by MAVS-Dependent Production of Type I and Type III Interferons
Anggakusuma,aAnne Frentzen,aEngin Gürlevik,bQinggong Yuan,b,cEike Steinmann,aMichael Ott,b,cPeter Staeheli,d Jonathan Schmid-Burgk,eTobias Schmidt,eVeit Hornung,eFlorian Kuehnel,bThomas Pietschmanna
Institute of Experimental Virology, Twincore Centre for Experimental and Clinical Infection Research, Hanover, Germanya
; Department of Gastroenterology, Hepatology and Endocrinology, Medical School Hanover, Germanyb
; Translational Research Group, Twincore Centre for Experimental and Clinical Infection Research, Hanover, Germanyc
; Department of Virology, University of Freiburg, Freiburg, Germanyd
; Institute for Clinical Chemistry and Clinical Pharmacology, University Hospital, University of Bonn, Bonn, Germanye
ABSTRACT
Hepatitis C virus (HCV) efficiently infects only humans and chimpanzees. Although the detailed mechanisms responsible for
this narrow species tropism remain elusive, recent evidence has shown that murine innate immune responses efficiently
sup-press HCV replication. Therefore, poor adaptation of HCV to evade and/or counteract innate immune responses may prevent
HCV replication in mice. The HCV NS3-4A protease cleaves human MAVS, a key cellular adaptor protein required for RIG-I-like
receptor (RLR)-dependent innate immune signaling. However, it is unclear if HCV interferes with mouse MAVS function
equally well. Moreover, MAVS-dependent signaling events that restrict HCV replication in mouse cells were incompletely
de-fined. Thus, we quantified the ability of HCV NS3-4A to counteract mouse and human MAVS. HCV NS3-4A similarly
dimin-ished both human and mouse MAVS-dependent signaling in human and mouse cells. Moreover, replicon-encoded protease
cleaved a similar fraction of both MAVS variants. Finally, FLAG-tagged MAVS proteins repressed HCV replication to similar
degrees. Depending on MAVS expression, HCV replication in mouse liver cells triggered not only type I but also type III IFNs,
which cooperatively repressed HCV replication. Mouse liver cells lacking both type I and III IFN receptors were refractory to
MAVS-dependent antiviral effects, indicating that the HCV-induced MAVS-dependent antiviral state depends on both type I and
III IFN receptor signaling.
IMPORTANCE
In this study, we found that HCV NS3-4A similarly diminished both human and mouse MAVS-dependent signaling in human
and mouse cells. Therefore, it is unlikely that ineffective cleavage of mouse MAVS
per se
precludes HCV propagation in
immu-nocompetent mouse liver cells. Hence, approaches to reinforce HCV replication in mouse liver cells (e.g., by expression of
essen-tial human replication cofactors) should not be thwarted by the poor ability of HCV to counteract MAVS-dependent antiviral
signaling. In addition, we show that mouse MAVS induces both type I and type III IFNs, which together control HCV
replica-tion. Characterization of type I or type III-dependent interferon-stimulated genes in these cells should help to identify key
mu-rine restriction factors that preclude HCV propagation in immunocompetent mouse liver cells.
H
epatitis C virus (HCV) infection is associated with chronic
liver disease, including hepatic steatosis, fibrosis, cirrhosis,
and hepatocellular carcinoma (
1
). Recent licensing of directly
act-ing antivirals (DAAs) has considerably improved therapeutic
op-tions, and novel drug combinations reach cure rates of more than
90% (
2
). However, natural or treatment-induced virus
elimina-tion does not prevent reinfecelimina-tion by HCV. Moreover, many of ca.
160 million infected individuals are not diagnosed, and the vast
majority of HCV patients have not been treated (
3
). Therefore,
development of a prophylactic vaccine that efficiently prevents
virus transmission is a major challenge for global control of
hep-atitis C. However, advances in HCV vaccine research are
ham-pered by a lack HCV-permissive, immunocompetent animal
models.
HCV, a plus-strand RNA virus and member of the family
Fla-viviridae
, has a narrow species tropism. Besides humans, only
chimpanzees are naturally susceptible to chronic HCV infection.
However, high costs and ethical concerns severely limit utilization
of chimpanzees for research purposes. Thus, understanding of the
mechanisms that limit HCV replication in other animals, for
in-stance, mice, is an important step to ultimately develop a robust
animal model for HCV. Encouragingly, recent advances have
highlighted key prerequisites for HCV infection of murine liver
cells. First, occludin and CD81 have been recognized to determine
HCV species tropism at the level of viral cell entry (
4
).
Impor-tantly, this blockade can be overcome by engineering mice to
ex-press human occludin and CD81 (
5
) or by adaptation of HCV to
usage of mouse CD81 (
6
). Second, HCV replication in murine
embryonic fibroblasts (
7
,
8
), in mouse liver-derived cells (
9
), and
Received27 October 2014Accepted12 January 2015
Accepted manuscript posted online21 January 2015
CitationAnggakusuma, Frentzen A, Gürlevik E, Yuan Q, Steinmann E, Ott M, Staeheli P, Hornung V, Kuehnel F, Pietschmann T. 2015. Control of hepatitis C virus replication in mouse liver-derived cells by MAVS-dependent production of type I and type III interferons. J Virol 89:3833–3845.doi:10.1128/JVI.03129-14. Editor:M. S. Diamond
Address correspondence to Thomas Pietschmann, [email protected].
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.03129-14
on November 7, 2019 by guest
http://jvi.asm.org/
in mouse livers
in vivo
(
10
) is heavily impaired by innate immune
signaling, since inactivation of host molecules involved in viral
RNA sensing, innate immune signaling, or responsiveness to
in-terferons substantially increases HCV replication. Therefore,
ab-lation of distinct innate immune signaling molecules combined
with overexpression of essential human entry factors has emerged
as a valid strategy to allow HCV propagation in mouse cells
in vitro
and
in vivo
(
9
,
10
). However, this environment is only partially
immunocompetent, thus limiting utility for immunological
stud-ies. Moreover, the efficiency of HCV propagation remains modest
either because additional immune control mechanisms curtail
HCV replication or because essential human replication cofactors
are lacking.
In human cells, the HCV protease NS3-4A interferes with
in-nate immune signaling by cleaving TRIF (TIR domain-containing
adaptor-inducing beta interferon [IFN-

]) (
11
) and MAVS
(mi-tochondrial antiviral signaling protein; also known as IPS-1,
VISA, or Cardif) (
12
), two critical adaptor proteins that link
cel-lular pattern recognition receptors with production of
interfer-ons. Nevertheless, viral interference in human cells is not
com-plete, as HCV infection of human liver cells triggers production of
both type I and III interferons which partially control HCV
repli-cation (
13–16
). Moreover, distinct human IFN-induced effector
proteins relevant for control of HCV replication have been
iden-tified (
17–19
). In contrast, little is known about murine
IFN-in-duced antiviral programs that limit HCV replication. Moreover,
the interferon-stimulated genes (ISGs) that establish antiviral
de-fenses against HCV replication in mouse cells are unknown.
Fi-nally, the level of interference of HCV with murine innate
im-mune signaling cascades is incompletely defined. Given the
importance of innate immunity for control of HCV replication in
both the human and murine systems, in this study, we wished to
better define the relevance of innate immune control and HCV
interference for propagation of HCV in mouse liver cells.
MATERIALS AND METHODS
Reagents.Mouse IFN-␣and IFN-3 were purchased from eBioscience
and R&D Systems, respectively. Boceprevir and 2=C-methyladenosine
(2=CMA) were gifts from Marc Windisch (Institute Pasteur Korea,
Seongnam, South Korea) and Tim Tellinghuisen (The Scripps Re-search Institute, FL), respectively. High-molecular-weight (HMW) poly(I·C) was purchased from InvivoGen. HCV subgenomic replicon
(HCV-SGR) RNA was generated in-house byin vitrotranscription as
described previously (9).
Cell culture and generation of cell lines.All cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented
with 2 mML-glutamine, nonessential amino acids, 100 U/ml of penicillin,
100g/ml of streptomycin, and 10% fetal calf serum (FCS) at 37°C and
5% CO2. MLT-MAVS⫺/⫺and MLT-IFNAR⫺/⫺cells were generated in a
previous study (9). Stable cell lines expressing miR122 were created by
lentiviral gene transfer as described earlier (9). MLT-MAVS⫺/⫺miR122
cells with stable restoration of human MAVS (hMAVS) or mouse MAVS (mMAVS) were created through lentiviral gene transduction followed by
passaging in the presence of antibiotic pressure (5g/ml of blasticidin) as
described previously (9).
The generation of HEK 293T-MAVS⫺/⫺cells was done by targeting
the critical exons of the MAVS gene in 293T cells using the following
transcription activator-like effector nuclease (TALEN) target site: 5=-TT
GCTGAAGACAAGACCTAT/AAGTATATCTGCCGC/AATTTCAGCA
ATTTTTGCAA-3=. TALEN plasmids were assembled as described in
ref-erence3. HEK 293T cells were plated at a density of 2⫻104cells per well
of a 96-well plate. The next day, TALEN plasmids were transfected using
GeneJuice transfection reagent (Merck Millipore) at a ratio of 1:1 (100 ng of each plasmid). Subsequently, all-allelic knockout clones were isolated. In short, 14 days after limiting dilution cloning, growing monoclones were selected by bright-field microscopy. Identified clones were trypsinized and expanded in two separate wells. One well was used to recover genomic DNA, and subsequently, the target region of interest was amplified in a two-step PCR and subjected to deep sequencing. Knockout cell clones were identified as cell clones harboring all-allelic frameshift
mutations using the OutKnocker webtool athttp://www.outknocker.org
/. To further confirm the knockout, Western blotting was performed.
Note that the faint band visible in Western blotting (seeFig. 2E) is a result of a nonspecific cross-reaction of the antibody used. This was confirmed by further Western blotting and genotyping, which verified proper knock-out of MAVS (data not shown). Genotypes of the respective knockknock-out cell lines are available upon request.
Mice. Normal C57BL/J6 and BALB/c mice were purchased from Charles River Labs. C57BL/6 mice lacking functional receptors for type I
and III IFN (20) were bred at University Medical Center Freiburg. Mouse
experiments were carried out using animals between 6 and 12 weeks of age. Mice were housed under specific-pathogen-free conditions at the Twincore animal facility, and all experiments were conducted according to institutional guidelines for animal care and use and in compliance with regulations of German animal welfare law.
In vivoimmortalization and generation of tumor cell lines.
Intrahe-patic tumors were induced in IFNAR⫺/⫺IFNLR1⫺/⫺double-knockout
mice (20) by retrograde liver transduction with oncogenic transposon
plasmids using hydrodynamic injection. For this purpose, mice received
30g of total plasmid DNA (8g of pT/KRas-G12V, 8g of pT3/EF1␣
-myrAkt1, 8g of pT3/EF1␣-shRp53, and 6g of pPGK-SB13). A more
detailed description of the used plasmids and plasmid construction can be
found elsewhere (9). The pPGK-SB13 plasmid for expression of Sleeping
Beauty SB13 was kindly provided by David A. Largaespada (University of Minnesota). For tumor induction, oncogenic transposon plasmids were injected into mice in 0.9% saline at a final volume of 10% of the animal’s body weight via tail vein injection within 5 s. Once tumors became palpa-ble (5 to 12 weeks postinjection), mice were sacrificed and tumors were harvested for subsequent isolation of immortalized cell lines. Isolated tu-mors were dissected and incubated for 30 min at 37°C in RPMI 1640 plus
GlutaMAX supplemented with 200g/ml of collagenase IA, collagenase
IV, and hyaluronidase IV, 300g/ml of dispase, and 50g/ml of DNase I
(Sigma-Aldrich). Separated cells were purified using a 40-m strainer,
washed once, and then cultivated in DMEM plus GlutaMAX with 10%
FCS and penicillin-streptomycin at 37°C in 5% CO2. Cells were then
cultivated as described previously (9). Deletions of IFNAR and IFNLR1
genes were confirmed by quantitative reverse transcription-PCR (qRT-PCR).
PMH isolation.Primary mouse hepatocytes (PMHs) were isolated from BALB/cJ and/or C57BL/6J mice by a modified 2-step Liberase
per-fusion (21,22). Briefly, mice were anesthetized using ketamine (Albrecht)
and Rompun (Bayer). The body cavity was opened and a catheter was placed into the portal vein and connected to a flow pump, which pumped media prewarmed at 37°C from a water bath into the catheter. The liver was first perfused with Earle’s balanced salt solution (EBSS) (GIBCO) solution containing 0.5 mM EGTA (Sigma-Aldrich) and 10 mM HEPES buffer (Sigma). Subsequently, EBSS supplemented with 10 mM HEPES
buffer and Liberase (Roche) at 100g/liter was applied for enzymatic
digestion of the tissue at 37°C. After digestion for 10 to 12 min, the liver was carefully disconnected and tissue was manually disrupted with a ster-ile scissors and scalpel in DMEM (GIBCO Germany) containing 10%
FCS. The suspended hepatocytes were passed through a 100-m nylon
filter into 50-ml Falcon tubes. The cell suspensions were centrifuged twice at 300 rpm for 5 min at 4°C, and the cell pellet was resuspended in ice-cold DMEM containing 10% FCS (PAN Biotech). Cell viability was tested by trypan blue (Fluka).
Anggakusuma et al.
on November 7, 2019 by guest
http://jvi.asm.org/
Transient HCV luciferase replication assay.Methods for
transfec-tion of HCV-SGR RNA are described elsewhere (9). Luciferase activity of
replicating HCV-SGR was analyzed as described elsewhere (9,23).
Lentiviral pseudoparticle production.Lentiviral, HIV-based
pseu-dotypes were created essentially as described previously (9). Briefly, HEK
293T cells were transfected with envelope glycoprotein expression
con-struct pcz-VSV-G, lentiviral gag-pol expression concon-struct pCMV⌬R.74,
and lentiviral genomic backbone for the desired gene expression using a polyethyleneimine (PEI) method (Carl Roth).
Plasmids.The lentiviral plasmid pWPI-JFH1-NS3-4A-BLR encodes the NS3-4A protease of HCV strain JFH1. The lentiviral plasmids pWPI-hMAVS-BLR, pWPI-mMAVS-BLR, pWPI-HA-hMAVS-GUN, and pWPI-HA-mMAVS-GUN encode human or mouse MAVS variants with or without a hemagglutinin (HA) tag. Lentiviral plasmid pWPI-mIFNLR1-BLR encodes the mouse ortholog of a high-affinity type III IFN receptor (NM174851.3). All pWPI constructs were cloned in-house with detailed cloning strategies that are available upon request. The plasmid pTRIP-pre-miR-122-puro was described previously and given as a kind
gift from Matthew Evans (Mount Sinai School of Medicine, NY) (24). The
IFN-promoter reporter plasmid (pGL3b-IFN-promoter-Luc) was
kindly provided by Stefan Lienenklaus (HZI, Braunschweig, Germany). Western blotting.Cells were washed with phosphate-buffered saline (PBS) and lysed in RIPA buffer (0.3 M NaCl, 20 mM Tris-HCl [pH 8], 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], and 1% Triton X-100) for 30 min on ice. Total protein content was determined by the Bradford assay. Equal protein amounts for each sample were mixed with
2⫻denaturing protein sample buffer (200 mM Tris-HCl [pH 8.8], 5 mM
EDTA, 0.1% bromophenol blue, 10% sucrose, 3.3% SDS, 2% 2-mercap-toethanol [2-ME]), heated for 5 min at 98°C, loaded onto an 11% SDS gel, and resolved by electrophoresis. Subsequently, proteins were transferred to a polyvinylidene difluoride membrane which was then blocked with 5% milk in PBS containing 0.5% Tween (PBS-T) for 1 h at room temper-ature. The membrane was then incubated with either anti-NS3337 tag (1:300), anti-HA tag (1:1,000; Covance), anti-FLAG tag (1:1,000; Sigma-Aldrich), hMAVS or mMAVS (both at 1:300; Santa Cruz),
anti-green fluorescent protein (anti-GFP, 1:1,000; Santa Cruz), or anti--actin
(1:1,000; Sigma-Aldrich), followed by incubation with secondary body coupled to horseradish peroxidase (Sigma-Aldrich). Bound anti-bodies were detected with the ECL Plus detection system (GE Healthcare). Quantitative reverse transcription-PCR.Total RNA was extracted using the Nucleospin RNA II kit (Macherey-Nagel) by following the
man-ufacturer’s protocol. Subsequently, 2l was reverse transcribed into
cDNA using the PrimeScript First Strand cDNA synthesis kit (TaKaRa), and PCR was carried out using a 400 nM concentration of primers
to-gether with SYBR PremixEx Taq(TaKaRa) and was quantified with a
LightCycler480 (Roche). Murine glyceraldehyde-3-phosphate dehydro-genase (mGAPDH) mRNA levels were always quantified in parallel.
Prim-ers used this study are as follows: for mouse IFN-, CTGCGTTCCTGCT
GTGCTTCTCCA (sense) and TTCTCCGTCATCTCCATAGGGATC
(antisense) (25); for mouse IFN-2/3, AGCTGCAGGCCTTCAAAAAG
(sense) and TGGGAGTGAATGTGGCTCAG (antisense) (26); for mouse
GAPDH, TTCACCACCATGGAGAAGGC (sense) and GGCATCGACT
GTGGTCATGA (antisense) (25); for mouse viperin, AACCCCCGTGAG
TGTCAACTA (sense) and AACCAGCCTGTTTGAGCAGAA (antisense)
(27); for mouse ISG15, AGCAATGGCCTGGGACCTAAA (sense) and
AGCCGGCACACCAATCTT (antisense) (28); and for mouse IFIT1, CA
GAAGCACACATTGAAGAA (sense) and TGTAAGTAGCCAGAGGA
AG (antisense) (29).
Immunofluorescence.HCV replication was visualized by indirect
im-munofluorescence as described previously (23). HCV antigen-expressing
cells were detected using the mouse monoclonal NS5A-9E10 antibody (kindly provided by Charles M. Rice, Center for the Study of Hepatitis C, The Rockefeller University, NY) at a dilution of 1:1,000. Bound primary antibodies were detected using goat anti-mouse IgG-specific secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 546
(Sigma-Aldrich) at a dilution of 1:1,000. Nuclear DNA was stained using 4=
,6-diamidino-2-phenylindole (DAPI) at a dilution of 1:3,000.
Statistical methods.GraphPad Prism 6 software was used for data analysis using a one-way analysis of variance (ANOVA) adjusted with
Bonferroni’s multiple-comparison test.Pvalues of⬍0.05 (single asterisks
in figures) were considered statistically significant, whereasPvalues of
⬍0.01 (double asterisks) and⬍0.001 (triple asterisks) were considered
highly significant.
RESULTS
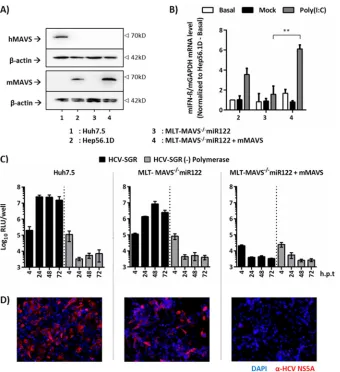
mMAVS restricts HCV replication in MLT cells.
Previously, we
have reported that HCV replicons and full-length RNAs efficiently
propagate in transfected mouse liver-derived tumor (MLT) cells
which lack endogenous expression of MAVS (MLT-MAVS
⫺/⫺miR122 cells) (
9
). These cells therefore provide a unique
environ-ment to test the importance of mouse MAVS (mMAVS) for
re-stricting HCV replication and to explore the ability of HCV to
interfere with mMAVS function.
To this end, we confirmed the absence of endogenous mMAVS
expression in these cells and subsequently restored mMAVS
ex-pression by lentiviral gene transfer. Murine Hep56.1D cells and
human Huh-7.5 cells served as controls for specific detection of
mMAVS in our protein expression analysis (
Fig. 1A
). Parental
MLT-MAVS
⫺/⫺miR122 cells were not responsive to transfection
of poly(I·C), a mimic of double-stranded RNA which triggers
RIG-I-like receptor (RLR [
30
])-dependent signaling via MAVS to
produce of IFN-

mRNA (
Fig. 1B
). In contrast, restoration of
mMAVS expression in these cells (
Fig. 1A
) resulted in
upregula-tion of IFN-

mRNA upon transfection of poly(I·C) to a level
similar to the one observed in poly(I·C)-transfected Hep56.1D
cells (
Fig. 1B
). Thus, we concluded that repair of mMAVS
expres-sion in MLT-MAVS
⫺/⫺miR122 cells restored the sensitivity of
these cells to RLR-dependent recognition of viral double-stranded
RNA comparable to that of cells expressing endogenous levels of
mMAVS (e.g., Hep56.1D). Next, we analyzed the influence of
re-introducing mMAVS on permissiveness for HCV replication. To
this end, cells were transfected with a subgenomic JFH1 replicon
expressing luciferase or a control luciferase replicon with an
inac-tivating mutation in the viral NS5B polymerase. HCV replication
in transfected cells was monitored by luciferase assays (
Fig. 1C
)
and indirect immunofluorescence (
Fig. 1D
). As reported
previ-ously (
9
), parental MLT-MAVS
⫺/⫺miR122 cells sustained
vigor-ous HCV RNA replication, as evidenced by rapid accumulation of
luciferase activity (
Fig. 1C
) and by detection of numerous
NS5A-expressing cells (
Fig. 1D
). In contrast, reintroduction of mMAVS
ablated HCV replication to background levels, with luciferase
ac-tivity indistinguishable from that of cells transfected with the
rep-lication-incompetent viral RNA (
Fig. 1C
) and with no
NS5A-pos-itive cells detectable (
Fig. 1D
). Therefore, repair of mMAVS
expression in MLT-MAVS
⫺/⫺miR122 cells efficiently restored
innate immune signaling and restricted HCV RNA replication.
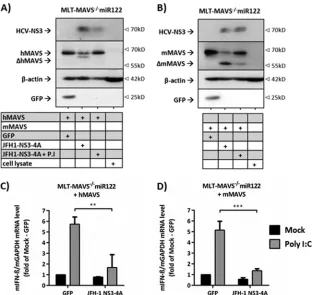
HCV interferes with mouse and human MAVS to similar
lev-els.
The above-described findings suggested that HCV replication
is efficiently restricted by mMAVS potentially because the HCV
NS3-4A protease fails to efficiently cleave and interfere with
mMAVS function. In turn, inefficient HCV interference with
mMAVS in mouse cells may explain why HCV is unable to
efficiently replicate in mouse cells with active innate immune
signaling.
To test these hypotheses, we compared HCV interference with
hMAVS and mMAVS. First, we stably expressed either human or
on November 7, 2019 by guest
http://jvi.asm.org/
mouse MAVS in MLT-MAVS
⫺/⫺miR122 cells (
Fig. 2
), followed
by transient expression of the NS3-4A protease via lentiviral gene
transduction. This method was chosen to mimic the expression of
the protease in viral invasion independent of constraints on HCV
RNA replication (e.g., due to lack of HCV replication-assisting
cofactors). Both hMAVS (
Fig. 2A
) and mMAVS (
Fig. 2B
) were
partially cleaved upon transient coexpression of NS3-4A in
MLT-MAVS
⫺/⫺miR122 cells. This is either due to incomplete
trans-duction of the cell populations with the HCV protease or due to
incomplete cleavage of overexpressed MAVS proteins under these
experimental conditions. Cleavage was specifically mediated by
the viral protease, since coexpression of GFP did not induce
cleav-age and as addition of a protease inhibitor reduced cleavcleav-age of
both hMAVS and mMAVS (
Fig. 2A
and
B
). When these cells were
transfected with poly(I·C), coexpression of the HCV NS3-4A
pro-tease clearly lowered both hMAVS- and mMAVS-dependent
production of IFN-

(
Fig. 2C
and
D
). Since the extents to
which IFN-

production was suppressed were comparable
be-tween NS3-4A coexpressed with hMAVS and mMAVS, we
con-clude that in this system the HCV protease interferes with the
function of these two MAVS proteins to similar levels. To
ex-clude that species-specific signaling activity of hMAVS in
mouse cells may mask differential HCV interference, we
con-ducted principally similar cotransfection experiments with
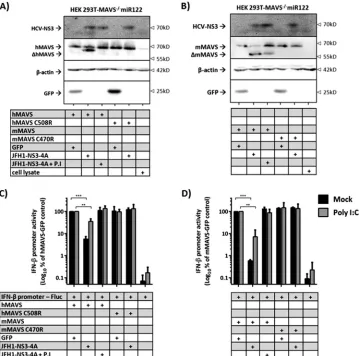
hu-man HEK 293T cells, which upon transcription activator-like
effector nuclease (TALEN)-mediated knockout of hMAVS lack
endogenous expression of the adaptor protein (
Fig. 3A
). Like in
the mouse cells, we noted HCV NS3-4A-dependent cleavage of
both MAVS species (
Fig. 3A
and
B
). The appearance of partial
cleavage of MAVS was mainly caused by the cotransfection
FIG 1mMAVS suppresses HCV replication in transfected mouse liver-derived tumor (MLT) cells. (A) Detection of mMAVS and hMAVS proteins in givenhuman and murine cell lines using MAVS-specific antibodies. Detection of-actin expression was used as a loading control. (B) Responsiveness of
MLT-MAVS⫺/⫺miR122 cells with or without mMAVS expression to transfection of poly(I·C). Hep56.1D mouse cells expressing endogenous levels of mMAVS served
as a control. A day after seeding, the indicated cells were either left untreated, mock transfected, or transfected with 10g/ml of poly(I·C). Four hours later, total
cellular RNA was collected and the expression of mIFN-mRNA was determined by RT-PCR. Expression of IFN-mRNA was normalized to endogenous levels
of mGAPDH and is given relative to the basal expression in Hep56.1D cells. Asterisks show a significant difference between the indicated data. (C) Transient
replication of HCV-JFH1 subgenomic replicon with luciferase reporter with an active NS5B polymerase (HCV-SGR) or a⌬GDD polymerase mutant [HCV-SGR
(⫺) Polymerase] in given cell lines. Transfected cells were harvested at the indicated time points, and HCV RNA replication was determined by luciferase assays. RLU, relative light units; h.p.t, hours posttransfection. (D) Detection of HCV NS5A expression (red) in transfected cells by indirect immunofluorescence (IF)
analysis 48 h posttransfection. Nuclei were stained with DAPI (blue). All graph data are shown as mean values⫾SDs from three independent experiments, and
images are representative of three individual experiments.
Anggakusuma et al.
on November 7, 2019 by guest
http://jvi.asm.org/
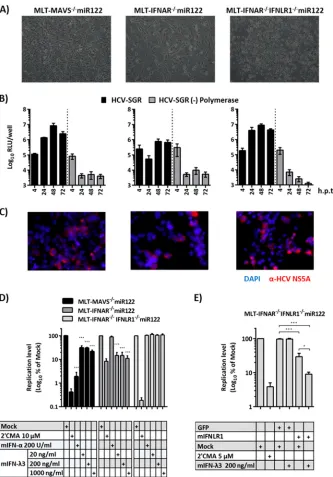
[image:4.585.125.465.64.436.2]efficiency issue, as not all cells were equally transfected with
equal amounts and ratios of plasmids. Finally, using an IFN-

promoter-dependent luciferase reporter plasmid, we observed
5- to 10-fold-greater suppression of poly(I·C)-dependent IFN-

promoter activity by expression of the NS3-4A protease in the case
of mMAVS than with hMAVS (
Fig. 3C
and
D
). Collectively, these
results indicate that the HCV NS3-4A protease cleaves and
inter-feres with the function of both mMAVS and hMAVS in human
and mouse cells. Moreover, interference with the murine MAVS
ortholog was not inferior to that with the human MAVS protein.
To investigate whether NS3-4A expressed from an
autono-mously replicating HCV RNA is sufficient to overcome restriction
by hMAVS or mMAVS in MLT-MAVS
⫺/⫺miR122 cells, we
trans-fected a subgenomic HCV luciferase replicon into parental
MLT-MAVS
⫺/⫺miR122 cells or derivative cell lines stably expressing
these MAVS variants. Irrespective of whether hMAVS or mMAVS
was present, luciferase activity did not rise over the background
level defined by the replication-inactive replicon mutant (
Fig. 4A
).
In parallel, only very inefficient cleavage of both MAVS variants
was observed (
Fig. 4B
), which likely results from initial
produc-tion of viral protease translated from the input of transfected viral
RNA. Taken together, these results indicate that HCV replication
was suppressed to background levels in cells overexpressing either
hMAVS or mMAVS.
Considering that overexpression of MAVS may preactivate
cellular innate immune responses prior to transfection, thus
ablating HCV replication, we chose an alternative
experimen-tal setting: MAVS proteins were introduced into
MLT-MAVS
⫺/⫺miR122 cells engineered to express a replicating
HCV luciferase replicon (
Fig. 4C
). In these experiments, the
degree of MAVS overexpression was modulated by serially
di-luting the lentiviral stocks used for transduction of MAVS
pro-teins (
Fig. 4D
). Interestingly, both mMAVS and hMAVS
re-stricted HCV replication in a dose-dependent fashion (
Fig.
4E
), as evidenced by ca. 85% reduction of luciferase activity at
the highest dose of MAVS expression compared with the
expression of GFP. Notably, the two MAVS proteins were
over-expressed to similar levels, as was evident from our protein
expression analysis utilizing FLAG-specific antibodies to detect
the FLAG-tagged hMAVS and mMAVS proteins transfected in
these experiments (
Fig. 4D
). Congruently, transfection of
wild-type, untagged hMAVS and mMAVS proteins resulted in
com-parable and dose-dependent repression of HCV replication in
MLT-MAVS
⫺/⫺miR122 replicon cells (data not shown).
Fi-nally, FLAG-tagged hMAVS and mMAVS variants that are
re-sistant to cleavage by the HCV NS3-4A protease due to
muta-tion of the cleavage site exerted a further increased repression
on HCV replication compared to that with the parental MAVS
FIG 2Comparison HCV NS3-4A-dependent cleavage of and interference with hMAVS and mMAVS in mouse cells. (A and B) Human (A) or mouse (B) MAVSwas expressed in MLT-MAVS⫺/⫺miR122 cells in the presence or absence of GFP or the HCV NS3-4A protease. Expression of proteins was monitored by Western
blotting using GFP-, actin-, mMAVS-, or hMAVS-specific antibodies. One day after lentiviral gene transduction, the medium was changed and the indicated cells were treated with addition of 10M boceprevir, a protease inhibitor (P.I). Twenty-four hours later, the cells were lysed for specific Western blot analysis. The
cleaved (⌬) products of hMAVS and mMAVS are indicated, and-actin expression was used as a loading control. Images are representative of two individual
experiments. (C and D) Effect of HCV NS3-4A protease expression on human (C) or mouse (D) MAVS-dependent type I IFN induction. One day after the cells were transduced, the cells were either mock transfected or transfected with 10g/ml of poly(I·C). Four hours later, total RNA was extracted and the relative levels
of mIFN-mRNA expression were determined by qRT-PCR. Data were normalized to the mGAPDH mRNA data. Asterisks show a significant difference
between the indicated data. Mean values⫾SDs from three independent experiments are given.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.136.455.65.360.2]proteins that are susceptible to cleavage by HCV (ca. 3-fold)
(
Fig. 4E
). These results indicate that the ability of HCV to
cleave MAVS facilitates HCV replication. Furthermore, since
the degrees of restriction exerted by parental MAVS and the
cleavage-resistant MAVS variant were in each case
indistin-guishable between the human and the mouse orthologs, we
conclude that HCV is capable of interfering with mouse MAVS
to a level similar to that of the human variant. As a
conse-quence, it is unlikely that inefficient viral cleavage of mouse
MAVS is the critical limitation that prevents HCV from
prop-agating in mouse liver cells with intact innate immune
sig-naling.
HCV replication in mouse liver tumor cells elicits
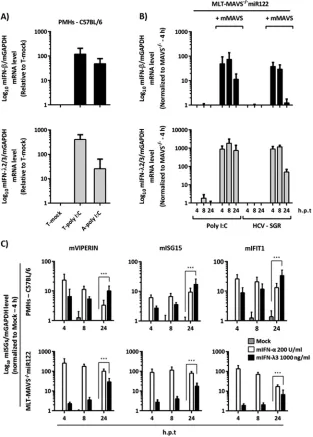
produc-tion of both type I and III IFNs in a MAVS-dependent fashion.
Next, we dissected the mMAVS-regulated downstream
signal-ing events responsible for limitsignal-ing HCV replication in mouse
cells. To this end, we first explored which type of IFN is
pro-duced upon administration of poly(I·C) or after transfection of
HCV RNA. Using mIFN-

(type I IFN)-specific and mIFN-
(type III IFN)-specific mRNA detection systems, we observed
that both types of IFNs were vigorously upregulated upon
ex-tracellular addition or transfection of poly(I·C) to primary
mouse hepatocytes (
Fig. 5A
). In the case of MLT-MAVS
⫺/⫺miR122 cells, repair of mMAVS expression led to rapid and
strong induction of mIFN-

and mIFN-
mRNA upon
trans-fection of poly(I·C) (
Fig. 5B
). Likewise, transfection of HCV
replicon RNA also induced both types of IFNs (
Fig. 5B
),
indi-cating that HCV replication in MLT-MAVS
⫺/⫺miR122 elicits
type I and III IFN production in a mMAVS-dependent
fashion.
FIG 3Comparison HCV NS3-4A-dependent cleavage of and interference with hMAVS and mMAVS in human cells. (A and B) Appearance of human (A) or
mouse (B) MAVS expression in the presence of HCV protease in human HEK 293T-MAVS⫺/⫺miR122 cells. The cells were reverse transfected with reporter
plasmids (100 ng) together with vector expressing each indicated gene (100 ng). A day after transfection, the medium was changed and the indicated cells were treated with addition of 10M boceprevir, a protease inhibitor (P.I). Twenty-four hours later, the cells were lysed and specific Western blotting detection using
the indicated antibodies was performed. The cleaved (⌬) products of hMAVS and mMAVS are indicated, and-actin expression was used as a loading control.
Images are representative of two individual experiments. (C and D) Effect of HCV protease presence on human (C) or mouse (D) MAVS-dependent IFN-
promoter regulation. HEK 293T-MAVS⫺/⫺miR122 cells were reverse transfected with reporter plasmids (100 ng) together with the each indicated expression
vector (100 ng). A day after the cells were reverse transfected, the medium was changed and the indicated cells were transfected with either water (mock) or 10
g/ml of poly(I·C) for 4 h. After that, the cells were treated with addition of water or 10M P.I. The cells were then lysed and assayed for luciferase activities 24 h later. Values were then normalized to those for MAVS-GFP-transfected cells. Asterisks show a significant difference between the indicated data. All graph data
are shown as mean values⫾SDs from three independent experiments, and Western blotting images are representative of two individual experiments.
Anggakusuma et al.
on November 7, 2019 by guest
http://jvi.asm.org/
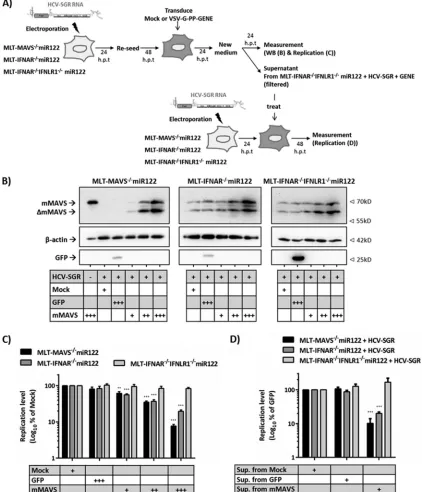
[image:6.585.113.472.67.423.2]FIG 4Control of HCV replication in mouse liver cells by human and mouse MAVS. (A) Replication of HCV in in MLT-MAVS⫺/⫺miR122 cells stably expressing
hMAVS or mMAVS. Luciferase replicon RNA was transfected into the given cell lines and RNA replication was determined at the given time points by way of
luciferase assays. A replicon with inactive polymerase transfected into MLT-MAVS⫺/⫺miR122 cells served as a control. (B) Cleavage of hMAVS and mMAVS by
replicon-encoded NS3-4A protease. MAVS cleavage was monitored 48 h after mock transfection (N) or HCV-SGR transfection (S) into MLT-MAVS⫺/⫺miR122
cells stably expressing hMAVS and mMAVS. (C) Schematic representation of the experimental setup used to titrate MAVS abundance. The HCV replicon
(HCV-SGR) was transfected into target cells (MLT-MAVS⫺/⫺miR122 cells) to establish HCV replication. Twenty-four hours later, cells were reseeded into
12-well plates. At 48 h posttransfection, the cells were transduced with equal titers of lentiviruses carrying the gene of interest.⫹, low-titer dose;⫹⫹, middle-titer
dose;⫹⫹⫹, high-titer dose. After another 48 h, expression of the gene of interest and HCV RNA replication were measured. (D) Dose-dependent expression of
FLAG-tagged MAVS variants. hMAVS C508R and mMAVS C470R represent hMAVS and mMAVS variants resistant to cleavage by the HCV NS3-4A protease
(E) Dose-dependent repression of HCV RNA replication by FLAG-tagged MAVS variants. Given MAVS proteins were introduced into MLT-MAVS⫺/⫺miR122
cells carrying a luciferase replicon.as described for panel C. HCV RNA replication was measured by luciferase activity and normalized to the data for cells that
were transduced to express GFP. Asterisks show a significant difference between the indicated data. All graph data are shown as mean values⫾SDs from three
independent experiments, and Western blotting images are representative of two individual experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
Both type I and III IFNs control HCV replication in mouse
liver tumor cells.
To assess if both types of IFN contribute to
control of HCV replication in these cells, we first investigated if
both primary mouse hepatocytes and MLT-MAVS
⫺/⫺miR122
cells are responsive to these IFNs. To this end, we incubated these
cells with exogenously added mouse IFN-
␣
or mouse IFN-
3 and
monitored upregulation of the mRNAs of typical
interferon-stim-ulated genes (ISGs). All three ISGs tested by us were clearly
in-duced upon administration of either mIFN-
␣
or mIFN-
,
indi-cating that these cells are responsive to both type I and III IFNs
(
Fig. 5C
). To explore the relevance of both types of IFNs for
con-trol of HCV replication in mouse liver-derived cells, we
estab-lished a novel MLT cell line lacking both type I and III IFN
recep-tors (MLT-IFNAR
⫺/⫺IFNLR1
⫺/⫺miR122) (
Fig. 6A
). When
transfected with an HCV luciferase replicon, these cells sustained
vigorous HCV RNA replication similar to that with
MLT-MAVS
⫺/⫺miR122 cells and somewhat greater than with MLT
cells lacking endogenous expression of the type I IFN receptor
FIG 5mMAVS-dependent induction of type I and III IFN mRNA in mouse liver cells. (A) Relative mRNA expression of mouse type I (mIFN-) and III IFN(mIFN-2/3) in wild-type C57BL/6 mouse-derived primary hepatocytes. The primary cells were treated with 50g/ml of poly(I·C) either by transfection (T-poly
I:C) or by direct addition to the culture medium (A-poly I:C). Eight hours later, total RNA from the each group of treated cells was extracted. (B) Induction of
mIFN-and mIFN-2/3 mRNA in MLT-MAVS⫺/⫺miR122 cells with or without mMAVS. The indicated cells were transfected with 50g/ml of poly(I·C) or
with 10g of HCV replicon RNA. At the indicated time points, transfected cells were collected and total RNA was extracted. The level of IFN mRNA expression
was normalized to that of mGAPDH mRNA as determined by qRT-PCR. h.p.t, hours posttransfection. (C) Induction of representative mouse
interferon-stimulated genes (mISGs) in wild-type C57BL/6 mouse PMHs and in MLT-MAVS⫺/⫺miR122 cells treated with the indicated doses of exogenous mouse type I
(mIFN-␣) or III (mIFN-3) IFN. At the indicated time points, cells were harvested and total RNA was extracted. mISG mRNA levels are expressed relative to
mGAPDH mRNA as determined by qRT-PCR. Asterisks show a significant difference between the indicated data. All graph data are shown as mean values⫾SDs
from three independent experiments.
Anggakusuma et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.136.449.70.507.2]alone (MLT-IFNAR
⫺/⫺miR122 [
9
]) (
Fig. 6B
). This observation
was also corroborated by analysis of NS5A expression in the
trans-fected cells showing larger numbers of NS5A-positive cells in the
case of MAVS knockout and IFNAR plus IFNLR1 double
knock-out than with the IFNAR knockknock-out alone (
Fig. 6C
). Therefore,
inactivation of MAVS or both type I and III IFN receptors together
renders MLT cells highly permissive to HCV RNA replication. To
confirm that the latter cells lack functional type I and III IFN
receptors, we treated them with exogenous mIFN-
␣
or different
doses of mIFN-
3 and used the nucleoside polymerase inhibitor
FIG 6Regulation of HCV-SGR replication in mouse cells by mouse type III IFNs. (A) Phenotypic appearances of all MLT cells. (B) Transient replication of theHCV luciferase replicon without polymerase activity in MLT-MAVS⫺/⫺miR122, MLT-IFNAR⫺/⫺miR122, and MLT-IFNAR⫺/⫺IFNLR1⫺/⫺miR122 cells. At
the indicated time points posttransfection, HCV RNA replication was determined by luciferase activity. (C) Indirect immunofluorescence analysis of NS5A expression in given cell lines 48 h posttransfection. The HCV-NS5A protein was detected by specific antibody (9E10), and cell nuclei were stained with DAPI. (D)
Effect of exogenously added mouse type I and III IFNs (mIFNa and mIFN-3, respectively) on HCV replication in given MLT cells. Forty-eight hours after
administration of the given drugs, HCV RNA replication was determined by luciferase assays. Luciferase activity was normalized to that measured in control cells that were mock treated. Asterisks indicate a significant difference compared to each related mock group. (E) Repair of type III IFN receptor expression restores
responsiveness of MLT-IFNAR⫺/⫺IFNLR1⫺/⫺miR122 cells to antiviral effect mediated by mIFN-3. Given MLT cells carrying a luciferase replicon were
transduced to express GFP or the mIFNLR1 cDNA. Subsequently, cells were treated as indicated with 2=CMA or mIFN-3. Luciferase activity was determined 48
h later and is expressed relative to that in mock-treated and untransduced cells. Asterisks show a significant difference between the indicated data. All graph data
are shown as mean values⫾SDs from three independent experiments; images are representative of two individual experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.126.459.64.541.2]2
=
C-methyladenosine (2
=
CMA) as a control. As expected, the
polymerase inhibitor reduced HCV replication in all three
above-mentioned cell lines (
Fig. 6D
). In contrast, mIFN-
3 was antiviral
in MLT-MAVS
⫺/⫺miR122 and MLT-IFNAR
⫺/⫺miR122 cells,
and mIFN-
␣
was active only in MLT-MAVS
⫺/⫺miR122 cells.
Importantly, HCV replication in MLT-IFNAR
⫺/⫺IFNLR1
⫺/⫺miR122 cells was fully resistant to exogenous administration of
both type I and III IFN, confirming the absence of both IFN
re-ceptors (
Fig. 6D
). Finally, we repaired expression of IFNLR1 in the
MLT cells with double knockout of type I and III IFN receptors
using lentiviral gene transfer and monitored HCV replication in
the presence or absence of mIFN-
3 (
Fig. 6E
). Reintroduction of
the type III IFN receptor chain reduced HCV replication ca.
3-fold. Moreover, it also restored responsiveness of these cells to
mIFN-
3, as addition of this type of IFN further repressed HCV
replication to a level ca. 10-fold lower than in mIFN-
3-treated
cells in the absence of IFNLR1 expression (
Fig. 6E
). Collectively,
these observations indicate that HCV replication in MLT cells is
susceptible to suppression by both exogenous type I and III IFNs.
Finally, we explored if MAVS-dependent upregulation of
en-dogenous type I and III IFNs limits HCV replication in the MLT
culture system by using the experimental setup detailed in
Fig. 7A
.
To this end, we transduced mMAVS into MLT-MAVS
⫺/⫺miR122,
MLT-IFNAR
⫺/⫺miR122,
or
MLT-IFNAR
⫺/⫺IFNLR1
⫺/⫺miR122 cells that had been transfected with an HCV
replicon. Subsequently, we monitored MAVS cleavage by HCV
(
Fig. 7B
) and the influence of mMAVS overexpression on
HCV replication by using luciferase assays (
Fig. 7C
). As expected,
lentiviral gene transfer resulted in dose-dependent expression of
mMAVS in all transduced cell lines and HCV NS3-4A partially
cleaved overexpressed or endogenous mMAVS (
Fig. 7B
).
Reintro-duction of mMAVS into MLT-MAVS
⫺/⫺miR122 cells, which
en-dogenously express both functional type I and III IFN receptors
(
Fig. 6
), suppressed HCV-dependent luciferase activity in a
dose-dependent fashion down to less than 10% at the highest dose of
mMAVS expression (
Fig. 7C
). Notably, mMAVS overexpression
also suppressed HCV replication in MLT cells lacking the type I
IFN receptor, albeit with modestly reduced efficacy (
Fig. 7C
). In
contrast, in cells that lack both type I and III IFN receptors,
over-expression of mMAVS no longer was antiviral, as evidenced by
stable high-level luciferase expression irrespective of the level of
overexpressed mMAVS (
Fig. 7C
). To examine if the antiviral
ac-tivity was caused by mMAVS-dependent secretion of both type I
and III IFNs, we assessed the bioactivity of the supernatants
col-lected from the mMAVS-overexpressing MLT-IFNAR
⫺/⫺IFNLR1
⫺/⫺miR122 cells. To this end, these supernatants were
filtered and passed onto MLT-MAVS
⫺/⫺miR122, MLT-IFNAR
⫺/⫺miR122, or MLT-IFNAR
⫺/⫺IFNLR1
⫺/⫺miR122 cells that
had been transfected with an HCV replicon (
Fig. 7D
). The
condi-tioned supernatants could suppress
de novo
HCV-dependent
lu-ciferase activity only in the MLT-MAVS
⫺/⫺miR122 and
MLT-IFNAR
⫺/⫺miR122 cells and not in the MLT cells lacking both IFN
receptors (
Fig. 7D
), confirming the secretion of bioactive type I
and III IFNs. Taken together, these data indicate that HCV
repli-cation in mouse-derived liver cells triggers mMAVS-dependent
signaling which upregulates both type I and III IFNs, which, in
turn, suppress HCV replication.
DISCUSSION
In this study, we showed that mMAVS-dependent innate immune
signaling potently restricts HCV replication in mouse liver cells by
way of inducing both type I and III IFN production. These results
refine and extend previous
in vitro
and
in vivo
studies highlighting
the fact that ablation of host proteins involved in innate immune
signaling facilitates HCV replication in mouse cells (
7–10
).
Nota-bly, mMAVS does potently restrict HCV replication in mouse cells
in spite of its susceptibility to cleavage by the CV NS3-4A protease,
which was reported recently (
31
,
32
). However, it remained
un-determined if the efficiency of cleavage paralleled that of hMAVS
and if expression of mMAVS poses a greater degree of restriction
on HCV replication than its human ortholog.
Therefore, we quantitatively and qualitatively compared the
cleavage efficiencies of hMAVS and mMAVS by the HCV NS3-4A
protease both in human and in mouse cells. Moreover, we
ex-plored to what level HCV interferes with the signaling activities of
both proteins and how efficiently these two orthologs restrict
HCV replication in mouse liver cells. Overexpressed HCV
NS3-4A similarly diminished hMAVS- and mMAVS-dependent
signaling in mouse (
Fig. 2
) and human (
Fig. 3
) cells. Moreover,
replicon-encoded protease cleaved similar fractions of
overex-pressed hMAVS and mMAVS, as was evident from detection of
these proteins via a FLAG tag (
Fig. 4
). Therefore, although
species-specific functioning of hMAVS and mMAVS in human and
mouse cells cannot be fully ruled out, these analyses strongly
sug-gest that in the human and mouse cells tested by us, cleavage and
viral interference with hMAVS and mMAVS were comparable.
These novel findings have implications relevant to the
devel-opment of immunocompetent mouse models for HCV. First, it is
unlikely that inefficient mMAVS cleavage by HCV
per se
prevents
efficient HCV replication in mouse liver cells. Therefore, MAVS
function apparently does not pose an insurmountable barrier to
HCV replication. Of note, in human cells, HCV NS3-4A also
cleaves human TRIF, which transmits Toll-like receptor 3
(TLR-3)-dependent signaling for production of IFN (
11
). Since there is
controversy about whether mouse TRIF is susceptible to cleavage
by HCV (
32
,
33
), it is possible that poor adaptation of HCV to
interfere with TRIF-dependent signaling prevents efficient HCV
replication in mouse cells. While resolving this question merits
further work, it is also possible that lack of human replication
cofactors limits replication and accumulation of HCV proteins to
establish sufficient interference with innate immunity. If these
cofactors were known, their ectopic expression in the mouse
en-vironment may reinforce HCV replication so that artificial
abla-tion of MAVS-dependent (and/or TRIF-dependent) innate
im-munity is not necessary. The cell lines described in this study
should be a useful platform to screen for such human replication
cofactors, for instance, by cDNA library-based approaches.
An alternative approach to establish
(pseudo)immunocompe-tent mouse models for HCV may be to selectively eliminate
dis-tinct IFN-expressed antiviral effector proteins to circumvent
global elimination of central innate immunity signaling
mole-cules. This study indicates that in the context of MLT cells, both
type I- and III-dependent antiviral effector mechanisms are
trig-gered by HCV replication which cooperate to efficiently suppress
HCV replication. This conclusion is based on our observations
that HCV RNA triggers both mouse IFN-

and IFN-
mRNA
expression (
Fig. 5
) and that HCV replication in MLT cells can be
Anggakusuma et al.on November 7, 2019 by guest
http://jvi.asm.org/
suppressed by exogenous administration of both type I and III
IFNs (
Fig. 6
). Moreover, we observed that MAVS
signaling-de-pendent suppression of HCV replication mediated by secreted
type I and type III IFNs was most pronounced in cells expressing
both type I and III receptors, was reduced when the type I IFN
receptor was lacking, and was fully ablated when both IFN
recep-tors were absent (
Fig. 7
). Although MAVS also directly activates
interferon-stimulated genes (reference
34
and data not shown),
FIG 7mMAVS-dependent secreted type I and III IFNs cooperatively restrict HCV replication in mouse liver cells. (A) Schematic representation of the experimental setup. First, the HCV replicon (HCV-SGR) was transfected into target cells to establish HCV replication. Twenty-four hours later, cells were reseeded into 12-well plates. Forty-eight posttransfection, the cells were mock transduced or challenged with matched titers of lentiviruses carrying either GFPor mMAVS using three different doses.⫹, low-titer dose;⫹⫹, middle-titer dose;⫹⫹⫹, high-titer dose. At the desired time point, expression of the gene of
interest was measured by Western blotting (B) and dose-dependent repression of HCV replication in given cell lines by mMAVS expression was measured by luciferase assays (C). Data are expressed relative to data for the mock-treated controls, and asterisks indicate a significant difference compared to each related
mock treatment. (D) To test the activity of secreted IFN, the supernatants from the mock-transduced or gene-of-interest (⫹⫹⫹)-transduced MLT-IFNAR⫺/⫺
IFNLR1⫺/⫺miR122 cells with replicating HCV-SGR were collected, filtered with a 0.45-mm filter, and then used to treatde novoHCV-SGR replication in the
indicated cells. Forty-eight hours later, the HCV RNA replication was measured by luciferase assays. Data are expressed relative to those for the mock-treated
controls, and asterisks indicate a significant difference compared to each related mock group. All graph data are shown as mean values⫾SDs from three
independent experiments, and Western blotting images are representative of two individual experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.80.502.70.562.2]this is apparently not sufficient to efficiently restrict HCV
replica-tion in the MLT cells lacking both IFN receptors (
Fig. 7
). These
data indicate that to mount a strong antiviral effect, it is necessary
for MAVS-dependent antiviral effector mechanisms to involve
signaling through type I and III IFN receptors. Further work will
be needed to find out which IFN-dependent mouse effector
pro-teins curtail HCV propagation. Evaluation of the type I- and III
IFN-dependent gene expression signatures should provide
inter-esting clues in this direction.
To what extent both type I and III IFNs limit HCV replication
in mouse livers
in vivo
remains to be shown. Notably, a recent
study concluded that type III IFN did not impact acute infection of
mouse livers by hepatotropic viruses due to the absence of
expres-sion of the type III IFN receptor (
35
). Given that expression of this
receptor is tightly controlled by epigenetic mechanisms, it is
pos-sible that this may change in the course of a long-term (
36
),
chronic infection. In any case, firm evidence derived from human
liver cells and from chimpanzees firmly establishes that HCV
in-duces both type I and III IFN responses which contribute to
con-trol of replication (
13
,
14
,
16
). In this regard, the novel MLT cells
described here may be useful not only to delineate species-specific
restrictions for HCV replication in mouse liver cells but also to
model both type I and III IFN-dependent HCV control
mecha-nisms.
ACKNOWLEDGMENTS
We are very grateful to Takaji Wakita for the gift of the JFH1 isolate, to Charles Rice for Huh-7.5 cells and 9E10 antibody, and to Matthew Evans for the miR122 expression construct. We also thank all members of the Institute for Experimental Virology at Twincore for helpful comments and discussions of this work.
This work was supported by a grant from the European Research Council, ERC-2011-StG_281473-(VIRAFRONT), and by a grant from the Helmholtz Association SO-024 to T.P.
Twincore Centre for Experimental and Clinical Infection Research is a joint venture between the Medical School Hanover and the Helmholtz Centre for Infection Research, Braunschweig.
REFERENCES
1.Maasoumy B, Wedemeyer H.2012. Natural history of acute and chronic
hepatitis C. Best Pract Res Clin Gastroenterol26:401– 412.http://dx.doi
.org/10.1016/j.bpg.2012.09.009.
2.Tse MT.2013. All-oral HCV therapies near approval. Nat Rev Drug Dis-cov12:409 – 411.http://dx.doi.org/10.1038/nrd4036.
3.Thomas DL.2013. Global control of hepatitis C: where challenge meets
opportunity. Nat Med19:850 – 858.http://dx.doi.org/10.1038/nm.3184.
4.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice
CM.2009. Human occludin is a hepatitis C virus entry factor required for
infection of mouse cells. Nature457:882– 886.http://dx.doi.org/10.1038
/nature07684.
5.Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, Schoggins JW, Catanese MT, Burton DR, Law M, Rice CM, Ploss
A.2011. A genetically humanized mouse model for hepatitis C virus
in-fection. Nature474:208 –211.http://dx.doi.org/10.1038/nature10168.
6.Bitzegeio J, Bankwitz D, Hueging K, Haid S, Brohm C, Zeisel MB, Herrmann E, Iken M, Ott M, Baumert TF, Pietschmann T. 2010. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog6:e1000978.http: //dx.doi.org/10.1371/journal.ppat.1000978.
7.Chang KS, Cai Z, Zhang C, Sen GC, Williams BR, Luo G. 2006. Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibro-blasts: protein kinase R (PKR)-dependent and PKR-independent mecha-nisms for controlling HCV RNA replication and mediating interferon activities. J Virol80:7364 –7374.http://dx.doi.org/10.1128/JVI.00586-06. 8.Lin LT, Noyce RS, Pham TN, Wilson JA, Sisson GR, Michalak TI,
Mossman KL, Richardson CD.2010. Replication of subgenomic hepati-tis C virus replicons in mouse fibroblasts is facilitated by deletion of inter-feron regulatory factor 3 and expression of liver-specific microRNA 122. J
Virol84:9170 –9180.http://dx.doi.org/10.1128/JVI.00559-10.
9.Frentzen A, Anggakusuma, Gurlevik E, Hueging K, Knocke S, Ginkel C, Brown RJ, Heim M, Dill MT, Kroger A, Kalinke U, Kaderali L, Kuehnel F, Pietschmann T.2014. Cell entry, efficient RNA replication, and pro-duction of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology59:78 – 88.http://dx.doi.org/10.1002/hep.26626. 10. Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T,
Vogt A, Catanese MT, Satoh T, Kawai T, Akira S, Law M, Rice CM, Ploss A.2013. Completion of the entire hepatitis C virus life cycle in
genetically humanized mice. Nature501:237–241.http://dx.doi.org/10
.1038/nature12427.
11. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM.2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor
protein TRIF. Proc Natl Acad Sci U S A102:2992–2997.http://dx.doi.org
/10.1073/pnas.0408824102.
12. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Barten-schlager R, Tschopp J.2005. Cardif is an adaptor protein in the RIG-I
antiviral pathway and is targeted by hepatitis C virus. Nature437:1167–
1172.http://dx.doi.org/10.1038/nature04193.
13. Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB.2011. Hepatitis C virus induces interferon-lambda
and interferon-stimulated genes in primary liver cultures. Hepatology54:
1913–1923.http://dx.doi.org/10.1002/hep.24580.
14. Park H, Serti E, Eke O, Muchmore B, Prokunina-Olsson L, Capone S, Folgori A, Rehermann B.2012. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection.
Hepa-tology56:2060 –2070.http://dx.doi.org/10.1002/hep.25897.
15. Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, Rice
CM.2014. Interferon lambda alleles predict innate antiviral immune
re-sponses and hepatitis C virus permissiveness. Cell Host Microbe15:190 –
202.http://dx.doi.org/10.1016/j.chom.2014.01.007.
16. Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. 2012. HCV infection induces a unique hepatic innate immune response associated with robust production of type III
interferons. Gastroenterology 142:978 –988.http://dx.doi.org/10.1053/j
.gastro.2011.12.055.
17.Metz P, Reuter A, Bender S, Bartenschlager R. 2013. Interferon-stimulated genes and their role in controlling hepatitis C virus. J Hepatol
59:1331–1341.http://dx.doi.org/10.1016/j.jhep.2013.07.033.
18. Schoggins JW.2014. Interferon-stimulated genes: roles in viral pathogen-esis. Curr Opin Virol6:40 – 46.http://dx.doi.org/10.1016/j.coviro.2014.03 .006.
19. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM.2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature472:481– 485.http://dx.doi.org/10 .1038/nature09907.
20. Cooper J, Fields JK.1988. A hospital wellness program: where challenge
meets opportunity. Home Healthc Nurse6:10 –16.
21. Rothe M, Rittelmeyer I, Iken M, Rudrich U, Schambach A, Glage S, Manns MP, Baum C, Bock M, Ott M, Modlich U. 2012. Epidermal growth factor improves lentivirus vector gene transfer into primary mouse
hepatocytes. Gene Ther 19:425– 434.http://dx.doi.org/10.1038/gt.2011
.117.
22. Yuan Q, Loya K, Rani B, Mobus S, Balakrishnan A, Lamle J, Cathomen T, Vogel A, Manns MP, Ott M, Cantz T, Sharma AD.
2013. MicroRNA-221 overexpression accelerates hepatocyte
prolifer-ation during liver regenerprolifer-ation. Hepatology57:299 –310.http://dx.doi
.org/10.1002/hep.25984.
23. Anggakusuma, Colpitts CC, Schang LM, Rachmawati H, Frentzen A, Pfaender S, Behrendt P, Brown RJ, Bankwitz D, Steinmann J, Ott M, Meuleman P, Rice CM, Ploss A, Pietschmann T, Steinmann E.2014. Turmeric curcumin inhibits entry of all hepatitis C virus genotypes into
human liver cells. Gut 63:1137–1149.http://dx.doi.org/10.1136/gutjnl
-2012-304299.
24. Narbus CM, Israelow B, Sourisseau M, Michta ML, Hopcraft SE, Zeiner GM, Evans MJ.2011. HepG2 cells expressing microRNA miR-122 sup-port the entire hepatitis C virus life cycle. J Virol85:12087–12092.http: //dx.doi.org/10.1128/JVI.05843-11.
25. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieber-Anggakusuma et al.
on November 7, 2019 by guest
http://jvi.asm.org/
man J.2010. The cytosolic exonuclease TREX1 inhibits the innate im-mune response to human immunodeficiency virus type 1. Nat Immunol
11:1005–1013.http://dx.doi.org/10.1038/ni.1941.
26. Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog4:e1000017.http://dx.doi.org /10.1371/journal.ppat.1000017.
27. Bossaller L, Rathinam VA, Bonegio R, Chiang PI, Busto P, Wespiser AR, Caffrey DR, Li Q Z, Mohan C, Fitzgerald KA, Latz E, Marshak-Rothstein A.2013. Overexpression of membrane-bound fas ligand (CD95L) exacerbates autoimmune disease and renal pathology in
pristane-induced lupus. J Immunol191:2104 –2114.http://dx.doi.org/10
.4049/jimmunol.1300341.
28. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR.
2006. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol80:4501– 4509.http://dx.doi.org/10.1128/JVI.80 .9.4501-4509.2006.
29. Fensterl V, White CL, Yamashita M, Sen GC.2008. Novel characteristics
of the function and induction of murine p56 family proteins. J Virol82:
11045–11053.http://dx.doi.org/10.1128/JVI.01593-08.
30. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S.2005. IPS-1, an adaptor triggering RIG-I- and
Mda5-mediated type I interferon induction. Nat Immunol6:981–988.
http://dx.doi.org/10.1038/ni1243.
31. Ahlen G, Derk E, Weiland M, Jiao J, Rahbin N, Aleman S, Peterson DL, Pokrovskaja K, Grander D, Frelin L, Sallberg M.2009. Cleavage of the
IPS-1/Cardif/MAVS/VISA does not inhibit T cell-mediated elimination of
hepatitis C virus non-structural 3/4A-expressing hepatocytes. Gut58:
560 –569.http://dx.doi.org/10.1136/gut.2007.147264.
32. Vogt A, Scull MA, Friling T, Horwitz JA, Donovan BM, Dorner M, Gerold G, Labitt RN, Rice CM, Ploss A. 2013. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology444:
1–11.http://dx.doi.org/10.1016/j.virol.2013.05.036.
33. Abe T, Kaname Y, Hamamoto I, Tsuda Y, Wen X, Taguwa S, Moriishi K, Takeuchi O, Kawai T, Kanto T, Hayashi N, Akira S, Matsuura Y.2007. Hepatitis C virus nonstructural protein 5A modu-lates the Toll-like receptor-MyD88-dependent signaling pathway in
macrophage cell lines. J Virol81:8953– 8966.http://dx.doi.org/10.1128
/JVI.00649-07.
34. Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M, Jr, Fruh K, Diamond MS.2013. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic
cells downstream of MAVS signaling. PLoS Pathog9:e1003118.http://dx
.doi.org/10.1371/journal.ppat.1003118.
35. Hermant P, Demarez C, Mahlakoiv T, Staeheli P, Meuleman P, Michiels T.2014. Human but not mouse hepatocytes respond to
inter-feron-lambda in vivo. PLoS One 9:e87906. http://dx.doi.org/10.1371
/journal.pone.0087906.
36. Ding S, Khoury-Hanold W, Iwasaki A, Robek MD. 2014. Epigenetic reprogramming of the type III interferon response potentiates antiviral
activity and suppresses tumor growth. PLoS Biol12:e1001758.http://dx
.doi.org/10.1371/journal.pbio.1001758.
on November 7, 2019 by guest
http://jvi.asm.org/