of CD4 and CD8 transcripts in BM-derived DCs.Subcon-fluent monolayers of DCs from naive WT,2m⫺
/⫺, PD-L1⫺/⫺, or CD8␣⫺/⫺ mice were harvested, total RNA was isolated, and TaqMan RT-PCR was per-formed using CD4- and CD8␣-specific primers as described in Materials and Methods. GAPDH was used as an internal reference control. Data represent mean copy numbers⫾standard errors of the means (SEM) (n⫽4). (A) Copy numbers for CD4 transcript. (B) Copy numbers for CD8␣transcript.
FIG 3HSV-1 infection of DCs isolated from different strains of mice. Sub-confluent monolayers of BM-derived DCs from WT,2m⫺
/⫺, PD-L1⫺/⫺, or CD8␣⫺/⫺mice were infected with 1 PFU/cell of LAT(⫹) or LAT(⫺) virus for 24 h as described in Materials and Methods. RNAs were isolated, and the copy numbers of HSV-1 gB transcripts were determined by TaqMan qRT-PCR. GAPDH was used as an internal reference control. Data represent means⫾ SEM (n⫽4).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.300.539.58.291.2] [image:5.585.43.284.65.503.2]DCs had lower levels of gB transcripts than LAT(
⫺
)-infected WT,

2m⫺/⫺, or CD8
␣
⫺/⫺DCs (
Fig. 3
) [
P
⬍
0.05 for LAT(
⫺
) virus].
Overall, the levels of gB transcripts were significantly lower in the
LAT(
⫺
)-infected WT,

2m⫺/⫺, and PD-L1
⫺/⫺DCs than in their
LAT(
⫹
)-infected counterparts (
Fig. 3
, compare left panel with
right panel). These results further bolster the notion that both
LAT and CD8
␣
⫹DCs contribute to an increase of nonproductive
HSV-1 infection in DCs.
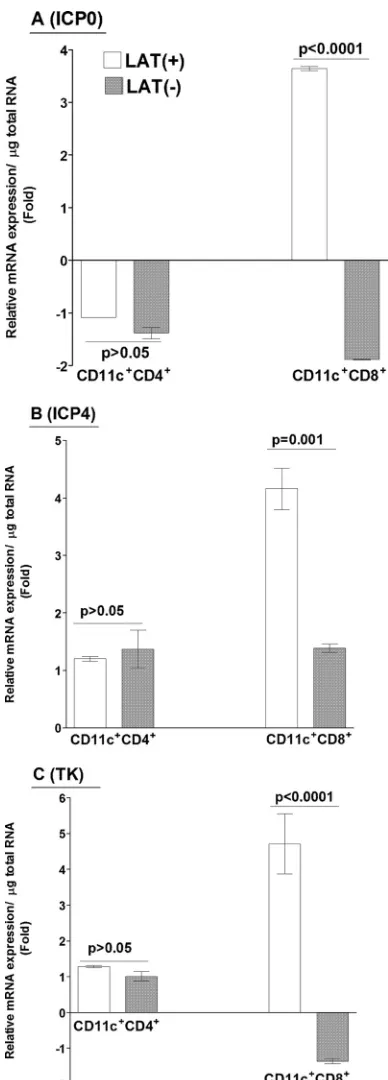
CD8
␣
ⴙDCs harbor more HSV-1 transcripts than do CD4
ⴙDCs
in vitro
.
As we have shown above (
Fig. 3
) that isolated DCs
from CD8
␣
⫺/⫺mice have less expression of gB transcripts than
DCs isolated from WT,

2m⫺/⫺, and PD-L1
⫺/⫺mice, which are
CD8
␣
⫹/⫹, we sought to determine whether CD8
␣
⫹DCs harbor
more virus than other subclasses of DCs do. DCs from naive, WT
mice were infected with 1 PFU/cell of LAT(
⫹
) or LAT(
⫺
) virus
for 24 h. At 24 h p.i., the DCs were harvested and sorted into
CD8
␣
⫹(CD4
⫺) and CD4
⫹(CD8
␣
⫺) subpopulations. Total RNA
was isolated from each cell population, and the expression of
var-ious viral transcripts was quantified by RT-PCR. ICP0 and ICP4
were used as indicators of
␣
genes, and TK was used as a

gene
marker. The relative expression levels of ICP0 (
Fig. 4A
), ICP4 (
Fig.
4B
), and TK (
Fig. 4C
) transcripts isolated from CD11c
⫹CD4
⫹cells infected with LAT(
⫹
) virus were not significantly different
from those from CD11c
⫹CD4
⫹cells infected with LAT(
⫺
) virus.
In contrast, the abundance of each of these three transcripts was
significantly higher in the CD11c
⫹CD8
␣
⫹cells infected with
LAT(
⫹
) virus than in those infected with LAT(
⫺
) virus (
Fig. 4
).
This suggested the possibility that these higher levels of HSV-1
transcripts in CD8
␣
⫹DCs may contribute directly or indirectly to
greater latency by inhibiting DC function, as suggested previously
(
32
). However, there was no LAT-dependent effect on the increase
of viral transcripts in CD4
⫹T cells.
FIG 4Expression of HSV-1 transcripts in CD8␣⫹DCs. Subconfluent mono-layers of BM-derived DCs from WT mice were infected with 1 PFU/cell of LAT(⫹) or LAT(⫺) virus as described in Materials and Methods. At 24 h p.i., infected DCs were fractionated into CD11c⫹ CD4⫺CD8␣⫹and CD11c⫹ CD4⫹CD8␣⫺subsets. Total RNA was isolated, and TaqMan RT-PCR was performed using ICP0-, ICP4-, and TK-specific primers as described in Mate-rials and Methods. GAPDH was used as an internal reference control. Data represent means⫾SEM (n⫽4) for two separate experiments. (A) ICP0; (B) ICP4; (C) TK.
FIG 5Expression of PD-L1 transcript in CD8␣⫹DCs. Subconfluent mono-layers of BM-derived DCs from WT mice were infected with 1 PFU/cell of LAT(⫹) or LAT(⫺) virus or mock infected as described forFig. 4. Infected and mock-infected DCs were fractionated into CD11c⫹ CD4⫺ CD8␣⫹ and CD11c⫹CD4⫹CD8␣⫺subsets. Total RNA was isolated, and TaqMan RT-PCR was performed using PD-L1-specific primers as described in Materials and Methods. Expression of PD-L1 in the CD11c⫹ CD4⫺ CD8␣⫹ and CD11c⫹CD4⫹CD8␣⫺subsets was normalized to that in their uninfected counterparts. GAPDH was used as an internal reference control. Data repre-sent means⫾SEM (n⫽4) for two separate experiments.
Mott et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.65.259.63.603.2] [image:6.585.309.529.65.252.2]More expression of PD-L1 transcript in CD8
␣
ⴙDCs infected
with WT HSV-1.
We showed previously that LAT expression and
enhanced latency were associated with more PD-1 expression
(
12
). Although PD-1 has two ligands, PD-L1 (
33
) and PD-L2 (
34
),
we found that PD-L1, not PD-L2, is associated with a higher level
of latency (
12
). Since DC function can be inhibited by HSV
infec-tion (
32
), we tested whether the higher expression levels of viral
transcripts in CD11c
⫹CD8
␣
⫹cells described above (
Fig. 4
)
con-tribute to more expression of PD-L1 transcripts. We found that
the abundance of the PD-L1 transcript was significantly greater in
both CD11c
⫹CD8
␣
⫹and CD11c
⫹CD4
⫹cells infected with
LAT(
⫹
) virus than in those infected with LAT(
⫺
) virus (
Fig. 5
).
Thus, LAT acts, at least in part, by contributing to more
expres-sion of PD-L1 in HSV-1-infected DCs.
Infected DCs from WT mice contribute to T-cell exhaustion
in vitro
.
PD-1 expression is not simply an indicator of exhaustion.
All T cells express PD-1 approximately 24 h after activation;
how-ever, this is usually followed by a rapid decline in expression. In
contrast, PD-1 expression remains high in cells that are
chroni-cally stimulated, such as during HSV-1 latency, and they become
refractory to stimulation or exhausted (
12
,
35
). To examine if
elevated expression of PD-L1 in LAT(
⫹
)-infected DCs is
associ-ated with greater T-cell exhaustion, we incubassoci-ated naive T cells
isolated from WT mice with WT DCs infected with 1 PFU/cell of
LAT(
⫹
) virus as described in Materials and Methods. Controls
included T cells alone (no DCs) and incubation of the T cells with
mock-infected DCs. Cocultures were carried out for 24, 48, 72,
and 96 h. At the indicated time points, cells were harvested,
stained with CD3, CD4, CD8, and PD-1 antibodies, and analyzed
using flow cytometry. The numbers of CD3
⫹CD8
⫹PD-1
⫹T cells
at 96 h were similar in the T cells that were cultured alone and
those that were cultured with mock-infected DCs. At this time
point, all of the CD8
⫹T cells that were cocultured with infected
DCs expressed PD-1 (
Fig. 6A
). The cumulative results from three
separate experiments are shown in
Fig. 6B
. There were no
differ-ences between the control groups (i.e., no DCs and mock-infected
DCs) from 24 to 96 h postcoculture (
Fig. 6B
). Similarly, there were
no differences in CD3
⫹CD8
⫹PD-1
⫹signals between T cells
in-cubated with infected DCs and the control groups at 24 and 48 h,
but at 72 h, the T cells that were cocultured with the infected DCs
had significantly higher levels of PD-1 expression than the control
groups (
Fig. 6B
).
FIG 6Expansion of exhausted T cells in the presence of infected DCs from WT mice. Subconfluent monolayers of BM-derived DCs from WT mice were infected with 1 PFU/cell of LAT(⫹) virus. At 24 h p.i., 1⫻106infected DCs were incubated with similar amounts of purified naive T cells isolated from WT mice. As
controls, a subset of T cells were cultured without any DCs or incubated with noninfected DCs. At 24, 48, 72, and 96 h postcoculture, cells were harvested, stained with anti-CD3, anti-CD4, anti-CD8, and anti-PD-1 monoclonal antibodies (MAbs), and analyzed by flow cytometry. A minimum of 104events was acquired on
a gate, including viable cells. (A) At 96 h postincubation, CD3 T cells were gated on expression of CD8 and PD-1. The number in each quadrant indicates the percentage of single- or double-positive T cells per treatment. (B) Mean percentages of CD3⫹PD-1⫹CD8⫹T cells at different times postcoculture are shown for each treatment from three experiments. (C) At 96 h postincubation, CD3 T cells were gated on expression of CD4 and PD-1. The number in each quadrant indicates the percentage of single- or double-positive T cells for each treatment condition. (D) Mean percentages of CD3⫹PD-1⫹CD4⫹T cells at different times postcoculture for each treatment are shown for three independent experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.70.515.67.384.2]In the same experiment, we examined the effects of DCs on the
PD-1 signal in CD3
⫹CD4
⫹cells (
Fig. 6C
and
D
). As we had found
for the CD8
⫹T cells (
Fig. 6A
and
B
), the numbers of CD3
⫹CD4
⫹PD-1
⫹T cells were similar for T cells that were cultured in the
absence of DCs (
Fig. 6C
, left panel) and those that were cultured
with mock-infected DCs (
Fig. 6C
, middle panel). However, when
the T cells were incubated with infected DCs, almost all of the
CD4
⫹T cells were PD-1
⫹at 96 h (
Fig. 6C
, right panel). The
cu-mulative results from three separate experiments are shown in
Fig.
6D
. The above results suggest that HSV-1-infected DCs
contrib-ute to increased PD-1 expression and T-cell exhaustion.
Since CD8
␣
⫹DCs harbor more viruses and CD8
␣
⫺/⫺mice
have smaller numbers of latent viral genomes than those in
con-trol strains, the greater propensity toward exhaustion of T cells
cultured with HSV-1-infected WT DCs (
Fig. 6A
to
D
) might be
attributable specifically to the CD8
␣
⫹subpopulation of DCs. To
more directly evaluate this, DCs from CD8
␣
⫺/⫺mice were
in-fected or mock inin-fected as described above and then incubated
with WT naive T cells for 96 h. In contrast to incubation of T cells
with HSV-1-infected WT DCs, incubation with infected
CD8
␣
⫺/⫺DCs did not affect PD-1 expression in either CD4
⫹or
CD8
⫹T cells (
Fig. 7
). Thus, HSV-1-infected CD8
␣
⫹DCs
contrib-ute to the increase in PD-1 expression in T cells.
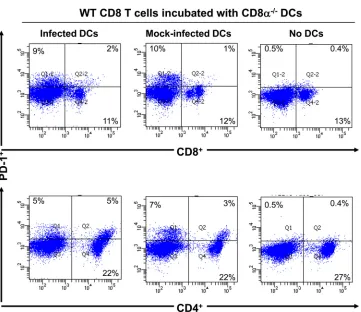
BM DCs from CD8
␣
ⴚ/ⴚmice influence latency.
Rag1
⫺/⫺mice do not have mature T cells or B cells (
36
), and it has been
reported that they exhibit a 10-fold reduction in the number of
DCs (
37
). DCs generated
in vitro
from Rag1
⫺/⫺thymic
progeni-tors have lymphoid characteristics, but CD8
␣
⫹DCs are absent
(
38
,
39
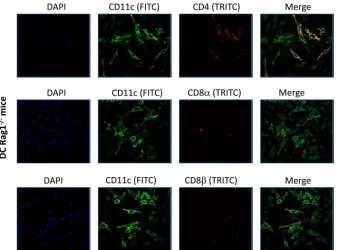
). We confirmed that CD11c
⫹CD8
␣
⫹or CD11c
⫹CD8

⫹DCs were not detectable in the bone marrow of Rag1
⫺/⫺mice,
although CD11c
⫹CD4
⫹cells were present (
Fig. 8
). Thus, to
fur-ther evaluate the contribution of CD8
␣
⫹DCs to the enhancement
of latent viral loads, we reconstituted Rag1
⫺/⫺mice with BM cells
isolated from WT or CD8
␣
⫺/⫺mice as described in Materials and
Methods. At 2 weeks posttransfer, mice were ocularly infected
with LAT(
⫹
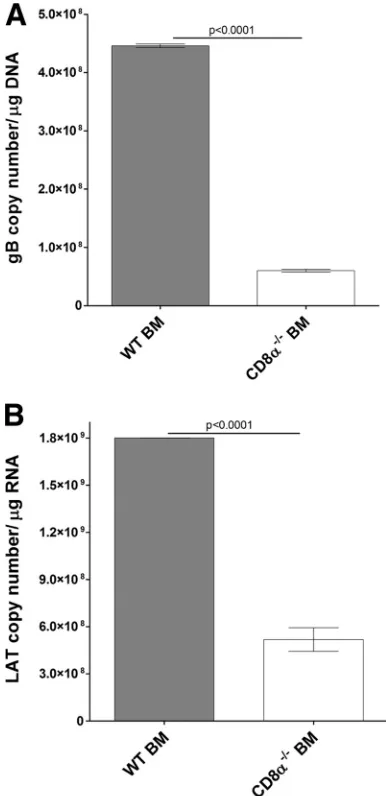
) virus, and the TG were harvested 30 days later. The
copy numbers of gB (
Fig. 9A
) and LAT (
Fig. 9B
) were significantly
lower in the TG of Rag1
⫺/⫺mice that received BM from CD8
␣
⫺/⫺mice than in the TG of Rag1
⫺/⫺mice that received BM from WT
mice (
Fig. 9
) (
P
⬍
0.0001). Thus, in line with our
in vitro
results,
reduced latency in Rag1
⫺/⫺mice appears to involve the absence of
CD8
␣
⫹DCs.
DISCUSSION
HSV infections are among the most frequent serious viral eye
infections in the United States and are a major cause of
virus-induced blindness (
8
,
9
,
40
). Because of the problems associated
with recurrent ocular infection, reducing the establishment of
la-FIG 7PD-1 expression was not altered in the presence of infected DCs from CD8␣⫺/⫺mice. Subconfluent monolayers of BM-derived DCs from CD8␣⫺/⫺mice were infected with 1 PFU/cell of LAT(⫹) virus as described in the text. At 24 h p.i., 1⫻106infected DCs were incubated with similar amounts of purified naiveT cells isolated from WT mice. As controls, a subset of T cells was cultured without any DCs or incubated with uninfected DCs. At 96 h postcoculture, cells were isolated, stained with anti-CD3, anti-CD4, anti-CD8, and anti-PD-1 MAbs, and analyzed by flow cytometry. A minimum of 104events was acquired on a gate,
including viable cells. Upper panels show CD3 T cells gated on expression of CD8 and PD-1, while lower panels show CD3 T cells gated on expression of CD4 and PD-1. The number in each quadrant indicates the percentage of single- or double-positive T cells for each treatment condition.
Mott et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.113.474.60.372.2]tency should be a major goal of a prophylactic vaccine against
ocular HSV-1. Previously, we showed that increased latency
cor-relates with higher levels of PD-1 expression and loss of T-cell
function (
12
). In addition, we have shown that CD8
␣
⫹DCs
en-hance HSV-1 latency in the TG of infected mice and that
immu-nization of mice with Flt3L, which increases the number of
CD8
␣
⫹cell-related DCs, increases the levels of latent virus in the
TG (
13
,
14
). In this study, we showed that depletion of DCs
sig-nificantly reduced T-cell exhaustion in mice ocularly infected with
LAT(
⫹
) virus. In contrast, depletion of DCs in mice infected with
LAT(
⫺
) virus did not alter the level of PD-1 expression. Thus, the
current study confirms and extends our previous finding that LAT
contributes to higher latent viral loads and to T-cell exhaustion
(
12
) by showing that LAT promotes T-cell exhaustion and that the
subsequent increase in latency is orchestrated by CD8
␣
⫹DCs.
DCs are essential for antigen presentation and the initiation of
protective T-cell responses and have been classified into several
subsets based on their immunophenotypes, resident locations,
and functional differences (
41
,
42
). Previously, we showed that
CD4
⫺CD8
␣
⫹DCs contribute to the promotion of HSV-1
la-tency, whereas CD4
⫹CD8
␣
⫺DCs contribute to a reduction in
latency (
13
,
14
). CD8
␣
⫹DCs can be induced in the absence of the
transcription factors Id2, Nfil3, and Batf3, but not IRF8 (
43
).
BXH2 mice have a spontaneous point mutation in the IRF8 gene,
similar to that in IRF8
⫺/⫺mice that blocks the generation of
CD8
␣
⫹DCs (
44
,
45
). Most recently, we showed that the levels of
both gB DNA and LAT RNA were significantly lower in BXH2
mice than in their WT counterparts (
17
). Notably, BXH2 mice
lack CD8
␣
⫹DCs but, in contrast to CD8
␣
⫺/⫺mice, have normal
CD8
⫹T cells.
In this study, we found that CD8
␣
⫹DCs harbor more virus
than other populations of DCs and that this higher susceptibility
of DCs is directly influenced by the presence of LAT. LAT has
antiapoptosis activity, and both
in vitro
and
in vivo
studies have
shown that infected cells have a greater propensity toward
apop-tosis and cell death in the absence of LAT (
27
,
46–50
).
Conse-quently, in the presence of LAT, a reduction in the levels of
apop-tosis and cell death may lead to more expression of viral
transcripts. Thus, we propose that infection of DCs with WT
HSV-1 may interfere with the function of DCs, either directly or
through a negative-feedback control mechanism, thus inhibiting
induction of antiviral responses and leading to T-cell exhaustion
and higher levels of latency. Similar to the results of this study, it
has been shown that CD8
␣
⫹DCs are more susceptible to
infec-tion (
51
,
52
). The infected CD8
␣
⫹DCs may lose function and/or
block the antigen-presenting function of other DC subtypes. This
may exacerbate viral latency and loss of T-cell function. It has been
reported that HSV-1 infection of human DCs results in functional
impairment of the infected cells (
53–55
). Previously, it was
dem-onstrated that the absence of resident corneal DCs did not alter
virus replication in the eye, blepharitis, corneal scarring (CS), or
survival of ocularly infected mice, but it was associated with an
approximately 5-fold reduction in the level of latent virus in the
TG (
13
). Thus, in contrast to the involvement of DCs at the level of
TG during latent but not acute HSV infection, resident corneal
DCs do not directly or indirectly alter acute infection in the eye or
TG. The potential clinical significance of our results is also
sug-gested by studies of vaccinia virus, which abortively infects both
mature and immature DCs and blocks their maturation, thereby
impairing T-cell activation (
56
). By inhibiting maturation
path-ways in DCs and inducing their death, vaccinia virus can subvert
the development of efficient antiviral T-cell immunity.
In the present study, we have shown that lower levels of CD8
␣
⫹transcripts in DCs are associated with less latency and T-cell
ex-FIG 8IHC of DCs isolated from Rag-1⫺/⫺mice. BM-derived DCs from Rag-1⫺/⫺mice were isolated and grown on Lab-Tex chamber slides. At 24 h postculture, DCs were fixed and stained with anti-CD11c and anti-CD4, anti-CD11c and anti-CD8␣, or anti-CD11c and anti-CD8antibodies, followed by incubation with relevant secondary antibody to each primary antibody as described in Materials and Methods. DAPI is shown as a nuclear counterstain.on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.126.463.65.315.2]haustion. Previously, it was shown that CD8
␣
⫺DCs drive
activa-tion of CD4
⫹T cells (
57
), can produce IFNs and IL-12 (
58
),
ac-quire the ability to cross-present antigen (
59
), and induce CD4
⫹cytotoxic regulatory T cells (
60–63
). In addition, as we have shown
here, CD8
␣
⫺DCs do not contribute to T-cell exhaustion. In this
study, we observed a discrepancy between more susceptibility of
DCs isolated from PD-L1
⫺/⫺mice, with less expression of CD8
␣
transcripts from their BM-derived DCs, and less latency and T-cell
exhaustion, as we reported previously (
12
). In contrast, DCs
iso-lated from CD8
␣
⫺/⫺mice had less susceptibility to viral infection
and also had less latency and T-cell exhaustion, as we reported
recently (
17
). Thus, it is possible that reduced latency in the
ab-sence of PD-L1 is due to the lack of interaction of PD-L1 with
PD-1, and therefore an increase in T-cell function, while in the
CD8
␣
⫺/⫺mice, despite the presence of PD-L1, the lower infection
level of DCs prevents functional impairment of T cells. We
re-cently reported that binding of CD80 expressed by a recombinant
HSV-1 to PD-L1 on the surfaces of DCs reduced T-cell exhaustion
in vitro
independent of CD28 (
64
).
We also found that CD8
⫹T-cell exhaustion was associated
with the presence of CD8
␣
⫹and/or LAT. In addition to this CD8
⫹T-cell exhaustion by CD8
␣
⫹and/or LAT, we now show that
CD4
⫹T cells are also exhausted in the presence of infected CD8
␣
⫹DCs. CD4
⫹T-cell exhaustion was reported recently (
12
), and it
has been reported that
in vivo
blockade of PD-L1 and lymphocyte
activation gene 3 restores CD4
⫹T-cell function (
65
). Along these
lines, our published studies have shown that transfer of CD11c
⫹CD8
␣
⫹DCs significantly enhances latency in the TG of
WT-in-fected mice, whereas transfer of CD11c
⫹CD8
␣
⫺DCs reduces the
level of latency (
14
). Furthermore, in the current study, we
showed that transfer of BM from WT but not CD8
␣
⫺/⫺mice to
recipient Rag1
⫺/⫺mice that also lack CD8
␣
⫹DCs induced higher
levels of latency, similar to that in WT mice infected with LAT(
⫹
)
virus.
A key finding in our study is that the increase in latency
asso-ciated with infection with LAT(
⫹
) virus compared with infection
with LAT(
⫺
) virus is due to CD8
␣
⫹DCs. This population of DCs
is infected more efficiently with nonreplicative LAT(
⫹
) virus, and
this leads to T-cell exhaustion and an increase in latency in
ocu-larly infected mice. Previously, it was shown that CD8
␣
⫹DCs
enhance susceptibility to infection in mice (
52
). We have also
shown that during HSV-1 latency, viral antigens in the presence of
PD-1 and PD-L1 contribute to CD8
⫹T-cell exhaustion, and that
WT mice infected with LAT(
⫹
) virus have a greater propensity for
latency, increased numbers of CD8
⫹T cells, and a greater
abun-dance of PD-1 and PD-L1 expression in the TG (
12
). Overall, our
results suggest that CD8
␣
⫹DCs play a nonredundant role in
in-creasing the number of latent viral genomes. Thus, we propose
that higher infection levels of CD8
␣
⫹DCs in the presence of LAT
reduce the antiviral response to infection and increase T-cell
ex-haustion, leading to higher latency levels. Based on our present
and published studies, this negative function of CD8
␣
⫹DCs can
potentially be altered by blocking PD-1–PD-L1 interaction by use
of CD80, as we showed recently (
64
), and/or by expanding the
CD8
␣
⫺DC population by use of GM-CSF, as shown previously
(
66
,
67
).
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants AI093941, EY13615, and T32 AI89553.
REFERENCES
1.Stevens JG.1989. Human herpesviruses: a consideration of the latent state. Microbiol. Rev.53:318 –332.
2.Wechsler SL, Nesburn AB, Watson R, Slanina S, Ghiasi H.1988. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J. Gen. Virol.69:3101–3106.http://dx.doi.org/10.1099/0022 -1317-69-12-3101.
3.Fraser NW, Valyi-Nagy T.1993. Viral, neuronal and immune factors which may influence herpes simplex virus (HSV) latency and reactivation. Microb. Pathog.15:83–91.http://dx.doi.org/10.1006/mpat.1993.1059.
FIG 9Effect of CD8␣expression on the level of latency in TG of latently infected mice. Rag1⫺/⫺mice received BM from WT or CD8␣⫺/⫺mice at a 1:1 ratio i.v. At 2 weeks posttransfer, recipient mice were ocularly infected with LAT(⫹) virus as described in Materials and Methods. On day 30 p.i., TG were harvested from the latently infected mice. Quantitative PCR and RT-PCR were performed on each individual mouse TG. In each experiment, an estimated relative copy number of gB or LAT was calculated using a standard curve generated from pGem-gB1 or pGEM-5317 (H. Ghiasi, unpublished data), re-spectively. Briefly, the DNA template was serially diluted 10-fold, such that 5
l contained 103to 1011copies of gB or LAT, and then subjected to TaqMan
PCR with the same set of primers. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standard, the copy number for each reaction was determined. GAPDH expression was used to normalize the relative expression of gB DNA or LAT RNA in the TG. Data represent means⫾ SEM for 10 TG. (A) gB DNA; (B) LAT RNA.
Mott et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.65.258.67.465.2]4.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL.1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol.61:3820 –3826.
5.Gordon YJ.1990. Pathogenesis and latency of herpes simplex virus type 1 (HSV-1): an ophthalmologist’s view of the eye as a model for the study of the virus-host relationship. Adv. Exp. Med. Biol.278:205–209.http://dx .doi.org/10.1007/978-1-4684-5853-4_21.
6.Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM.2005. HSV-1 DNA in tears and saliva of normal adults. Invest. Oph-thalmol. Vis. Sci.46:241–247.http://dx.doi.org/10.1167/iovs.04-0614. 7.Steiner I.1996. Human herpes viruses latent infection in the nervous
system. Immunol. Rev. 152:157–173. http://dx.doi.org/10.1111/j.1600 -065X.1996.tb00915.x.
8.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA.1994. Herpetic Eye Disease Study. A controlled trial of oral acyclovir for herpes simplex stro-mal keratitis. Ophthalmology101:1871–1882.
9.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA.1996. Risk factors for herpes simplex virus epithelial keratitis recur-ring durecur-ring treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br. J. Ophthalmol.80:969 –972.
10. Hill JM, Sedarati F, Javier RT, Wagner EK, Stevens JG.1990. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology174:117–125.http://dx.doi.org/10.1016/0042-6822(90)90060-5. 11. Wechsler SL, Nesburn AB, Watson R, Slanina SM, Ghiasi H.1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alter-native splicing produces distinct latency-related RNAs containing open reading frames. J. Virol.62:4051– 4058.
12. Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmo-hamed L, Ahmed R, Wechsler SL, Ghiasi H.2011. The role of LAT in increased CD8⫹T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J. Virol.85:4184 – 4197.http: //dx.doi.org/10.1128/JVI.02290-10.
13. Mott KR, Ghiasi H.2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin. Vaccine Immunol. 15:1859 –1867. http://dx.doi.org/10.1128/CVI .00318-08.
14. Mott KR, UnderHill D, Wechsler SL, Ghiasi H.2008. Lymphoid-related CD11c⫹CD8a⫹dendritic cells are involved in enhancing HSV-1 latency. J. Virol.82:9870 –9879.http://dx.doi.org/10.1128/JVI.00566-08. 15. Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL.2000. CD8(⫹) T
cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med.191:1459 –1466.http://dx.doi.org /10.1084/jem.191.9.1459.
16. Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. 2003. Herpes simplex virus-specific memory CD8⫹T cells are selectively acti-vated and retained in latently infected sensory ganglia. Immunity18:593– 603.http://dx.doi.org/10.1016/S1074-7613(03)00112-2.
17. Mott KR, Allen SJ, Zandian M, Konda B, Sharifi BG, Jones C, Wechsler SL, Town T, Ghiasi H.2014. CD8a dendritic cells drive establishment of HSV-1 latency. PLoS One 9:e93444. http://dx.doi.org/10.1371/journal .pone.0093444.
18. Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H.
2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8⫹T cells in trigeminal ganglia of latently infected mice. J. Virol. 83:2246 –2254. http://dx.doi.org/10 .1128/JVI.02234-08.
19. Osorio Y, Ghiasi H.2003. Comparison of adjuvant efficacy of herpes simplex virus type 1 recombinant viruses expressing TH1 and TH2 cyto-kine genes. J. Virol.77:5774 –5783.http://dx.doi.org/10.1128/JVI.77.10 .5774-5783.2003.
20. Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL.1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol.68:8045– 8055. 21. Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu XL, Trinchieri G, O’Garra A, Liu YJ.2002. The development of murine plasmacytoid den-dritic cell precursors is differentially regulated by FLT3-ligand and gran-ulocyte/macrophage colony-stimulating factor. J. Exp. Med.195:953–958. http://dx.doi.org/10.1084/jem.20020045.
22. Mott KR, Underhill D, Wechsler SL, Town T, Ghiasi H.2009. A role for the JAK-STAT1 pathway in blocking replication of HSV-1 in dendritic cells and macrophages. Virol. J. 6:56. http://dx.doi.org/10.1186/1743 -422X-6-56.
23. Osorio Y, La Point SF, Nusinowitz S, Hofman FM, Ghiasi H.2005. CD8⫹-dependent CNS demyelination following ocular infection of mice with a recombinant HSV-1 expressing murine IL-2. Exp. Neurol.193:1– 18.http://dx.doi.org/10.1016/j.expneurol.2004.12.004.
24. Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL.1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res.22:25–39. 25. Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A
recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J. Virol.81:12962–12972.http://dx.doi.org/10.1128/JVI.01442-07. 26. Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi
H.2007. The corneas of naive mice contain both CD4⫹and CD8⫹T cells. Mol. Vis.13:1802–1812.http://www.molvis.org/molvis/v13/a201/. 27.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1
latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J. Virol.79:9019 –9025.http://dx.doi.org/10.1128/JVI.79 .14.9019-9025.2005.
28. Liu T, Tang Q, Hendricks RL.1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol.70:264 –271.
29. Birmanns B, Reibstein I, Steiner I.1993. Characterization of an in vivo reactivation model of herpes simplex virus from mice trigeminal ganglia. J. Gen. Virol.74:2487–2491.http://dx.doi.org/10.1099/0022-1317-74-11 -2487.
30. Rutebemberwa A, Ray SC, Astemborski J, Levine J, Liu L, Dowd KA, Clute S, Wang C, Korman A, Sette A, Sidney J, Pardoll DM, Cox AL.
2008. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and re-quire preservation of cognate antigen during chronic infection. J. Immu-nol.181:8215– 8225.http://www.jimmunol.org/content/181/12/8215. 31. Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G,
Ferrari C.2006. PD-1 expression in acute hepatitis C virus (HCV) infec-tion is associated with HCV-specific CD8 exhausinfec-tion. J. Virol.80:11398 – 11403.http://dx.doi.org/10.1128/JVI.01177-06.
32. Stefanidou M, Ramos I, Mas Casullo V, Trepanier JB, Rosenbaum S, Fernandez-Sesma A, Herold BC.2013. Herpes simplex virus 2 (HSV-2) prevents dendritic cell maturation, induces apoptosis, and triggers release of proinflammatory cytokines: potential links to HSV-HIV synergy. J. Virol.87:1443–1453.http://dx.doi.org/10.1128/JVI.01302-12.
33. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T.2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte acti-vation. J. Exp. Med.192:1027–1034.http://dx.doi.org/10.1084/jem.192.7 .1027.
34. Sharpe AH, Freeman GJ. 2002. The B7-CD28 superfamily. Nat. Rev. Immunol.2:116 –126.http://dx.doi.org/10.1038/nri727.
35. Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L.2011. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8⫹T cells in latently infected trigeminal ganglia: a novel immune evasion mecha-nism. J. Virol.85:9127–9138.http://dx.doi.org/10.1128/JVI.00587-11. 36. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S,
Papa-ioannou VE.1992. RAG-1-deficient mice have no mature B and T lym-phocytes. Cell 68:869 – 877. http://dx.doi.org/10.1016/0092-8674(92) 90030-G.
37. Shortman K, Vremec D, Corcoran LM, Georgopoulos K, Lucas K, Wu L.1998. The linkage between T-cell and dendritic cell development in the mouse thymus. Immunol. Rev.165:39 – 46.http://dx.doi.org/10.1111/j .1600-065X.1998.tb01228.x.
38. Saunders D, Lucas K, Ismaili J, Wu L, Maraskovsky E, Dunn A, Shortman K.1996. Dendritic cell development in culture from thymic precursor cells in the absence of granulocyte/macrophage colony-stimulating factor. J. Exp. Med.184:2185–2196.http://dx.doi.org/10.1084 /jem.184.6.2185.
39. Rodewald HR, Brocker T, Haller C.1999. Developmental dissociation of
on November 7, 2019 by guest
http://jvi.asm.org/
thymic dendritic cell and thymocyte lineages revealed in growth factor receptor mutant mice. Proc. Natl. Acad. Sci. U. S. A.96:15068 –15073. http://dx.doi.org/10.1073/pnas.96.26.15068.
40. Dawson CR.1984. Ocular herpes simplex virus infections. Clin. Derma-tol.2:56 – 66.http://dx.doi.org/10.1016/0738-081X(84)90066-X. 41. Shortman K, Naik SH.2007. Steady-state and inflammatory
dendritic-cell development. Nat. Rev. Immunol.7:19 –30.http://dx.doi.org/10.1038 /nri1996.
42. Belz GT, Nutt SL.2012. Transcriptional programming of the dendritic cell network. Nat. Rev. Immunol.12:101–113.http://dx.doi.org/10.1038 /nri3149.
43. Seillet C, Jackson JT, Markey KA, Brady HJ, Hill GR, Macdonald KP, Nutt SL, Belz GT.2013. CD8alpha⫹DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood121:1574 –1583.http: //dx.doi.org/10.1182/blood-2012-07-445650.
44. Turcotte K, Gauthier S, Mitsos LM, Shustik C, Copeland NG, Jenkins NA, Fournet JC, Jolicoeur P, Gros P.2004. Genetic control of myelopro-liferation in BXH-2 mice. Blood 103:2343–2350. http://dx.doi.org/10 .1182/blood-2003-06-1852.
45. Tailor P, Tamura T, Morse HC, 3rd, Ozato K.2008. The BXH2 muta-tion in IRF8 differentially impairs dendritic cell subset development in the mouse. Blood111:1942–1945.http://dx.doi.org/10.1182/blood-2007-07 -100750.
46. Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL.2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science287:1500 –1503. http://dx.doi.org /10.1126/science.287.5457.1500.
47. Carpenter D, Hsiang C, Brown DJ, Jin L, Osorio N, BenMohamed L, Jones C, Wechsler SL.2007. Stable cell lines expressing high levels of the herpes simplex virus type 1 LAT are refractory to caspase 3 activation and DNA laddering following cold shock induced apoptosis. Virology369:12– 18.http://dx.doi.org/10.1016/j.virol.2007.07.023.
48. Jin L, Peng W, Perng GC, Brick DJ, Nesburn AB, Jones C, Wechsler SL.
2003. Identification of herpes simplex virus type 1 latency-associated tran-script sequences that both inhibit apoptosis and enhance the spontaneous reactivation phenotype. J. Virol.77:6556 – 6561.http://dx.doi.org/10.1128 /JVI.77.11.6556-6561.2003.
49. Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, Wechsler SL, Jones C.2004. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J. Neurovirol.10:260 –265.http://dx.doi .org/10.1080/13550280490468690.
50. Inman M, Perng GC, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. 2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J. Virol.75:3636 –3646.http://dx .doi.org/10.1128/JVI.75.8.3636-3646.2001.
51. Alaniz RC, Sandall S, Thomas EK, Wilson CB.2004. Increased dendritic cell numbers impair protective immunity to intracellular bacteria despite augmenting antigen-specific CD8⫹T lymphocyte responses. J. Immunol.
172:3725–3735.http://www.jimmunol.org/content/172/6/3725. 52. Edelson BT, Bradstreet TR, Hildner K, Carrero JA, Frederick KE, Kc W,
Belizaire R, Aoshi T, Schreiber RD, Miller MJ, Murphy TL, Unanue ER, Murphy KM.2011. CD8alpha(⫹) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immu-nity35:236 –248.http://dx.doi.org/10.1016/j.immuni.2011.06.012. 53. Salio M, Cella M, Suter M, Lanzavecchia A.1999. Inhibition of dendritic
cell maturation by herpes simplex virus. Eur. J. Immunol.29:3245–3253. 54. Kruse M, Rosorius O, Kratzer F, Stelz G, Kuhnt C, Schuler G, Hauber
J, Steinkasserer A.2000. Mature dendritic cells infected with herpes sim-plex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol.
74:7127–7136.http://dx.doi.org/10.1128/JVI.74.15.7127-7136.2000. 55. Stefanidou M, Ramos I, Mas Casullo V, Trepanier JB, Rosenbaum S,
Fernandez-Sesma A, Herold BC.2013. Herpes simplex virus 2 (HSV-2) prevents dendritic cell maturation, induces apoptosis, and triggers release of pro-inflammatory cytokines: potential links to HSV-HIV synergy. J. Virol.87:1443–1453.http://dx.doi.org/10.1128/JVI.01302-12.
56. Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox WI, Stein-man RM, Bhardwaj N.1999. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J. Immu-nol.163:6762– 6768.
57. Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM.2008. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity29:795– 806.http://dx.doi.org/10 .1016/j.immuni.2008.08.013.
58. Maldonado-Lopez R, Moser M.2001. Dendritic cell subsets and the regulation of Th1/Th2 responses. Semin. Immunol.13:275–282.http://dx .doi.org/10.1006/smim.2001.0323.
59. den Haan JM, Bevan MJ.2002. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(⫹) and CD8(⫺) den-dritic cells in vivo. J. Exp. Med.196:817– 827.http://dx.doi.org/10.1084 /jem.20020295.
60. Kawamura K, Kadowaki N, Kitawaki T, Uchiyama T. 2006. Virus-stimulated plasmacytoid dendritic cells induce CD4⫹cytotoxic regula-tory T cells. Blood107:1031–1038.http://dx.doi.org/10.1182/blood-2005 -04-1737.
61. Brown DM, Lee S, Garcia-Hernandez ML, Swain SL.2012. Multifunc-tional CD4 cells expressing gamma interferon and perforin mediate pro-tection against lethal influenza virus infection. J. Virol.86:6792– 6803. http://dx.doi.org/10.1128/JVI.07172-11.
62. Zhou Y, Callendret B, Xu D, Brasky KM, Feng Z, Hensley LL, Guedj J, Perelson AS, Lemon SM, Lanford RE, Walker CM.2012. Dominance of the CD4(⫹) T helper cell response during acute resolving hepatitis A virus infection. J. Exp. Med. 209:1481–1492. http://dx.doi.org/10.1084/jem .20111906.
63. Lee HK, Zamora M, Linehan MM, Iijima N, Gonzalez D, Haberman A, Iwasaki A.2009. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J. Exp. Med.206:359 –370. http://dx.doi.org/10.1084/jem.20080601.
64. Mott KR, Allen SJ, Zandian M, Akbari O, Hamrah P, Maazi H, Wechsler SL, Sharpe AH, Freeman GJ, Ghiasi H. 2014. Inclusion of CD80 in HSV targets the recombinant virus to PD-L1 on DCs and allows productive infection and robust immune responses. PLoS One9:e87617. http://dx.doi.org/10.1371/journal.pone.0087617.
65. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, Harty JT.2011. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol.13:188 –195.http://dx.doi.org/10.1038/ni.2180. 66. Daro E, Pulendran B, Brasel K, Teepe M, Pettit D, Lynch DH, Vremec D, Robb L, Shortman K, McKenna HJ, Maliszewski CR, Maraskovsky E.2000. Polyethylene glycol-modified GM-CSF expands CD11b(high)CD11c(high) but not CD11b(low)CD11c(high) murine dendritic cells in vivo: a compara-tive analysis with Flt3 ligand. J. Immunol.165:49 –58.http://www.jimmunol .org/content/165/1/49.
67. Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR.1999. Distinct dendritic cell subsets differentially regu-late the class of immune response in vivo. Proc. Natl. Acad. Sci. U. S. A.
96:1036 –1041.http://dx.doi.org/10.1073/pnas.96.3.1036.
Mott et al.
on November 7, 2019 by guest
http://jvi.asm.org/