Products
Yuchen Nan,aYing Yu,aZexu Ma,aSunil K. Khattar,bBrenda Fredericksen,c*Yan-Jin Zhanga
Molecular Virology Laboratoryaand Virology Laboratory,bVirginia-Maryland College of Veterinary Medicine and Maryland Pathogen Research Institute, University of Maryland, College Park, Maryland, USA; Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, Maryland, USAc
ABSTRACT
Hepatitis E virus (HEV) causes both endemic and epidemic human hepatitis by fecal-oral transmission in many parts of the
world. Zoonotic transmission of HEV from animals to humans has been reported. Due to the lack of an efficient cell culture
sys-tem, the molecular mechanisms of HEV infection remain largely unknown. In this study, we found that HEV replication in
hepa-toma cells inhibited poly(I·C)-induced beta interferon (IFN-

) expression and that the HEV open reading frame 1 (ORF1)
prod-uct was responsible for this inhibition. Two domains, X and the papain-like cysteine protease domain (PCP), of HEV ORF1 were
identified as the putative IFN antagonists. When overexpressed in HEK293T cells, the X domain (or macro domain) inhibited
poly(I·C)-induced phosphorylation of interferon regulatory factor 3 (IRF-3), which is the key transcription factor for IFN
induc-tion. The PCP domain was shown to have deubiquitinase activity for both RIG-I and TBK-1, whose ubiquitination is a key step
in their activation in poly(I·C)-induced IFN induction. Furthermore, replication of a HEV replicon containing green fluorescent
protein (GFP) (E2-GFP) in hepatoma cells led to impaired phosphorylation of IRF-3 and reduced ubiquitination of RIG-I and
TBK-1, which confirmed our observations of X and PCP inhibitory effects in HEK293T cells. Altogether, our study identified the
IFN antagonists within the HEV ORF1 polyprotein and expanded our understanding of the functions of several of the HEV
ORF1 products, as well as the mechanisms of HEV pathogenesis.
IMPORTANCE
Type I interferons (IFNs) are important components of innate immunity and play a crucial role against viral infection. They also
serve as key regulators to evoke an adaptive immune response. Virus infection can induce the synthesis of interferons; however,
viruses have evolved many strategies to antagonize the induction of interferons. There is little knowledge about how hepatitis E
virus (HEV) inhibits induction of host IFNs, though the viral genome was sequenced more than 2 decades ago. This is the first
report of identification of the potential IFN antagonists encoded by HEV. By screening all the domains in the open reading frame
1 (ORF1) polyprotein, we identified two IFN antagonists and performed further research to determine how and at which step in
the IFN induction pathway they antagonize host IFN induction. Our work provides valuable information about HEV-cell
inter-action and pathogenesis.
H
epatitis E virus (HEV) is a viral pathogen transmitted by the
fecal-oral route that causes acute hepatitis with a mortality
rate at or below 3% in young adults and up to 30% in pregnant
women in the third trimester (
1
,
2
,
54
). While previously thought
to be a public health problem only for developing countries,
hep-atitis E has now been recognized frequently in industrialized
countries (
1
). Isolation of HEV from pig, chicken, mongoose,
rab-bit, rat, ferret, bat, fish, and deer has been reported (
3–5
).
Zoo-notic transmission of HEV from animals to humans has been
documented (
1
) and is considered a major transmission route for
sporadic cases in the industrialized countries.
HEV contains a 7.2-kb single-stranded positive-sense RNA
ge-nome, which is capped and polyadenylated (
6
,
54
). It has been
classified as the sole member of the genus
Hepevirus
in the family
Hepeviridae
(
2
,
6
). There are four major genotypes and a single
known serotype for HEV (
3
,
7
). There are three open reading
frames (ORFs) in the HEV genome (
8
). ORF1 encodes a
polypro-tein that has all the nonstructural propolypro-teins for HEV replication.
ORF2 encodes the capsid protein of the HEV virion. ORF3
en-codes a small multifunctional protein with a molecular mass of 13
kDa (vp13).
As an invader, HEV faces host innate immune responses,
which are mainly induced by activation of host pattern
recogni-tion receptors. For recognirecogni-tion of RNA viruses, those receptors
include RIG (retinoic-acid-inducible gene)-I-like receptors
(RLRs) and Toll-like receptors (TLRs). Stimulation of the RLR
and TLR signaling pathways leads to activation of transcription
factors, such as interferon-regulatory factor 3 (IRF-3), IRF-7, and
NF-
B. These transcription factors mediate expression of type I
interferons (IFNs) and inflammatory cytokines, which not only
lead to an antiviral state of the neighboring uninfected cells, but
also serve as regulators to evoke an adaptive immune response.
Thus, viruses have evolved many strategies to evade host innate
immune responses. Little is known about how HEV evades host
Received2 July 2014 Accepted31 July 2014 Published ahead of print6 August 2014 Editor:G. McFadden
Address correspondence to Yan-Jin Zhang, [email protected].
* Present address: Brenda Fredericksen, AIDS Review Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, DHHS, Bethesda, MD, USA.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01935-14
on November 7, 2019 by guest
http://jvi.asm.org/
IFN induction. Microarray analysis of hepatitis C virus
(HCV)-and HEV-infected chimpanzees showed that HEV evoked a lesser
magnitude of IFN response than HCV, indicating that HEV must
employ an effective strategy to dampen host innate immune
re-sponses (
9
).
The objective of this study was to elucidate the mechanism of
HEV interference with type I IFN induction. We found that HEV
replication in S10-3 hepatoma cells inhibited IFN-

induction
stimulated by poly(I·C), a double-stranded RNA (dsRNA)
homo-logue. Further studies identified two putative domains (X and
PCP) of ORF1 polyprotein as the IFN antagonists. The X domain
(also known as the macro domain) inhibited poly(I·C)-induced
IRF-3 phosphorylation, while the PCP domain led to
deubiquiti-nation of both RIG-I and TBK-1. These findings were also
con-firmed in hepatoma cells with HEV replication. Our findings
pro-vide valuable information about the function of the HEV ORF1
product and improve our understanding of HEV pathogenesis.
MATERIALS AND METHODS
Cells, transfection, viruses, and chemicals.HEK293T and HEK293 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supple-mented with 10% fetal bovine serum (FBS). S10-3 cells, a subclone of Huh-7 hepatoma cells (10), were maintained in DMEM-reduced serum (DMEM-RS) supplemented with 3% FBS. Transfection of HEK293T and HEK293 cells with plasmid DNA was performed by using FuGeneHD (Promega, Madison, WI) according to the instructions of the manufac-turer.
Sendai virus (ATCC VR-907) was obtained from the ATCC. Propaga-tion of Sendai virus was done in 10-day-old embryonated specific-patho-gen-free (SPF) chicken eggs. The allantoic fluid was harvested 3 days postinfection, and the Sendai virus titer was determined by hemaggluti-nation assay, as described previously (11).
Full-length RNAs of HEV and HEV-green fluorescent protein (GFP) were obtained byin vitrotranscription from plasmids E2 and pSK-E2-GFP (12), respectively, using an AmpliCap-Max T7 High Yield Mes-sage Maker Kit (Cellscript, Madison, WI). Transfection of S10-3 cells with RNA was performed using an optimized protocol with DMRIE-C reagent (Invitrogen, Grand Island, NY). Briefly, after S10-3 cells reached 70% confluence in a 12-well plate, the medium was discarded and the mono-layer cells were washed twice with phosphate-buffered saline (PBS), pH 7.4, followed by addition of 0.5 ml serum-free Opti-MEM to each well. For each well, 1g RNA was added to 50l Opti-MEM and mixed evenly with 4l DMRIE-C. After incubation at room temperature for 20 min, the mixture was added to the S10-3 cells. One milliliter DMEM-RS with 3% FBS was added to each well 5 h posttransfection. The cells were cul-tured at 34.5°C.
HEK293 cells stably expressing VenusC1-IRF-3 (see below) (HEK293-IRF-3) were established by transfection of the cells with VenusC1-IRF-3 and selection under the antibiotic G418. The surviving cells were cloned by limited dilution and cell sorting by flow cytometry.
Poly(I·C) (low molecular weight), a synthetic analog of dsRNA (Invi-vogen, San Diego, CA), was used to induce interferon production. The 293T and S10-3 cells were transfected with poly(I·C) at a concentration of 1g/ml and incubated for 12 h before being harvested for further analysis. Plasmids.Putative ORF1 domains were PCR amplified and cloned into the vector pCAGEN (Addgene; plasmid number 11160) with a hem-agglutinin (HA) tag at the N terminus as previously reported (13,14). VenusC1-IRF-3 was constructed in house by subcloning IRF-3 from pFLAG-CMV-IRF-3 (a gift from Michael Gale, Jr.). Full-length RIG-I cDNA from S10-3 cell RNA was cloned into the KpnI site in the pCMV-Flag-MAT-Tag-1 vector (Sigma). All primers used for plasmid construc-tion are listed inTable 1. All in-house-constructed plasmids were sub-jected to DNA sequencing to confirm the inserts.
HEV replicons pSK-E2 and pSK-E2-GFP were described previously
(12,15,16). The construction of Myc-RIG-I(N) (N-terminal domain) (17), MDA-5(N) (18), FLAG-TBK-1 (19), and FLAG-IKKε(20) plasmids was described previously.
IFA.An immunofluorescence assay (IFA) was carried out as previ-ously reported (21) by using chimpanzee antibody against the HEV capsid protein. Specific antibody-capsid reactions were detected with DyLight 549 goat anti-human immunoglobulin G (IgG) conjugate (Rockland Im-munologicals, Gilbertsville, PA). The cover glass was mounted on a slide using SlowFade Gold antifade reagent containing 4=6=-diamidino-2-phe-nylinodole (DAPI) (Invitrogen) and observed using fluorescence micros-copy.
Western blot analysis.Cells were lysed in Laemmli sample buffer. The whole proteins in the lysate were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting as previously described (21,22). Antibodies against GFP (Santa Cruz Bio-technology, Santa Cruz, CA), phosphorylated IRF-3-S396 (Cell Signaling Technology, Danvers, MA), HA (Thermo Fisher Scientific, Waltham, MA), IRF-3 (Santa Cruz), and tubulin (Sigma-Aldrich, St. Louis, MO) were used in the blotting. The chemiluminescence signal was recorded digitally using a ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). Digital signal acquisition and densitometry analyses were conducted using the Quantity One Program, version 4.6 (Bio-Rad).
Reverse transcription and real-time quantitative PCR (RT-qPCR). Total RNA was isolated from cells with TRIzol reagent (Invitrogen). RNase-free DNase was used to remove carryover DNA from the RNA isolation procedure. Reverse transcription using avian myeloblastosis vi-rus (AMV) reverse transcriptase, along with oligo(dT) and a random 15-mer, was conducted. Real-time PCR with SYBR green (Invitrogen) detec-tion of IFN-was done as described previously (23). Transcripts of RPL32 (ribosomal protein L32) were also detected from the same samples to serve as an internal control for normalization. Gene expression was quan-tified by the 2⫺⌬⌬CTmethod (24). Primers for IFN-and RPL32 were
described previously (25).
[image:2.585.299.543.81.324.2]Ubiquitination assay.Immunoprecipitation (IP) was conducted as previously described (13, 26) with modifications. Briefly, S10-3 and TABLE 1Primers used in this study
Primera Sequence (5=to 3=)b
Target gene
H1F1 GCGAATTCGAGGCCCATCAGTTTATCAAGG Met H1R7 CCCTCGAGTTAAATCCAGGAGCGCAGGTTGG H1F3 GCGAATTCAGAACCACTAAGGTTACCGG Y H1R6 CCCTCGAGTTACTGAGCGTAGAACTCCAAC
H1F4 GCGAATTCTGTAGGCGCTGGCTCTCGGC PCP H1R5 CCCTCGAGTTAGAGATTGTGGCGCTCTGG
H1F5 GCGAATTCTCTTTTGATGCCAGTCAGAG HPX H1R4 CCCTCGAGTTAGCCGGCACAGGCCCGGCCG
H1F6 GCGAATTCTGTCGAGTCACCCCCGGCG Hel H1R3 GCGATATCTTAACCAGCAAGGAAAAAGTTATTA H1F7 GCGAATTCGGCGAAATTGGCCACCAGCG RdRp H1R2 GCGATATCTCATTCCACCCGACACAGAAT
H1F10 CGCTCGAGCTTTTGATGCCAGTCAGAG HV
H1R16 GCGAATTCTAAACAGCATCAACCTCCGAC
H1F9 GCGAATTCCCTAGTCCAGCCCAGCCCG Pro H1R18 GCGAATTCTAGCGATGCCGGGCCGTCTGG
H1F8 GCGAATTCCCGGATGGCTCTAAGGTG X H1R13 CCCTCGAGTGCTGTCCGCGCAACATCC
IRF3-F2 CGGAATTCGGGAACCCCAAAGCCACGGATC IRF-3 IRF3-R2 CCGGTACCTCAGCTCTCCCCAGGGCCCTG
RIG-I-F1 TTAGGTACCATGACCACCGAGCAGCGAC RIG-I RIG-I-F2 GGAGGTACCTCATTTGGACATTTCTGCTG
aF, forward primer; R, reverse primer. “H1” before a primer name indicates the primer
is based on sequences of HEV ORF1.
bThe italicized letters indicate restriction enzyme cleavage sites for cloning.
on November 7, 2019 by guest
http://jvi.asm.org/
HEK293T cells were lysed with lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.2 mM EDTA, 2 mM EGTA, 0.5% IGEPAL CA-630, 10% glycerol, 1 mM sodium vanadate) as described previously (13), with supplementa-tion with a protease inhibitor cocktail (Sigma) as recommended by the manufacturer andN-ethylmaleimide (NEM) (Sigma) at a final concen-tration of 50M. The lysate was clarified by centrifugation at 14,000⫻g for 5 min at 4°C. Antibodies against RIG-I or TBK-1 were added to the supernatant. IP with protein G agarose (KPL Inc., Gaithersburg, MD) was done following the manufacturer’s instructions. The IP samples were sub-jected to immunoblot analysis with antibody against ubiquitin.
Reporter assay and luminescent cell viability assay.HEK293T cells were transfected with the reporter pGL3.0-IFN-promoter and testing plasmids. TheRenillavector pRL-TK (Promega) was also transfected for normalization. At 48 h after transfection, the cells were lysed for luciferase activity assay of firefly andRenillaluciferases by following the manufac-turer’s instructions (Promega). Lysate of the cells transfected with empty vector was used as a control for calculation of the IFN-promoter acti-vation level. The relative firefly luciferase activity is shown after normal-ization with theRenillalevel as described previously (27). A cell viability assay was conducted with a CellTiter-Glo Luminescent Cell Viability As-say kit by following the manufacturer’s instructions (Promega).
Statistical analysis.Differences in indicators between treatment sam-ples, such as the IFN-mRNA level between the group in the presence of HEV replication and the control sample, were assessed by the Studentt test. A two-tailedPvalue of less than 0.05 was considered significant.
RESULTS
HEV replication inhibits IFN-

expression induced by
poly(I·C) transfection.
We first tested whether HEV replication
would interfere with interferon induction. S10-3 cells, a subclone
of Huh-7 cells that is more susceptible to transfection with whole
HEV genomic RNA (
28
), were used. HEV genomic RNA was
tran-scribed from the pSK-E2 plasmid containing whole cDNA of the
Sar55 (genotype I) HEV genome, as previously described (
12
).
Full-length HEV RNA generated from the pSKE2-GFP construct,
in which insertion of GFP disrupts the expression of ORF2 and
ORF3 (
12
), was included for comparison. S10-3 cells were
trans-fected with the two different HEV RNAs. HEV replication was
detected by immunofluorescence assay with HEV antibody (
Fig.
1A
). The cells were transfected with poly(I·C) 10 days post-HEV
RNA transfection to stimulate IFN production. RT-qPCR was
then conducted to detect IFN-

transcript.
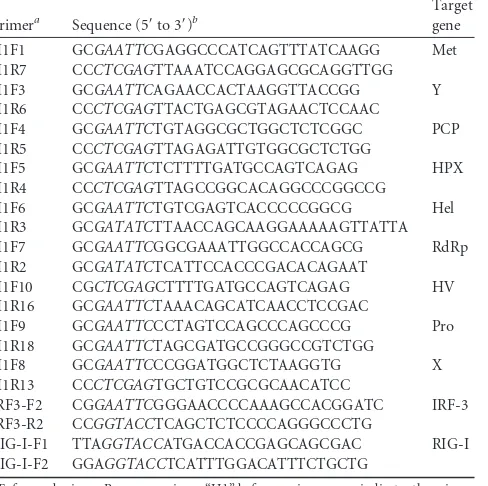
Compared with uninfected cells, S10-3 cells with HEV
replica-tion (from pSK-E2 RNA) had a significantly lower level of IFN-

transcript (3.2-fold) (
Fig. 1B
), which indicates that HEV
replica-tion reduced IFN expression evoked by poly(I·C). Similarly, the
cells transfected with RNA from pSKE2-GFP had a significantly
lower level of IFN-

transcript (4.8-fold). This result suggests that
the ORF1 product was at least partially responsible for the
inhibi-tion, as GFP insertion interrupts the expression of both ORF2 and
ORF3 in pSKE2-GFP.
To ensure that the impaired IFN-

mRNA level was not due to
a cytotoxic effect induced by poly(I·C) or HEV replication, we
performed a cell viability assay of mock-infected or HEV-infected
S10-3 cells with or without poly(I·C) stimulation. The results
showed that HEV replication or poly(I·C) stimulation had a
min-imal effect on the viability of S10-3 cells (
Fig. 1C
), which suggests
the IFN-

mRNA reduction was due to inhibition by HEV
repli-cation.
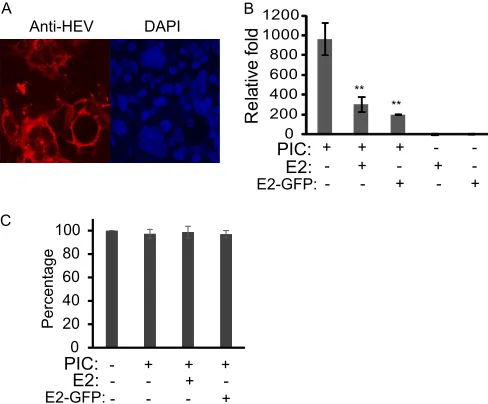
The HEV ORF1 product inhibits IFN production.
After
find-ing that the inhibition of IFN induction was potentially due to the
ORF1 product, we cloned 6 fragments of ORF1 into the pCAGEN
plasmid with an HA tag at the N terminus according to a previous
analysis of the putative domains (
29
) (
Fig. 2A
). They were the
methyltransferase domain (Met); the Y domain (Y); papain-like
cysteine protease (PCP); a fragment covering the hypervariable
region, the proline-rich domain, and the X domain (HPX);
heli-case (Hel); and RNA-dependent RNA polymerase (RdRp).
Pro-tein expression of these HA-tagged fragments in HEK293T cells
transfected with the plasmids was confirmed with Western
blot-ting using HA antibody (
Fig. 2B
). All the proteins except HPX
appeared to be one band. The HPX lane had two bands, indicating
excision of the product.
Based on the size of the excised HPX band, it appeared that the
excision occurred in the X domain. Recently, a highly conserved
“glycine triad” (G815-G816-G817) was identified in the macro
domain of HEV, which is homologous to the rubella virus
pro-tease substrate (G1299-G1300-G1301) (
30
). It is possible that the
two bands of HPX product in HEK293T cells were due to the
cellular protease-mediated cleavage at this glycine triad.
Next, we examined the effects of the proteins on IFN
expres-sion in HEK293T cells. An empty vector and porcine reproductive
and respiratory syndrome virus (PRRSV) nonstructural protein
1

(nsp1

), which is known to inhibit IFN induction (
31
), were
included as controls. At 48 h after transfection, the cells were
transfected with poly(I·C) to stimulate IFN induction. RT-qPCR
was conducted to detect the IFN-

mRNA level. Compared with
the cells transfected with empty vector, PCP and HPX led to
sig-nificant reductions in IFN-

expression (2.6- and 7.6-fold,
respec-A B
PIC: E2:
Rel
a
ti
v
e f
o
ld
** **
0 200 400 600 800 1000 1200
E2-GFP:
Anti-HEV DAPI
C
0 20 40 60 80 100
Percentage
PIC: E2: E2-GFP:
- + + +
- - +
-- - - +
-+ +
+
- - +
-- - - +
-+
+
FIG 1HEV replication in S10-3 cells (a subclone of Huh-7 cells) reduces IFN production induced by poly(I · C). (A) HEV replication in S10-3 cells detected by immunofluorescence assay. The cells were transfected with full-length HEV genomic RNA from pSK-E2. The red fluorescence (left) indicates detection of the HEV ORF2 protein. Nuclear DNA (right) was counterstained using DAPI. (B) Reduction of IFN-expression in HEV-infected S10-3 cells detected by RT-qPCR. The cells were transfected with poly(I · C) (PIC). Relative levels (fold) of IFN-mRNA in comparison to control cells without HEV RNA transfection and without PIC stimulation are shown. The graph represents averages of three independent experiments. Significant differences from the HEV-negative group are denoted by asterisks (**,P⬍0.01). (C) Cell viability assay comparing S10-3 cells with or without HEV replication and PIC trans-fection. Relative percentages are shown in comparison with normal S10-3 cells without PIC treatment. The error bars indicate standard error.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.299.543.68.270.2]tively) (
Fig. 2C
). Likewise, PRRSV nsp1

led to a 4.6-fold
reduc-tion. However, the other fragments of ORF1 did not have an
inhibitory effect on IFN-

induction, indicating that ORF1 PCP
and HPX were IFN antagonists.
To exclude the possibility of a cytotoxic effect of HPX and PCP,
we conducted cell viability assays for HEK293T cells transfected
with empty vector and HEV ORF1 domains. The results showed
that transfection with PCP and HPX plasmids had an
undetect-able cytotoxic effect (
Fig. 2D
). This also confirmed that the
re-duced IFN-

mRNA level in the cells with PCP and HPX was due
to the antagonist functions of the two proteins.
To further confirm that HEV PCP and HPX are real IFN
an-tagonists, we used Sendai virus, a well-known natural inducer of
type I IFNs, to stimulate the HEK293T cells transfected with PCP
and HPX, along with IFN-

reporter plasmids. The reporter assay
showed that PCP and HPX significantly inhibited Sendai
virus-induced IFN-

promoter activation (3.8- and 5.5-fold) (
Fig. 2E
).
Taken together, our data indicate than HEV PCP and HPX
func-tion as antagonists to type I IFN inducfunc-tion.
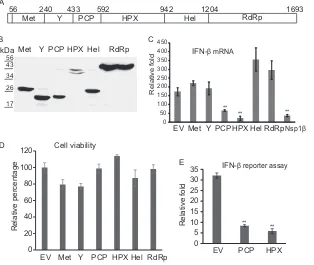
The HEV X domain inhibits IFN induction by blocking
phos-phorylation of IRF-3.
After finding that the two ORF1 products
inhibit IFN induction, we first selected HPX to determine its
in-terference mechanism. Since overexpression of signal molecules
of RLR pathways, such as RIG-I (N-terminal domain), MDA-5
(N-terminal domain), TBK-1, and IKK
ε
, leads to the activation of
the IFN-

promoter (
31–34
), HEK293T cells were transfected
with plasmids encoding the IFN-

reporter, HPX, and the specific
components of the RIG-I signaling pathway. Overexpression of
the individual components was needed to determine which step in
the RLR signaling HEV HPX blocks. Compared with the cells
transfected with empty vector, HPX expression resulted in a
sig-nificant reduction of RIG-I- and MDA-5-induced IFN-

reporter
expression (1.8- and 2.2-fold, respectively) (
Fig. 3A
). Similarly,
when the cells were transfected with a TBK-1 or IKKε
plasmid,
HPX reduced the luciferase yield significantly (2.26- and
2.35-fold, respectively) (
Fig. 3B
). PRRSV nsp1

reduced the luciferase
yield of the IFN-

reporter, as expected. These results indicated
that HPX interfered with IFN induction via the RLR pathway.
As HPX inhibits the RLR pathway, we tested the activation
status of IRF-3 in cells expressing HPX. Since the phosphorylation
level of endogenous IRF-3 in HEK293T cells after poly(I·C)
stim-ulation is hard to detect, we cotransfected the cells with
FLAG-IRF-3 and HPX plasmids. At 40 h after transfection, the cells were
transfected with poly(I·C) and harvested 8 h later for Western
blotting. Compared with cells transfected with empty vector, the
cells with HPX had much lower levels of IRF-3 phosphorylation
Met A
B
0 50 100 150 200 250 300 350 400 450
Rel
a
ti
v
e f
o
ld
**
** **
RdRp Hel
56 240 433 592 942 1204 1693
Met Y PCP HPX
C RdRp Hel HPX PCP Y kDa
17 26 34 43 56
Nsp1β
0 20 40 60 80 100 120
EV Met Y PCP HPX Hel RdRp
R
e
la
ti
v
e
percentage
D
0 5 10 15 20 25 30 35
EV PCP HPX
Re
la
ti
v
e
f
o
ld
E
EV Met Y PCP HPX Hel RdRp
** ** IFN-β mRNA
IFN-β reporter assay Cell viability
FIG 2Screening of ORF1 products for potential IFN antagonists. (A) Schematic illustration of ORF1 products. Met, methyltransferase domain; Y, Y domain; PCP, papain-like cysteine protease; HPX, hypervariable region, proline-rich domain, and X domain; Hel, helicase; RdRp, RNA-dependent RNA polymerase. The numbers above the boxes indicate the numbers of amino acids of the ORF1 polyprotein. (B) Cloning and expression of ORF1 fragments in HEK293T cells detected by Western blotting. The fragments were expressed as HA-tagged proteins. (C) Inhibition of poly(I · C)-induced IFN-expression by ORF1 fragments in HEK293T cells detected by RT-qPCR. The cells were transfected with plasmids of the ORF1 fragments and then transfected with poly(I · C). At 12 h after poly(I · C) treatment, the cells were harvested for RNA isolation and real-time PCR to detect IFN-transcript. PRRSV nsp1was included as a control. Relative levels (fold) in comparison with untreated control cells are shown. EV, empty vector. Significant differences from the EV control are denoted by asterisks (**,P⬍ 0.01). The graph represents averages of three independent experiments. The error bars indicate standard errors. (D) Cell viability assay comparing HEK293T cells transfected with HEV ORF1 domains. Relative percentages are shown in comparison with normal cells transfected with EV. (E) Inhibition of Sendai virus-evoked IFN-expression by HEV PCP and HPX domains. HEK293T cells were transfected with PCP and HPX plasmids, along with IFN-luciferase reporter and a Renillacontrol plasmid. The cells were infected with Sendai virus at an MOI of 1 48 h after transfection. Luciferase activity was assayed 48 h after infection and is shown as a relative level (fold) in comparison with the EV control. The graph represents the averages of three independent experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.136.450.69.334.2](
Fig. 4A
), while the levels of total IRF-3 and tubulin were similar
between the samples. This result indicated that HPX interfered
with IRF-3 activation.
The HPX region contains 3 putative domains, the
hypervari-able (HV), proline-rich (Pro), and X domains. To determine
which domain was capable of antagonizing IRF-3 activation, we
cloned the three domains individually into the pCAGEN
expres-sion vector, which encodes an HA tag at the N terminus of the
target protein. When the plasmids were coexpressed with
FLAG-IRF-3 in 293T cells, expression of FLAG-IRF-3 was very low. Thus,
FLAG-IRF-3 expression was interfered with in the cells transfected
with the plasmids with HV and Pro domains, while endogenous
IRF-3 expression was not affected (data not shown). To resolve
the issue of plasmid interference in coexpression, we established
HEK293 cells stably expressing VenusC1-IRF-3, which makes it
easy to observe IRF-3 expression and to select positive clones.
Phosphorylation of VenusC1-IRF-3 after poly(I·C) stimulation in
the stable cells in the presence of the HV, Pro, or X domain was
examined by Western blotting. The X domain was identified as the
inhibitor of IRF-3 phosphorylation, while the HV and Pro
do-mains had minimum effect (
Fig. 4B
).
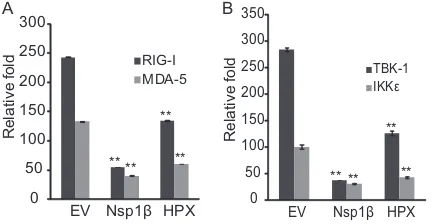
The PCP domain deubiquitinates both RIG-I and TBK-1.
During our screening for the IFN antagonists from the ORF1
do-mains, the papain-like cysteine protease domain also inhibited
poly(I·C)-induced IFN expression. It was reported that the HEV
Met-PCP polyprotein had deubiquitination activity for
ubiqui-tin-, SUMO-, and ISG15-conjugated cellular proteins (
35
). We
reasoned that PCP could potentially lead to deubiquitination of
both RIG-I and TBK-1, as ubiquitination of these two molecules is
essential for their activation (
36
). To test this, we cotransfected
293T cells with plasmids expressing PCP and RIG-I or TBK-1,
since the levels of endogenous RIG-I and TBK-1 are too low to be
detected in Western blotting. Samples were immunoprecipitated
with antibodies to RIG-I or TBK-1, and the ubiquitination status
was assessed by Western blotting with ubiquitin antibody.
De-creased ubiquitination levels of both RIG-I and TBK-1 were
ob-served in cells cotransfected with PCP (3.8 and 3.2-fold,
respec-tively) in comparison with the empty-vector control (
Fig. 5A
and
B
), while the total RIG-I and TBK-1 levels were not affected in
whole-cell lysate. These results suggested that PCP may inhibit
RIG-I and TBK-1 activation via its deubiquitinase activity.
HEV replication in hepatoma cells inhibits IRF-3
phosphor-ylation and ubiquitination of RIG-I and TBK-1.
The results
de-scribed above showed that overexpression of the X and PCP
do-mains of HEV ORF1 in HEK293T cells led to inhibition of IRF-3
phosphorylation and RIG-I and TBK-1 ubiquitination,
respec-tively. We reasoned that the HEV ORF1 product could perform
the same functions during whole-virus infection in hepatoma
cells. To test this, we employed the HEV replicon pSK-E2-GFP.
S10-3 cells that were transfected with RNA from E2-GFP and
cultured for 10 days were stimulated with poly(I·C) for 10 h.
Com-pared to the cells without HEV RNA, the cells with E2-GFP
repli-cation had much lower poly(I·C)-induced IRF-3
phosphoryla-tion, while the total IRF-3 level had minimum change (
Fig. 6A
).
This result was consistent with the findings for X domain
expres-sion in HEK293 cells.
In the S10-3 cells transfected with E2-GFP RNA, the
ubiquiti-nation levels of RIG-I and TBK-1 were also examined. IP results
showed that both RIG-I and TBK-1 in S10-3 cells transfected with
E2-GFP RNA had lower ubiquitination levels than the control
without HEV replication (
Fig. 6B
and
C
). Again, the results were
consistent with the observation of PCP overexpression in
HEK293T cells. Endogenous RIG-I in S10-3 cells was also
detect-able, and no change in its expression was observed between
con-trol cells and E2-GFP RNA-transfected cells (
Fig. 6B
). There was
no change between the samples for the total cellular
ubiquitina-tion level in the whole-cell lysate. However, endogenous TBK-1 in
the whole-cell lysate of S10-3 cells was below detection level in
Western blotting, though it was detectable after being enriched by
IP (
Fig. 6C
). These results demonstrated that HEV replication
inhibited poly(I·C)-induced IFN production by blocking
phos-phorylation of IRF-3 and ubiquitination of RIG-I and TBK-1.
DISCUSSION
Type I IFNs, such as IFN-
␣
and IFN-

, are critical for innate
immunity against viral infection and contribute to the
modula-tion of adaptive immunity (
37
). Viruses have developed a variety
DNA: PIC:
pIRF-3
HPX IRF-3
Tubulin
PIC: - + + + +
Plasmid: EV EV HV Pro X
p-IRF-3
IRF-3
Tubulin
HA-tagged
A B
kDa
10 15 25
EV EV HPX
- + +
FIG 4Inhibition of IRF-3 phosphorylation by the X domain. (A) Expression of an HPX fragment inhibits IRF-3 phosphorylation. HEK293T cells were transfected with IRF-3 and empty vector (EV) or HPX plasmids and then transfected with poly(I · C) (PIC). At 8 h post-PIC treatment, the cells were harvested for detection of IRF-3 phosphorylation by Western blotting with antibody against phosphorylated IRF-3 (pIRF-3). Total IRF-3 and HPX ex-pression was detected with IRF-3 and HA antibodies, respectively. (B) Identi-fication of the X domain in inhibition of IRF-3 phosphorylation. HEK293-VenusC1-IRF-3 stable cells were transfected with HV, Pro, or X domain plasmids, and Western blotting was done as for panel A. The molecular mass marker for the HA blot is shown on the right.
0 50 100 150 200 250 300
EV Nsp1β HPX
Re
la
ti
v
e
f
o
ld RIG-I
MDA-5
** ** **
**
A B
0 50 100 150 200 250 300 350
EV Nsp1β HPX
TBK-1 IKKε
R
e
la
tiv
e
f
o
ld
** ** **
**
FIG 3Reporter assay showing that the ORF1 HPX product inhibits IFN- induction. (A) The HPX product inhibits RIG-I- or MDA-5-activated IFN- induction. HEK293T cells were transfected with IFN- reporter plasmid, along with RIG-I(N) (N-terminal domain) or MDA-5(N) and nsp1or HPX DNA. Firefly andRenillaluciferase activities were measured at 48 h posttrans-fection. PRRSV nsp1was included as a positive control. EV, empty vector. Significant differences from the EV control are denoted by asterisks (**,P⬍ 0.01). The graph represents the averages of three independent experiments. The error bars indicate standard errors. (B) HPX fragments inhibit TBK-1- or IKKε-activated IFN-induction. Transfections were done as for panel A, with the replacement of RIG-I or MDA-5 with TBK-1 or IKKε. Calculations and presentation are done similarly to those in panel A.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.54.271.67.179.2] [image:5.585.299.546.69.159.2]of strategies to subvert or evade the innate immune response.
IRF-3, a critical transcription factor, is frequently targeted by
vi-ruses to interfere with IFN induction. VP35 of Ebola virus inhibits
IRF-3 activation, and this inhibition is related to the viral
viru-lence (
38
). PRRSV nsp1

inhibits IRF-3 activation (
31
). Npro of
classical swine fever virus induces proteasome-mediated
degrada-tion of IRF-3 (
39
,
40
). In this study, we demonstrated that HEV
replication inhibited IFN production. Furthermore, the ORF1
product was responsible for this inhibition. Out of 8 putative
do-mains in the HEV ORF1 product, the PCP and X dodo-mains were
demonstrated to inhibit RIG-I-mediated signaling in different
steps. Sendai virus was used to evoke IFN induction, and the
re-sults confirm the inhibition of the HEV domains. PCP mediates
the deubiquitination of RIG-I and TBK-1, while the X domain
inhibits IRF-3 phosphorylation. A model is proposed to illustrate
the HEV inhibition in the RLR pathway of IFN induction (
Fig. 7
).
PCP: - + PCP: - +
IP: RIG-I WCL
WB: Ub
RIG-I
WB: Ub
RIG-I
PCP
Tubulin
A B
PCP: - + PCP:
-IP: TBK-1 WCL
WB: Ub
WB: Ub
TBK-1 PCP Tubulin
+
TBK-1 kDa
15 25 35 40 55 70 100
130 kDa
15 25 35 40 55 70 100 130
Relative: 1.0 Relative: 1.0 0.24
0.26
FIG 5Deubiquitination of RIG-I and TBK-1 by the PCP domain. (A) RIG-I deubiquitination by PCP. HEK293T cells were transfected with PCP and RIG-I plasmids. At 48 h posttransfection, the cells were harvested for immunoprecipitation with FLAG antibody to pull down RIG-I. Western blotting (WB) with antibodies against ubiquitin (Ub) and RIG-I was conducted. The relative levels of ubiquitination signal intensity are shown below the blots after normalization with RIG-I. Whole-cell lysate (WCL) was used to detect ubiquitin, RIG-I, PCP, and tubulin. The molecular mass marker for the WCL blot is shown on the right. (B) TBK-1 deubiquitination by PCP. The experiment was done as for panel A with the exception that the TBK-1 plasmid was used.
PIC: - + - + E2-GFP: - - + +
A B
E2-GFP: - + E2-GFP: - +
IP: RIG-I WCL
WB: Ub
RIG-I
WB: Ub
RIG-I GFP Tubulin E2-GFP: - +
IP: TBK-1
WB: Ub
TBK-1 C
pIRF-3
GFP
IRF-3
Tubulin
kDa
15 25 35 40 55 70 100 130
Relative: 1.0 0.60
Relative: 1.0 0.32
FIG 6HEV replication leads to downregulation of IRF-3 phosphorylation and deubiquitination of RIG-I and TBK-1 in S10-3 cells. (A) Reduction of PIC-induced IRF-3 phosphorylation in S10-3 cells transfected with HEV RNA from the pSK-E2-GFP replicon. The cells were treated with PIC to induce IRF-3 activation. At 8 h post-PIC treatment, the cells were harvested for de-tection of IRF-3 phosphorylation by Western blotting with antibody against pIRF-3. Total IRF-3, GFP, and tubulin were also detected. (B) Deubiquitina-tion of RIG-I in S10-3 cells with HEV replicaDeubiquitina-tion. The cells were harvested for immunoprecipitation with RIG-I antibody. Western blotting with anti-bodies against ubiquitin and RIG-I was conducted. The relative levels of ubiquitination signal intensity are shown below the blots after normaliza-tion with RIG-I. Whole-cell lysate (WCL) was used to detect ubiquitin, RIG-I, PCP, and tubulin. The molecular mass marker for the WCL blot is illustrated on the right. (C) Deubiquitination of TBK-1 in S10-3 cells with HEV replication. The relative levels of ubiquitination signal intensity are shown below the blots after normalization with TBK-1. The ubiquitin level in the WCL is the same as in panel B.
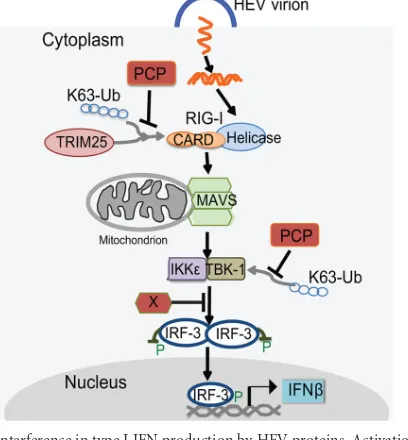
FIG 7Interference in type I IFN production by HEV proteins. Activation of the RLR pathway and signaling by viral dsRNA are shown. Viral dsRNA is generated during HEV replication. “P” indicates phosphorylation. The red boxes indicate HEV proteins identified in this study as inhibiting the signaling steps indicated. The HEV X domain inhibits IRF-3 phosphorylation. HEV PCP inhibits K63-linked polyubiquitination of RIG-I and TBK-1. MAVS, mi-tochondrial antiviral signaling.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.136.450.68.229.2] [image:6.585.41.287.399.562.2] [image:6.585.319.523.436.656.2]The X domain of HEV is known as a macro domain due to its
homology with the C-terminal nonhistone domain of histone
macroH2A, and such domains have been identified in a variety of
bacterial, archaeal, and eukaryotic organisms (reviewed in
refer-ence
41
). Studies of human macro domains indicate they possess a
DNA binding motif and are involved in DNA repair, chromatin
remodeling, and transcriptional regulation. Little information
about viral macro domains is available. The coronavirus macro
domains were shown to possess relatively poor ADP-ribose 1
⬙
-phosphohydrolase activities, implying they are functionally
differ-ent from their human homologues (
42
).
In our study, the HEV X domain was found to inhibit IRF-3
phosphorylation, which appears to be a new function for a viral
macro domain. However, we were unable to detect interaction
between the X domain and IRF-3 or the upstream kinase TBK-1 in
IP experiments (data not shown). Thus, the mechanism through
which the X domain inhibits IRF-3 phosphorylation needs to be
further elucidated. In addition, we found that the X domain of
HEV could bind tightly with chromosomal DNA when
overex-pressed in HEK293T cells (data not shown). The human macro
domains have a DNA binding motif and are involved in
down-regulation of gene activation, suggesting that the HEV X domain
could potentially regulate host gene expression, such as
IFN-stim-ulated genes, which may further dampen IFN signaling. However,
due to the lack of an X antibody and an efficient cell culture system
for HEV, we do not know whether the X domain performs the
proposed function during HEV replication.
Another IFN antagonist we identified from HEV ORF1 is the
PCP domain, which has moderate similarity to the protease
do-main of rubella virus (
29
). While the PCP domain encoded by
rubella virus has been shown to be responsible for the proteolytic
processing of its nonstructural protein (
43
), the role of the HEV
PCP in proteolytically processing the ORF1 product into small
subunits has yet to be confirmed. Recently, the connection of
ubiquitination and activation of the IFN induction pathway was
defined (reviewed in reference
44
). Since cysteine proteases
rep-resent a large family of deubiquitinases (
45
), this implies that
vi-rus-encoded cysteine proteases could function as deubiquitinases
to inhibit ubiquitination-dependent activation of host IFN
induc-tion pathways. Evidence from other viruses shows that
virus-en-coded cysteine proteases, such as arterivirus papain-like protease 2
(
46
) and the leader proteinase of foot-and-mouth disease virus
(
47
), indeed possess a deubiquitinase function and inhibit host
innate immunity.
In a previous report, the HEV Met-PCP polyprotein, which
contains the Met, Y, and PCP domains, was found to act as a
deubiquitinase for ubiquitin-, SUMO-, and ISG15-linked
pro-teins in a cell-free system (
35
). However, Met-PCP was unable to
cleave an LXGG motif, which should be recognized by cellular or
viral PCP. In this study, we demonstrate that the PCP domain
could act as the deubiquitinase for RIG-I and TBK-1 in HEK293T
cells. In addition, our data indicate that HEV replication in S10-3
cells could lead to deubiquitination of RIG-I and TBK-1. A study
of the foot-and-mouth disease virus indicates the
deubiquiti-nase activity of the viral leader protease does not rely on its
proteolytic activity (
47
). It is not known whether HEV PCP
deu-biquitinase activity relies on its protease activity. Since a
Zn-bind-ing fZn-bind-inger is required for protease activity of PCP (
48
), a recent
bioinformatics study identified the putative Zn-binding finger in
the PCP domain as Cys457-His458-Cys459 and Cys481-Cys483
(
49
). Moreover, a recent report determined that the HEV PCP
domain acts as a chymotrypsin-like protease when purified and
renatured from a bacterial expression system (
50
).
What is more, the severe acute respiratory syndrome (SARS)
virus processes a papain-like cysteine protease that is capable of
cleaving ubiquitin- and ISG15-conjugated proteins and helping
the virus evade host innate immunity (
51
,
52
). One study
demon-strated that targeting the viral cysteine protease with a
virus-spe-cific inhibitor blocks SARS virus replication while not affecting
cellular deubiquitinases (
53
). This implies that the deubiquitinase
function of viral PCP could be a potential therapeutic target.
In conclusion, our study identified the PCP and X domains of
HEV ORF1 polyprotein as the IFN antagonists, and they inhibit
RIG-I-mediated signaling in different steps, ubiquitination of
RIG-I and TBK-1 and IRF-3 phosphorylation, respectively. This
provides valuable information for understanding HEV
pathogen-esis and facilitating future development of antiviral drugs.
ACKNOWLEDGMENTS
We thank Suzanne Emerson in the NIH for plasmids E2 and pSK-E2-GFP and S10-3 cells. We also thank Constance L. Cepko for sharing pCAGEN plasmids via Addgene.
Y. Nan, Z. Ma, and Y. Yu were partially supported by the China Schol-arship Council. This study was partially supported by the University of Maryland and NIH grant 1R21AI068881 to Y. Zhang.
REFERENCES
1.Khuroo MS.2011. Discovery of hepatitis E: the epidemic non-A, non-B hepatitis 30 years down the memory lane. Virus Res.161:3–14.http://dx
.doi.org/10.1016/j.virusres.2011.02.007.
2.Kamar N, Dalton HR, Abravanel F, Izopet J.2014. Hepatitis E virus infection. Clin. Microbiol. Rev. 27:116 –138.http://dx.doi.org/10.1128
/CMR.00057-13.
3.Haqshenas G, Shivaprasad HL, Woolcock PR, Read DH, Meng XJ. 2001. Genetic identification and characterization of a novel virus related to human hepatitis E virus from chickens with hepatitis-splenomegaly syndrome in the United States. J. Gen. Virol.82:2449 –2462.
4.Meng XJ, Purcell RH, Halbur PG, Lehman JR, Webb DM, Tsareva TS, Haynes JS, Thacker BJ, Emerson SU.1997. A novel virus in swine is closely related to the human hepatitis E virus. Proc. Natl. Acad. Sci. U. S. A. 94:9860 –9865.http://dx.doi.org/10.1073/pnas.94.18.9860.
5.Li TC, Chijiwa K, Sera N, Ishibashi T, Etoh Y, Shinohara Y, Kurata Y, Ishida M, Sakamoto S, Takeda N, Miyamura T.2005. Hepatitis E virus transmission from wild boar meat. Emerg. Infect. Dis.11:1958 –1960.
http://dx.doi.org/10.3201/eid1112.051041.
6.Emerson SU, Purcell RH.2003. Hepatitis E virus. Rev. Med. Virol.13: 145–154.http://dx.doi.org/10.1002/rmv.384.
7.Mizuo H, Suzuki K, Takikawa Y, Sugai Y, Tokita H, Akahane Y, Itoh K, Gotanda Y, Takahashi M, Nishizawa T, Okamoto H.2002. Polyphyletic strains of hepatitis E virus are responsible for sporadic cases of acute hep-atitis in Japan. J. Clin. Microbiol. 40:3209 –3218.http://dx.doi.org/10
.1128/JCM.40.9.3209-3218.2002.
8.Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, Fry KE, Reyes GR.1991. Hepatitis E virus (HEV): molecular cloning and sequenc-ing of the full-length viral genome. Virology185:120 –131.http://dx.doi
.org/10.1016/0042-6822(91)90760-9.
9.Yu C, Boon D, McDonald SL, Myers TG, Tomioka K, Nguyen H, Engle RE, Govindarajan S, Emerson SU, Purcell RH.2010. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: similarities and differences. J. Virol. 84:11264 –11278. http://dx.doi.org/10.1128/JVI
.01205-10.
10. Graff J, Torian U, Nguyen H, Emerson SU.2006. A bicistronic sub-genomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol.80:5919 –5926.http://dx.doi.org/10.1128/JVI.00046-06. 11. Giles RE, Ruddle FH.1973. Production of Sendai virus for cell fusion. In
vitro9:103–107.http://dx.doi.org/10.1007/BF02616007.
12. Emerson SU, Nguyen H, Graff J, Stephany DA, Brockington A, Purcell RH.2004. In vitro replication of hepatitis E virus (HEV) genomes and of
on November 7, 2019 by guest
http://jvi.asm.org/
an HEV replicon expressing green fluorescent protein. J. Virol.78:4838 – 4846.http://dx.doi.org/10.1128/JVI.78.9.4838-4846.2004.
13. Wang R, Nan Y, Yu Y, Zhang YJ. 2013. Porcine reproductive and respiratory syndrome virus Nsp1beta inhibits interferon-activated JAK/ STAT signal transduction by inducing karyopherin-alpha1 degradation. J. Virol.87:5219 –5228.http://dx.doi.org/10.1128/JVI.02643-12.
14. Wang R, Nan Y, Yu Y, Yang Z, Zhang YJ.2013. Variable interference with interferon signal transduction by different strains of porcine repro-ductive and respiratory syndrome virus. Vet. Microbiol.166:493–503.
http://dx.doi.org/10.1016/j.vetmic.2013.07.022.
15. Emerson SU, Zhang M, Meng XJ, Nguyen H, St Claire M, Govindarajan S, Huang YK, Purcell RH.2001. Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. U. S. A.98:15270 –15275.http://dx
.doi.org/10.1073/pnas.251555098.
16. Graff J, Nguyen H, Kasorndorkbua C, Halbur PG, St Claire M, Purcell RH, Emerson SU.2005. In vitro and in vivo mutational analysis of the 3=-terminal regions of hepatitis E virus genomes and replicons. J. Virol. 79:1017–1026.http://dx.doi.org/10.1128/JVI.79.2.1017-1026.2005. 17. Gale M, Jr, Foy EM.2005. Evasion of intracellular host defence by
hep-atitis C virus. Nature 436:939 –945. http://dx.doi.org/10.1038/nature
04078.
18. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T.2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol.175:2851– 2858.http://dx.doi.org/10.4049/jimmunol.175.5.2851.
19. Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, Kaneko Y, Nimura Y, Motoyama N, Ikeda K, Karin M, Nakanishi M. 2000. NAK is an IkappaB kinase-activating kinase. Nature404:778 –782.
http://dx.doi.org/10.1038/35008109.
20. Peters RT, Liao SM, Maniatis T.2000. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell5:513–522.http://dx
.doi.org/10.1016/S1097-2765(00)80445-1.
21. Kannan H, Fan S, Patel D, Bossis I, Zhang YJ.2009. The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J. Virol.83:6375– 6382.http://dx.doi.org/10.1128
/JVI.02571-08.
22. Zhang YJ, Wang KY, Stein DA, Patel D, Watkins R, Moulton HM, Iversen PL, Matson DO.2007. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res.73:12–23.
http://dx.doi.org/10.1016/j.antiviral.2006.05.017.
23. Patel D, Opriessnig T, Stein DA, Halbur PG, Meng XJ, Iversen PL, Zhang YJ.2008. Peptide-conjugated morpholino oligomers inhibit por-cine reproductive and respiratory syndrome virus replication. Antiviral Res.77:95–107.http://dx.doi.org/10.1016/j.antiviral.2007.09.002. 24. Livak KJ, Schmittgen TD.2001. Analysis of relative gene expression data
using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods25:402– 408.http://dx.doi.org/10.1006/meth.2001.1262. 25. Nan Y, Wang R, Shen M, Faaberg KS, Samal SK, Zhang YJ. 2012.
Induction of type I interferons by a novel porcine reproductive and respi-ratory syndrome virus isolate. Virology432:261–270.http://dx.doi.org/10
.1016/j.virol.2012.05.015.
26. Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ.2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol.84: 11045–11055.http://dx.doi.org/10.1128/JVI.00655-10.
27. Nan Y, Ma Z, Wang R, Yu Y, Kannan H, Fredericksen B, Zhang YJ. 2014. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol.88:8696 – 8705.http://dx.doi.org/10.1128/JVI.01228-14. 28. Shukla P, Nguyen HT, Torian U, Engle RE, Faulk K, Dalton HR,
Bendall RP, Keane FE, Purcell RH, Emerson SU.2011. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infec-tious virus-host recombinant. Proc. Natl. Acad. Sci. U. S. A.108:2438 – 2443.http://dx.doi.org/10.1073/pnas.1018878108.
29. Koonin EV, Gorbalenya AE, Purdy MA, Rozanov MN, Reyes GR, Bradley DW.1992. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. U. S. A.89:8259 – 8263.http://dx.doi.org/10.1073/pnas.89
.17.8259.
30. Parvez MK.2013. Molecular characterization of hepatitis E virus ORF1
gene supports a papain-like cysteine protease (PCP)-domain activity. Vi-rus Res.178:553–556.http://dx.doi.org/10.1016/j.virusres.2013.07.020. 31. Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK,
Osorio FA.2010. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol.84:1574 –1584.http://dx.doi.org/10
.1128/JVI.01326-09.
32. Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M, Jr.2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U. S. A.102:2986 –2991.http://dx
.doi.org/10.1073/pnas.0408707102.
33. Lu LL, Puri M, Horvath CM, Sen GC.2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for in-hibitor of kappaB kinase epsilon (IKKe)/TBK1. J. Biol. Chem.283:14269 – 14276.http://dx.doi.org/10.1074/jbc.M710089200.
34. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S.2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol.6:981–988.
http://dx.doi.org/10.1038/ni1243.
35. Karpe YA, Lole KS.2011. Deubiquitination activity associated with hep-atitis E virus putative papain-like cysteine protease. J. Gen. Virol.92: 2088 –2092.http://dx.doi.org/10.1099/vir.0.033738-0.
36. Maelfait J, Beyaert R.2012. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev.76:33– 45.http://dx.doi.org/10
.1128/MMBR.05012-11.
37. Takaoka A, Yanai H.2006. Interferon signalling network in innate de-fence. Cell. Microbiol.8:907–922.http://dx.doi.org/10.1111/j.1462-5822
.2006.00716.x.
38. Hartman AL, Bird BH, Towner JS, Antoniadou ZA, Zaki SR, Nichol ST. 2008. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of Ebola virus. J. Virol.82:2699 –2704.http://dx.doi.org/10.1128
/JVI.02344-07.
39. Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N.2007. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol.81: 3087–3096.http://dx.doi.org/10.1128/JVI.02032-06.
40. Gottipati K, Ruggli N, Gerber M, Tratschin JD, Benning M, Bellamy H, Choi KH.2013. The structure of classical swine fever virus N(pro): a novel cysteine autoprotease and zinc-binding protein involved in subversion of type I interferon induction. PLoS Pathog.9:e1003704.http://dx.doi.org
/10.1371/journal.ppat.1003704.
41. Han W, Li X, Fu X.2011. The macro domain protein family: structure, functions, and their potential therapeutic implications. Mutat. Res.727: 86 –103.http://dx.doi.org/10.1016/j.mrrev.2011.03.001.
42. Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. 2006. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J. Virol.80:8493– 8502.http://dx
.doi.org/10.1128/JVI.00713-06.
43. Marr LD, Wang CY, Frey TK.1994. Expression of the rubella virus nonstructural protein ORF and demonstration of proteolytic processing. Virology198:586 –592.http://dx.doi.org/10.1006/viro.1994.1070. 44. Liu X, Wang Q, Chen W, Wang C.2013. Dynamic regulation of innate
immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Fac-tor Rev.24:559 –570.http://dx.doi.org/10.1016/j.cytogfr.2013.07.002. 45. Amerik AY, Hochstrasser M.2004. Mechanism and function of
deubiq-uitinating enzymes. Biochim. Biophys. Acta1695:189 –207.http://dx.doi
.org/10.1016/j.bbamcr.2004.10.003.
46. van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, Snijder EJ, Mark BL, Kikkert M.2013. Deubiquiti-nase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc. Natl. Acad. Sci. U. S. A. 110:E838 –E847.http://dx.doi.org/10.1073/pnas.1218464110.
47. Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H, Chen Z, Xiao S.2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol.85:3758 –3766.http://dx.doi.org/10.1128
/JVI.02589-10.
48. Herold J, Siddell SG, Gorbalenya AE.1999. A human RNA viral cysteine proteinase that depends upon a unique Zn2⫹-binding finger connecting the two domains of a papain-like fold. J. Biol. Chem.274:14918 –14925.
http://dx.doi.org/10.1074/jbc.274.21.14918.
on November 7, 2019 by guest
http://jvi.asm.org/
49. Parvez MK, Khan AA.2014. Molecular modeling and analysis of hepatitis E virus (HEV) papain-like cysteine protease. Virus Res.179:220 –224.
http://dx.doi.org/10.1016/j.virusres.2013.11.016.
50. Paliwal D, Panda SK, Kapur N, Varma SP, Durgapal H.2014. Hepatitis E virus (HEV) protease: a chymotrypsin-like enzyme that processes both non-structural (pORF1) and capsid (pORF2) protein. J. Gen. Virol.95: 1689 –1700.http://dx.doi.org/10.1099/vir.0.066142-0.
51. Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD.2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A.103:5717–5722.http://dx.doi.org/10.1073/pnas.0510851103.
52. Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Menard R.2005. The papain-like protease from the severe acute respira-tory syndrome coronavirus is a deubiquitinating enzyme. J. Virol.79: 15199 –15208.http://dx.doi.org/10.1128/JVI.79.24.15199-15208.2005. 53. Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri
R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. 2008. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A.105:16119 – 16124.http://dx.doi.org/10.1073/pnas.0805240105.
54. Jameel S.1999. Molecular biology and pathogenesis of hepatitis E virus. Expert Rev. Mol. Med.1999:1–16.
on November 7, 2019 by guest
http://jvi.asm.org/