Vol. 51, No. 2 JOURNALOFVIROLOGY, Aug. 1984, p.470-478
0022-538X/84/080470-09$02.00/0
Copyright ©)1984, AmericanSociety for Microbiology
Murine
Leukemia Virus Mutant with
a
Frameshift in the
Reverse
Transcriptase
Coding Region: Implications for
pol
Gene Structure
JUDITH G. LEVIN,l* STELLA C. HU,1 ALAN REIN, LESLEY I. MESSER,'t ANDBRENDA I. GERWIN3
Laboratory of Molecutlar Genetics, National Instituite ofChild Health andHutman Development, Bethesda, Maryland
202051; Laboratory ofMoleculalr Virology and Carcinogenesis, Litton Bionetics, Inc.-Basic ResearchProgram, National CancQer Instituite, Frederick CancerResearch Faicility, Frederick, Maryland 217012; andLaboratory ofHuman
Carcinogenesis, NationalCancer Institutte, Bethesda, Ma,Iyland202053 Received 7February 1984/Accepted 12 April 1984
The mQlecular defect in the nonconditional B-tropic MuLV pol mutant, clone 23 (Gerwinet al., J. Virol. 31:741-751, 1979), has been characterized by recombinantDNA technology. The entiremutant genome was
cloned fromanEcoRIdigest of integratedcellular DNA intobacteriophage A Charon4Aand then subclonedat theEcoRI siteof pBR322. NIH-3T3 cells transfected with the plasmid clone, termed pRTM (RTM, reverse transcriptase mutant), reproduced the properties of clone 23 virus-infected cells.Invivo ligation experiments involvingcotransfectionof subclones of pRTM and wild-type murine leukemia virus localizedthedefectin the clone 23 genometo an approximately 400-base-pair region in thepol gene between the Sail andXhoI sites.
Sequence analysis of this region in the wild-type and mutant genonies revealed that the mutant has one
additionalC residue located 231 bases downstream ofthe last base ofthe Sall recognition site. This 1-base insertion bringsthreeTGA termination codons into phase. Thus, the mutation in clone 23 leadstopremature termination of translation,explaining thepresenceinclone23 virionsofatruncated polymerasewith low levels
ofenzymatic activity. Itwaspreviously shownthatthegagprecursoriscleaved normally in clone23-infected cells;therefore, ifavirus-codedproteaseis involved in this cleavage, itmustbeencoded bysequencesupstream of the reverse transcriptase region of thepolgene. Thisconsideration, coupled with the observed molecular
weight of themutantpolymeraseandourprecise determinationof its C terminus, have ledtoaproposalfor the
geneticorganizationof the murineleukemia viruspolgene.
Themurineleukemia virus (MuLV)polgenecodesfor the reversetranscriptase enzyme. Thisproteinisasingle 70,000-to80,000 (70 to8QK)-dalton polypeptide (60) which, like the avian retrovirus enzyme (60), exhibits RNA- and
DNA-dependent DNA polymerase activityand RNase Hactivity
(30, 42, 61). Although polymerase and RNase H reside on
the same polypeptide, eachof these activities is thought to
have adifferent active site. This conclusion is based on the
differential sensitivities of thetwoactivitiestovarious types of inhibitors (6, 11, 18, 40, 41, 60) and on the fact that
proteolytic cleavage ofreverse transcriptase gives rise to a
protein
product that has RNase H but not polymerase activity (15, 29). It has been suggested (28, 57) that anMuLV-associated endonuclease protein of approximately
40Kcaltons(28, 43), which haspropertiessimilartothoseof
the avian retrovirus-encoded endonuclease pp32 (20) and
which
shares methionine-containing tryptic peptides with MuLV polymerase precursor molecules (28), may also beencodedby the pol gene.
The present study was undertaken to further define the
relationship between the genetic structure of the pol gene anditsassociatedactivities. Ourapproachwasto character-ize the molecular defect inanonconditional MuLV
polymer-ase mitant, clone 23 (16), whose phenotype was already known. Earlier work onclone23 has demonstratedthatit is
a replication-defective virus with a defect in the pol gene (16). Cells producing clone 23 are missing the gag-pol
precursor, Pr180g'w'1""/ (27), buthave been shown tocontain
*Correspondingauthor.
tPresent address: Houghton Poultry Research
Station.
Houghton, Huntingdon, Cambridgeshire PE17 2DA, Great Britain.
smaller proteins of 147K and 117K daltons which could be immunoprecipitated with anti-polymerase sera (16). Other
experimnents
haveindicatedthatmutantvirionsareunabletosynthesize high-molecular-weight viral DNAin vivo (1) and have only 2 to 5% of the wild-type level of polymerase
activitywhenassayedwithasynthetic primer-templatesuch
as poly(rA)
oligo(dT)12>18
(16). This activity is associatedwith a truncated enzyme sedimenting with a molecular weight of 47K in glycerol gradients (16). Recent work has also shown that endogenousreversetranscriptase activityis very low and that the only products synthesized in the
endogenousreactionaresmall minus-strand DNA
intermedi-ates no larger than 2.5 kilobases (kb) (L. I. Messer, K. M.
Currey, J. B. O'Neill, J. V. Maizel, Jr., J. G. Levin, and
B. I. Gerwin, manuscript in preparation). Despite its
re-duced size,themutantenzymeretains RNase Hactivityand isindistinguishable from the wild-typeenzymewith respect
to primer-template and metal ion preferences and immune reactivity towards antibodydirected against purified MuLV
polymerase (16).Interestingly, clone 23virions containallof the usual gagstructuralproteins(16), afunctional ecotropic
glycoprotein (16), normal levelsof thetRNAPrO primer(33), and a70S RNA complex that canbe heat denatured to 35S
RNA subunits (16).
In this study, a molecular clone of the entire mutant
genomewas isolated in bacteriophage A Charon 4A (4) and
then subcloned at the EcoRI site ofpBR322. The plasmid
clone is referred to as pRTM (RTM, reverse transcriptase
mutant). Analysis of pRTM showsthatthedefect inclone23
is aframeshift mutation resulting from insertion ofone base
nearthe middleof the pol gene. This frameshift bringsthree
TGAtermination codons into phase and leadstopremature termination of translation. Knowledge of the exact position
470
on November 10, 2019 by guest
http://jvi.asm.org/
ofthe genetic defect inclone 23. coupled with the previous characterization of the mutant phenotype (16). suggests a
mapfor the organization of the MuLVpolgene. MATERIALS AND METHODS
Materials.
32P-labeled
nucleoside triphosphates were pur-chased from Amersham Corp. (Arlington Heights, Ill.).[3H]TTP was obtained from New England Nuclear Corp. (Boston, Mass.). Restriction endonuclease enzymes were
obtained from New England BioLabs (Beverly, Mass.) or
from Bethesda Research Laboratories (Gaithersburg, Md.). G418was agift from the Schering Corp. (Bloomfield, N.J.).
Cells and viruses. Conditions for growth of the SC-1i
NIH-3T3, mink (CCL64), 3T3FL (17), and clone 23 (16) cell lines have been described previously(16, 31, 32). AKR-L1 MuLV was obtained from Marilyn Lander and was propagated in SC-1 cells (32).
Transmission of the pol mutant to mink cells. The replica-tion-defective MuLV was rescued from clone 23 cells by superinfecting the cells with amphotropic MuLV 1504A as
described previously (52). Culture fluids from the superin-fected culturewere initially assayed in the complementation plaque assay (51) and then used to infect mink cells at a
multiplicity of infection of 0.2 complementation PFU per cell. The mink cells were cloned in microtest wells the
followingday: cloneswere thenpicked and screened forthe
presence of a defective ecotropic MuLV by testing their
abilitytofuse XC cells after cocultivation with cells produc-ing Moloney mink cell focus-forming MuLV (50). Clones that were positive in this preliminary screen were then
superinfected with amphotropic MuLV, Moloney mink cell
focus-forming MuLV, or feline leukemia virus, and the supernatants of these cultures were tested in the
comple-mentation plaque assay (51, 52).
Viral probes and hybridization procedures. An ecotropic
MuLV-specific plasmid probe (generous gift of Sisir K.
Chattopadhyay and Douglas Lowy), containing a
400-base-pair(bp)SinatI fragmentderived from theenvl regionofAKR
MuLV (7), was used for hybridization to
high-molecular-weightcellular DNA.Forusein screening molecularclones.
the400-bpfragmentwas separated from thevector. In some
experiments, an insert probe containing the entire AKR MuLVgenome wasprepared by isolating the 11-kb HinzdIII-EcoRI fragment of the recombinant plasmid 623P (see be-low). Probeswerenick translated(53)to aspecificactivityof
1 x 108to 2 x 108
cpm/jig.
Hybridization wascarriedout at40°C for 20 h essentially as described by Wahl et al. (65), except that the prehybridization step was omitted and the buffer was7 mM Tris-hydrochloride(pH 7.5). After
hybrid-ization, filterswerewashed threetimesatroomtemperature with 2x SSC (lx SSC is0.15M NaCl plus 0.015 M sodium citrate) containing0.1% (wt/vol)sodium dodecyl sulfate and five timesat520C with
0.lx
SSC. Bands were visualized byautoradiographyat -80°C for24h, usingKodak XAR-5film with a Lightning-Plus intensifying screen.
Molecular cloning procedures. A 4-mg sample of high-molecular-weightDNA (31) isolated from clone 23-infected mink cellswasdigested with EcoRI.Thedigestwas fraction-ated by preparative gel electrophoresis (47), and fractions
were tested for the presence of ecotropic MuLV DNA by hybridization to the ecotropic virus-specific plasmid probe
described above. Peak fractions, approximately 10 kb in size,werepooled and packagedinvitro(25) inbacteriophage A Charon 4A (4). Phage were plated on
Escherichia
coli LE392, and recombinants were screened by in situ hybrid-ization (3), usingthe 400-bp ecotropic MuLV-specific insertprobe (see above). A clone 23-K recombinant was isolated
and then subcloned and amplified as described previously (22). To subclone the mutant genome into pBR322, DNA
fromthe A recombinant clone was digested with EcoRI and
ligated with T4 ligase to EcoRI-digested pBR322. The
liga-tion mixture was used totransformE. coli RRI, and
recom-binants were screened by colony hybridization (21), using
the 11-kb insertprobederivedfrom 623P.Aclone designated pRTM was isolated that contained the entire clone 23
genome.including two long terminal repeat units (see Fig. 2)
andflankingcellularsequences of0.8and 0.3kbatthe 5'and 3'termini, respectively, of the viral DNA insert. To increase the yields of plasmid DNA. pRTM was transferred to E. coli HB1I1, which was used as the host for all subsequent subcloning experiments.
Plasmids. pSV2 Neo (59) was agenerous gift from Gray
Crouse. pKC7 (49) was kindly provided by Bernard Weiss. Amt8-1 (8),generously supplied by SisirK. Chattopadhyay, contains thewild-type amphotropic MuLVgenomeinserted at the EcoRI site of pBR322. It has aSalI site at the same
position as ecotropic MuLV and one long terminal repeat, but is permuted in the env gene. pWB-5 (5) was agenerous
giftfrom Larry Boone and containsthe entire wild-type
B-tropic MuLV genome inserted in permuted form at the
HindlIl site of pBR325. A subclone of pWB-5 was isolated that has the 1.6-kbHindIII-XhofragmentofMuLVinserted into the HinidlIl and X/Iol sites ofpKC7. The subclones of
pRTM were asfollows. pRTM-1 and pRTM-2containthe 5' Eco-Sal and 3' Sal-Eco fragments of pRTM, respectively, inserted attheEcoRI andSa/I sites ofpBR322. pRTM-3 and pRTM-4 contain the 5' Eco-X/ho and 3' X/ho-Eco fragments ofpRTM,respectively, insertedatthe EcoRl and Xliol sites of pKC7. 623P is a recombinant plasmid subclone of the
bacteriophage X623 clone (34), which contains the entire
AKR MuLV genome. The HindIll and EcoRI sites in 623P
are located in the 5' and 3' flanking cellular sequences,
respectively (34, 48), and were used as cloning sites for insertion into pBR322 (B. I. Gerwin, unpublished data). Subclones of 623P were as follows: 623P-2, generously provided by Douglas Lowy, has the 3' Sal-Eco fragment of 623P inserted at the SallI and EcoRI sites of pBR322 (44); 623P-3 contains the 5' HinidIII-Xliofragmentof 623P insert-ed at the HinzdlIl and X/iol sites of pKC7; and 623P-4 contains the 3' X/ho-Eco fragments of623P inserted at the EcoRl andXhoI sites ofpKC7.
Isolation of plasmid DNA. Plasmids were grown in LB broth containing 25 pLg of ampicillin per ml. Plasmid DNA
wasisolated essentially as described by Boone et al. (5). with the exception of the sodium dodecyl sulfate step, and was
then treated with ribonuclease (50 pLg/ml) and banded two
times incesium chloride-ethidium bromidegradients.
Cotransfection ofpRTM with pSV2 Neo. The pRTM and
pSV2 Neo (59) plasmids were digestedwith EcoRI. Then 3
pLgof eachdigest wasco-precipitated(eitherwith orwithout 20
p.g
of calf thymus DNA) with calcium phosphate, and theprecipitates were applied to NIH-3T3 cells as described
previously (9, 19). Two days later, the cells were cloned in microtest plates in medium containing 400 p.g of the drug G418 per ml. Clones resistant to G418 were picked and
screenedforthe presence ofadefective ecotropic MuLV as
described above, approximately 20% ofthe G418-resistant clones werefound to contain the MuLV.
Transfection of NIH-3T3 cells withplasmidsubclones.
NIH-3T3 cellswereseededat adensity of 8 x
105
cells per60-mmpetri dish. One day later, 0.5
pLg
of each plasmid DNA,previouslydigested witheitherSallI orX/iol. wasappliedto
on November 10, 2019 by guest
http://jvi.asm.org/
472 LEVIN ET AL.
the cells as described previously (9, 19), in the absence of
added carrier DNA. The next day, transfected cells were
trypsinized and dividedbetweentwoT75flasks. Since some
ofthe transfections were expected to yield virions with the
hostrangeofB-tropicMuLV, it was necessary toprovide a
permissive host for virus amplification. Thus, 2 days after
transfection, the transfected NIH-3T3 cells were treated
with mitomycinC (31) to preventfurther cell division. Each flask then received 5 x 105 3T3FL cells (17), which support
replication ofboth N-and B-tropic MuLV. When these cells
reached confluency, they were divided among several T75
flasks. Ten days after transfection, the supernatant fluids
werecollected and assayed forreversetranscriptaseactivity (32) and infectivity (see the legends toFig. 3 and4).
Sequencing procedures. Fragments to be sequenced were
end labeled with [a-32P]TTP, using the Klenowfragment of
DNA polymerase (35), denatured by heating in dimethyl
sulfoxide, and electrophoresed in 5% polyacrylamide gels
under strand-separating conditions (37). Separated strands were excised, recovered from polyacrylamide, and subject-ed to sequencing by base-specific chemical cleavages as
detailed in reference 37. The guanine-plus-adenine reaction
was carried out as described by Mark and Rapp(36). RESULTS
Molecular cloningof the clone 23mutantgenomefrom mink
cells.Earlier workonthe clone23viralmutantdemonstrated
that it hasa defect in thepolgene(16). To characterize the
defect in clone 23 with respect to its genomic structure, it
was necessarytomakeamolecular cloneof the viralmutant. Since the presence of other murine viral genomes could
interfere with thescreening procedures usedin cloning, the clone23genome wastransferred biologically from mouseto minkcells, asdescribed above. Mink cellclones containing
defective ecotropic MuLV were identified by the
comple-mentation plaque assay (51). One of these clones was
selected for further study.
Analysis of the mink cell genomic DNA by restriction endonuclease digestion and Southern blotting (58) (Fig. 1)
was carried out by using a mouse ecotropic virus-specific
probe (7). Digestion with PstI yielded an 8.2-kb fragment thatcomigrated withaDNA band producedby PstI cleavage
ofgenomicDNAfrom cellsinfected with the closely related wild-type AKR MuLV (Fig. 1). Since PstI is known to
remove only 0.6 kb from the longterminal repeat regions of
the complete 8.8-kb AKR viral genome (48), the results
suggested thatafull-length copyofthe mutantviralgenome is present in mink cells. Digestion of mink cell DNA with EcoRI, which doesnot cut mostecotropicMuLV DNAs (7), gave onefragment approximately 10kbin size, presumably
representing the viral genome plus flanking cellular
se-quences.Thisresult also showed that the infected minkcells
containonly one copy ofecotropic MuLV.
Since restriction with EcoRI leaves the clone 23 genome
intact(Fig. 1),DNA ofinfected mink cellswasdigested with
this enzymein preparation for molecularcloning. The
clon-ingprocedures wereperformedasdetailed above. A recom-binantcloneofbacteriophage A Charon4A (4)containingthe entire mutant viral genome and 1.1 kb offlanking cellular
sequences wasisolated,andthe insert was subclonedatthe EcoRI site ofthe plasmid pBR322.
To test the biological activity of the plasmid clone
(pRTM), NIH-3T3cells werecotransfectedwith pRTM and
anotherplasmid, pSV2 Neo (59), which carriesthe genefor
neomycinresistance. Cell cloneswerefirstselected for their
abilitytogrowinthe presenceof theneomycinanalogG418,
Eco
RI
Pst
I
CL
CL
23 AKR 23
-
23.6
-
9.4
-6.7
FIG. 1. Southern blot analysisofgenomic DNA digested with E(oRI andPsul. High-molecular-weight cellularDNA was isolated from clone 23 virus-infected mink cells (CL 23) and from AKR MuLV-infected SC-1 cells (AKR) as described previously (31). DNAsamples were digested withEcoRI (clone 23) and Pstl(AKR and clone23), subjectedtoelectrophoresisin a0.6%agarosegel (25
V. 24h). transferred to a nitrocellulose filter(58).andhybridizedto a 32P-labeled plasmid probe containing a 400-bp fragment specific forecotropic MuLV (7). as described in the text. The molecular weight markers are Hindlll fragments ofbacteriophage X and are given in kb.
and thenwerescreened for the presence of defective
ecotro-pic MuLV by thecomplementation plaque assay (51). Two cell clones, referred to as C5 and D3, gave positive results since supernatant fluids from these clones formed comple-mentation plaques only after superinfection of the cells with Moloney mink cell focus-forming MuLV, an XC-negative helper virus (Table 1). Polyacrylamide gel analysis showed that bothof these clones contained the gag precursorprotein
Pr65c""I
(26) and two proteins with molecular weights of120K and 110K, respectively; no
Pr180g'''
(27) was de-tected. Control NIH-3T3 cells transfected with pSV2 Neo (59) alone did not contain any of the virus-specific proteins found in either mutant or wild-type MuLV-infected cells (data not shown). These results demonstrated that cellsTABLE 1. Presence ofa replication-defective ecotropic MuLV in cells transfected withpRTM
Cell clone" Superinfectingvirus CPFU/ml'
C5 None 0-1
MoloneyMCF" 2 x 10i
D3 None <1
MoloneyMCF 8 x 104
NIH-3T3clones were isolated aftercotransfectionwithpRTMandpSV2 Neo(59)andselectionin mediumcontainingthedrugG418.
MCF,Mink cellfocus-forming.
CPFU,Complementation plaque-formingunits. The titerwasmeasured 9 daysaftersuperinfection.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.322.560.624.687.2]- Y
Ll
E U)
mU CUnX/)i-mco
m
~~~m
m 0- c c nmx _
11 10.
E
cn
Pol
, , 1 , , . .
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 8.8
KILOBASE
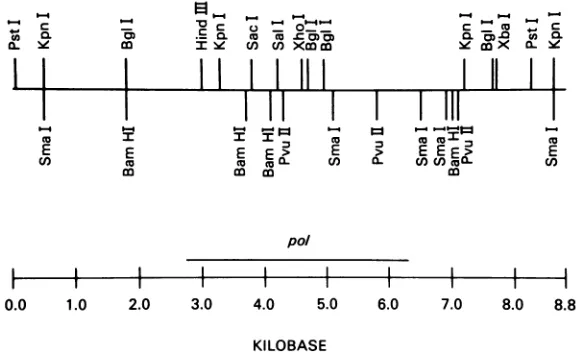
FIG. 2. Restrictionmapofclone 23 genome. The restriction mapoftheclone23polmutantgenomewasderivedby analysis of the plasmid clone pRTM and the Hirt supernatant DNAfraction(24) prepared from mink cells infected with clone23(baboonvirus)pseudotypevirions. The plasmid was mapped by visualization ofrestriction fragments with ethidium bromide; the Hirt supernatant DNA was mapped by Southernblotting (58)andhybridizationwith the 623P AKR MuLV genomeasprobe. This probe does not react with baboon virus under the conditions used here(data not shown). Distances between sites are givenin kb. The boundaries of thepolgene aretakenfromthe nucleotide sequenceofAKRMuLV (23).
transfected with pRTM have the same properties as the original clone 23 mouse cells(16; Table 1).
Thegenetic structure of the clone 23 genome wasinitially probed by subjecting the plasmid clone to restriction endo-nuclease analysis. Theresulting restriction map is shown in Fig. 2. This map is consistent with the published map of
AKR MuLV (48), differing only by the presence of aPillII site in the enm' gene at map position 7.1 kb. The plasmid pRTM and the clone 23 genome in mink cells also share the
same restriction sites (Fig. 2). This was demonstrated by superinfectingthe mink cells with baboonendogenousvirus,
cocultivatingthese cells with uninfected minkcells,and then
preparingHirt supernatant DNA(24) foranalysis by restric-tion endonucleasedigestion and Southernblotting(58) (data
Plasmids Utilized
Cloned Viral DNA Inserts
not shown). The restriction map of pRTM was also
com-pared with that of a plasmid clone, pWB-5, containing the wild-type B-tropic MuLVgenomein permuted form (5). No differences between the mutantandwild-typegenomeswere
revealed in theseexperiments (data not shown).
Localization of thedefectin the clone23 genome. Thefact thatthe mutant and wild-typegenomes had identical restric-tion maps made it possible to perform in vivo ligation
experimentstolocalize the defectinclone 23. Thisapproach involved transfection of NIH-3T3 cells with combinations of
plasmid DNAs containing either mutant or wild-type viral
DNA inserts. Subclones were constructed that extended from the cellularflankingsequenceat the 5' or3' endof the genome to the Sail site inpol(mapposition4.2 kb r48; Fig.
Polymerase Activity Infectivity
[3H]dTMP incorporated PFU/ml (pmol)
pRTM-1 + 623P-2 Dz zz 70.9 6x 105
Amt8-1 + pRTM-2 L+tZZZZZZZZ4o <0.1 <2x 100
pRTM-1 + pRTM-2
L=='+L'i[
0.1 <2x 100Amt8-1 1 <0.1 <2x 10°
623P-2 + 0 0.2 <2x 100
Amt8-1 + 623P-2 7.0 1.4x 105
4= SalISite [1= LTR WildType r7777,-= Mutant
FIG. 3. Reconstitution of polymerase activity and infectivity by transfection withplasmids containing mutant or wild-type viral DNA cleavedatthe Sall site. Allplasmids were digested with Sall before transfection. Procedures for transfection ofNIH-3T3cells are given in the
text. Polymerase activity was assayed with poly(rA)
oligo(dT)12-1s
as described previously (32); the data are given for 10-,ul portions of concentratedsupernatantfluid(totalvolume,100,ul). Infectivity wasdeterminedby the XC assay (54). For simplicity, only the biologically relevant DNA,i.e.,the viral DNAinsertincluding the long terminal repeat segment, is shown in the schematic representation of column 2. The plasmidAmt8-1 (8) isdepicted schematically as a 5' subclone since only the 5' portion of the viral DNA is unpermuted and therefore biologically activeafter Sallcleavage. Foramore completedescription of the plasmids used, see the text.on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.166.455.76.253.2] [image:4.612.157.470.496.664.2]474 LEVIN ET AL.
PlasmidsUtilized
Cloned Viral
DNA Inserts Polymerase Activity
[3H]dTMPincorporated FlU/ml (pmol)
623P-3 + pRTM-4 O +ZZz O 342.1 2x 105
pRTM-3 + 623P-4
aZ=j+Lzzt=
0.3 < 2 x100pRTM-3 + pRTM-4 DlzzzzItz 0.1 <2x 10°
623P-3 <0.1 <2x1&0
623P-4
t-
<0.1 <2x100623P-3 + 623P-4 °
+L
350.8 2x 106+= XhoI Site O= LTR
Wild Type V11111M, Mutant
polymerase activity aind infectivity by transfection with plasmids containing mutant or wild-type viral
cleavedatthe Xliolsite.Theexperimentalproceduures werethesame asthosedescribed inthelegendtoFig. 3.exceptthat theplasmidswere
digested with Xliol before transfection and infectivity wasdeterminedby the S'L focus assay (2).
21).Plasmidsweredigested with Soll, andpairwise
combina-tions of 5'and 3'subclones werethencotransfected, relying
oncellular ligation (39, 45;A. Rein andR. J. Mural,
unpub-lished data) to generate acomplete genome. Production of
nondefective MuLV from combinations in which neither
subclone contained a defect was monitored by the
appear-ance ofreverse transcriptase activity and infectious virions
intheculture fluidsapproximately10days aftertransfection. Figure 3 shows the results of thisexperiment. Details on the plasmids used (column 1) are given above and in the
legend to Fig. 3. The biologically relevant pieces of the
cloned viral DNA insertsareshown schematicallyincolumn
2.AdditionofplasmidDNAscontainingthe5'portion ofthe mutant genome and the 3' portion of the wild-type (line 1)
gave rise to infectious virus with reverse transcriptase activity. In contrast, combinations involving the 3' portion
of the mutantgenome andthe 5' portion of either the
wild-type (line 2) or mutant (line 3) were inactive. Control
experiments showed that plasmids containing the 5' and 3' portions of the wild type were not active (lines 4 and 5.
respectively) unless both DNAs were transfected together
(line 6). These results demonstrated that the clone 23
genome has nodefectupstream ofthe Sall site.
A similarexperiment (Fig. 4)wasundertaken with respect to the XhoI site (map position 4.6 kb [48; Fig. 2]). In this case, the plasmid containingthe3' Xiho-Eco fragmentofthe mutant (line 1) was biologically active, whereas the 5'
mutantviral DNAinsert spanningthe Sail site wasnot(line
2). Negative controls included transfection of 5' and 3' portionsofthe mutantgenome (line 3)oreitherofthe
wild-typesubclones alone(lines4and 5). AsobservedintheSall
experiment (Fig. 3), cotransfection of both wild-type sub-clones generated infectious virus with polymerase activity
(line 6). These results demonstrated that the clone 23
genome has no defect downstream from the XIloI site. Takentogether,theresults inFig.3and 4showthatclone23
is defective only in the 400-bp region between the Still and XhoI sites.
Sequence analysis of the Sal-Xho region ofpol gene. The
Maxam-Gilbert method (37) was used to determine the
sequenceofthe Sili-XIoa regions of themutantand wild-type pol genes. Figure 5 shows a portion oftwo 8%
polyacryl-amide-urea gels illustrating the critical difference between
the mutant andwild-typegenomes. Thewild-type sequence had a run offive C residues, whereas the parallel mutant
sequence containedanadditional C (Fig.5).Thepresenceof
an extra C in the mutant sequence was confirmed by
sequence analysis of an M13 clone (38) containing the mutant Sal-Xhlo fragment (data not shown).
The entire nucleotide sequence ofthe Sal-Xho region is given in Fig. 6. The sequences of the mutant and wild-type coding strands were compared by using the NUCALN programof Wilburand Lipman (66); nucleotide 1 is the last
base ofthe Still recognition site. The data show that
inser-tionof the additional C residue inthe mutantsequence after
nucleotide 231 bringsaTGAterminationcodonintophaseat position 233. Additional TGA termination codons are also
WILDTYPE
G A-G A>C T C C A
A
G
C
A
ci
MUTANT
(1 A-G A>C T>C C
.:,..M,-. _*
iEi__ ~~~~dmd
~%w4
__Q
I
_.-;.
NW
I P
A A c
A
---T
-c'
-<Cl
N-\C
Cz
FIG. 5. Maxam-Gilbert sequencegels. showing aportionofthe wild-typeandclone23mutantsequenceintheSilI-Xlio regionof the
polgene.To obtain thewild-type fragment usedforsequencing,the
Hinidlll-Xhlosubclone (seethetext)ofpWB-5(5) wasdigestedwith
SollandXliol;the MuLV-specificSal-Xhofragmentwas recovered froma5%polyacrylamide gelasdescribedpreviously (37).The
Sal-Xlio fragment of the mutant was isolated by digestion ofpRTM-2
(see the text) with Still andXlhol, followed by preparative electro-phoresis in a 1.0% agarose gel and recovery of the DNA by adsorptiontoglassbeads(62). Sequenceanalysiswasperformedas
described in thetextandin reference37. The nucleotidesequences
aredisplayedon8% polyacrylamide-urea gels.
Infectivity
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.153.464.78.251.2] [image:5.612.318.555.485.617.2]20 40
MUT CGAGAAGCAGGGCTACGCCAAAGGCGTCCTAACGCAAAAACTGGGACCTTGGCGTCGGCC
WT CGAGAAGCAGGGCTACGCCAAAUGCGTCCTAACGCAAAAACTGGGACCTTGGCGTCGGCC
20 40
80 100
CGTGGCCTACCTGTCCAAAAAGCTAGACCCAGTGGCAGCTGGGTGGCCCCCTTGTCTACG
CGTGGCCTACCTGTCCAAAAAGCTAGACCCAGTGGCAGCTGGGTGGCCCCCTTGTCTACG
80 100
140 160
GATGGTAGCAGCCATTGCCGTTCTGACAAAAGATGCAGGCAAGCTAACTATGGGACAGCC
GATGGTAGCAGCCATTGCCGTTCTGACAAAAGATGCAGGCAAGCTAACTATGGGACAGCC
140 160
200 Val 220 Pro ProEnd
GCTAGTCATCCTGGCCCCCCATGCAGTGGAGGCACTGGTCAAGCAACCCCCCTGACCGCT
GCTAGTCATCCTGGCCCCCCATGCAGTAGAGGCACTGGTCAAGCAACCCCC-TGACCGCT
200 Val 220 Pro Pro Asp
260 280 End
GGCTATCCAACGCCCGCATGACCCACTACCAGGCAATGCTCCTAGACACTGACCGAGTTC
GGCTATCCAACGCCCGCATGACCCACTACCAGGCAATGCTCCTAGACACTGACCGAGTTC
260 280
320 340
AGTTCGGACCAGTGGTGGCCCTCAATCCTGCCACCTTACTCCCTCTCCCGGAAGAAGGAG AGTTCGGACCAGTGGTGGCCCTCAATCCTGCCACCTTACTCCCTCTCCCGGAAGAAGGAG
320 340
End 370 CCCCCOATGATTGCC
CCCCCCATGATTGCC 370
FIG. 6. Nucleotide sequence of the entire
Sal-Xho
region of the mutant and wild-type pol genes. Sequencing was by the method of Maxam andGilbert(37). The sequence of the coding strands of the clone 23 mutant and the wild-type Sal-Xho fragments was compared by using the NUCALN programofWilbur and Lipman (66). Nucleotide 1 is the last base of theSallrecognition site. MUT, mutant; WT,wild type.generated at positions 290 and 368 as a result of this
insertion. Thus, themutationinclone23 leadstopremature
termination of translation.
Theonly other
nucleotide
difference between themutant and wild-type sequence occurs atposition
208. This basechange is silent since GTG (mutant)and GTA(wild type)are both valine codons. Interestingly, N- and
B-tropic
MuLV share the same nucleotide sequence in the Sal-Xhoregion,except at position 88, where AKR MuLV has a T residue
(23),butboth themutantandwild-typeB-tropicviruses have
aC.Thisdifferencedoesnotaffect theamino acid sequence
ofthe enzyme,however, since GATand GAC both code for thesame amino acid (asparticacid).
DISCUSSION
In the present study, the molecular defect in a naturally arising MuLV pol mutant, clone 23 (16), has been precisely
defined in terms of the genetic structure of the mutant
genome.Analysis ofamolecular cloneoftheentire clone 23 genome demonstrated that the mutant is defective in the approximately 400-bp sequence between the SalI andXhoI sites (Fig. 2) of the pol gene. No defects were present elsewhere in the viral genome (Fig. 3 and 4).
Sequence analysis of the Sal-Xho regions of the mutant and wild-type genomes revealed that the mutant has one
additional C located 231 bases downstream from the last
base of the Sall recognition site (Fig. 5 and 6). The only
otherdifference between themutantand wildtype occurs at
position 208,butthis changedoes notaffect the amino acid sequence(Fig. 6).The consequences of the 1-baseinsertion,
however,arequite striking. Three TGAtermination codons
are brought into phase, the first immediately after the
insertion (position 233) and the other two at positions 290 and368,respectively (Fig. 6).Thus,themutationinclone 23
produces a shift in readingframe that results in premature
termination of translation.
Two lines of evidence establish that termination occurs
within the structural gene for reverse transcriptase. First, the mutantenzyme has beenpurifiedand shown to havean
abnormally low sedimentation rate in glycerol gradients,
consistent with a molecular weight of 47K (16). Second, recent experiments using a competition immunoassay
indi-catethat thespecific activity of the mutant enzyme molecule isatleast10-fold lower than that of the wild type (Messeret
al., in preparation).
Correlation of the nucleotide position of the clone 23
defect with the mutant phenotype allowed us to propose a
scheme for the genetic organization of the MuLV pol gene
(Fig. 7). This scheme involves calculations that are based uponthegenomicsequenceof the very closely relatedAKR
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.146.481.70.428.2]476 LEVIN ET AL.
Reverse Transcriptase RNase H
70-80K
l
Endonuclease
40-50K
4500- 4776
Map Position 1kb) 2.73 3.09 4.98- 5.26 6.32
FIG. 7. Organization of the MuLV polgene.Theproposedmapforthe MuLVpolgene isbasedonconsiderations discussed in thetext. The molecular weights ofpol-associated proteins are given in kilodaltons. The numbering system of Herr for the AKR MuLV genomic
sequence(23)wasusedtodesignatenucleotide position.Mappositionwascalculated by adding 482 bp. thesizeof the AKR U3region(23). to
eachof thenucleotide positions aindisgiveninkb. Theuncertaintyin theCterminusofreversetranscriptase isindicatedonthe line drawing
by thebrackets around the predicted boundaries of theC-terminal region. The polopenreadingframeisterminatedbyanochre termination
codonat nucleotide positions 5841 to5843 (23).
MuLV (23), anduses the numbering system of Herr (23). Termination at the first TGA brought into frame by the mutation locates the C terminus ofthe truncated clone 23
reverse transcriptase molecule at nucleotide 3956. The
pre-cise locationof the Cterminus, coupled with thepreviously determined (16) molecular weight (47K) of the mutant en-zyme, places the N terminus at approximately nucleotide 2691 and predicts a mutant protein of 421 amino acid residues (23). However, since the 5' boundary of the pol gene is at nucleotide 2253 (23), a protein encoded by nucleotides 2253 to 3956 would have a molecular weight of ca.63K(23). Althoughthemolecular weightestimate of 47K was made under nondenaturing conditions (16), it seems
unlikely that it should underestimate the true molecular weight by as much as 25%. We therefore propose that reversetranscriptase isnotthe N-terminal proteinof the pol
gene product. This conclusion is consistent with recent
findings of Copeland, Gerard. and Oroszlan (T. D. Cope-land, G.Gerard,and S. Oroszlan. personal communication). who have used amino acid sequencing to localize the N
terminusof Moloney MuLV reversetranscriptaseto nucleo-tide2598 (accordingto the numberingsystemofShinnicket al. [571). Inspection of the nucleotide sequences of the
Moloney (57) and AKR (23) pol genes suggests that the
reverse transcriptase molecules of these viruses share the
same N termini and places the N terminus of the AKR
enzyme at nucleotide 2613(23).
Aproteolytic activity associatedwith MuLV particleshas been reported (67). Previous characterization of clone 23
virions has demonstrated that all of the gaig structural proteins are present in the correct proportions (16),
indicat-ing that the gag precursor is cleaved normally. As shown
above, clone 23 terminates gaig-pol synthesis within the reversetranscriptase coding region. Thus, if thecleavageof
the gag precursor is catalyzed by a virus-coded protease, thisenzymecannotbeencodedtothe 3' sideof the mutation in theclone 23genome, butmustbeencodedtothe 5' side of
reverse transcriptase. Localization of the N terminus of reverse transcriptase to nucleotide 2613 leaves 360 bp of
geneticinformationatthe 5' endofpolwhichcould encodea
protein of 13K daltons (23). Such a protein would
corre-spondinsizetotheavianretrovirus-codedprotease p15(12, 56) and would also show sequencehomologyand similarity
in predicted secondary structure to avianp15 (S. Oroszlan,
personal communication). We therefore suggestthat the N-terminal portion ofthepolgeneproduct encoded by
nucleo-tides2253to2612 isa13K proteaseinvolved inthe cleavage
ofgag proteins. The existence ofan MuLV-codedprotease upstreamof the reversetranscriptase codingregion has also
been considered by Oroszlan and co-workers (S. Oroszlan,
personal communication).
Furtherinformation about theorganization of the polgene can be derived by locating the C-terminus of wild-type reverse transcriptase. If the wild-type reverse transcriptase
molecule is encoded by a sequence beginning at nucleotide position 2613and has amolecular weight of 70to 80K(60). then its C terminus would be encoded approximately at
nucleotides 4500 to 4776 (23). However, the open reading
frame of the polgene of AKR MuLV extendsto nucleotide position 5840(23), givingthisgene additional coding
capaci-ty ofapproximately 40to 50K daltons. It is likely that this regionatthe 3'end of the polgeneencodes the endonuclease
described byKopchicketal.(28) and Nissen-Meyerand Nes
(43).
One striking feature of the scheme proposed in Fig. 7 is
that itplacesthe MuLVpol-associatedproteins inthe same
relative positionson thegenetic mapas inthe avian retrovi-ruses, despite the fact that the polyprotein precursors are
cleaved quite differently in the two virus groups. Thus, in
both cases, the protein encoded just downstream from the
firstfourgagproteins is thegagcleavageenzyme;however, inthe avianviruses,this protein(p15)(13, 63, 64)is thefifth
gag protein (12, 56), whereas in MuLV it is the first pol protein. This protease is followed by reverse transcriptase.
Downstream from reverse transcriptase is the virus-coded
endonuclease;this enzymeis theC-terminal portion of the
subunit ofavian reversetranscriptase (10, 14),but
apparent-ly is cleaved away from reverse transcriptase during
proc-essingof the MuLVpolgene product.
Thecurrentdata, in addition togeneratingaproposal for
the structure ofthe pol gene, also suggest some inferences
concerning structure-function relationships in the wild-type
enzyme molecule. As mentioned above, the MuLV reverse
transcriptasemolecule isasinglepolypeptidewhich has both RNase H and polymerase activities (30, 42, 61). These activitiesarethoughttohavedifferentactive sites(6, 11, 15, 18, 29, 40, 41, 60), but the location ofthe sites has notbeen
identified. Thepresent workon clone 23 indicates that both
ofthese sites, as well as the antigenic determinants of the enzyme recognized by heterologous goat antisera (16), lie
within the N-terminal portion of the polymerase protein.
This conclusion is in accord with a recent suggestion by
Crouch and Dirksen that the active site for the avian retrovirus RNase H is at or near the N terminus of the a-subunit of reverse transcriptase (11). It is intriguing that
despitethe lossofapproximatelyonethird of the moleculeat the C-terminal end, the clone 23 enzyme can stillcarry out
some of the reactions required for viral DNA synthesis,
Protease
13K
5' I
Nucleotide Position 2253 2613
-
3'
5840 J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.79.542.79.157.2]including synthesis
of minus-strong stop DNA (60) andpartial elongationof minus-strand DNA after thejumptothe 3' end of the
template
(Messer et al., in preparation). However, theenzymecannotmakeanyplus-strandDNAorfull-length
minus-strand DNA and terminatesDNAsynthe-sis near a
major
wild-type pause site (Messer et al.. inpreparation).
Futureexperiments
will bedesigned to assess the role of the C-terminalportion
of the enzyme in these reactions,The fact that clone 23 has normal amounts of tRNAPI' in
viral 4S RNA
(33)
is also of interest. Othernonconditionalpol mutants in both the avian and murine systems have
reduced levelsoftRNATrP(46,
55)
andtRNAPr,(33).
respec-tively,
and based on these observations, it was concludedthat reverse
transcriptase
is themajor
determinant in theselective
encapsidation
ofprimer
tRNA that occurs duringvirus
assembly.
The clone 23 data suggest thattRNA-polymerase
interactions that form the basis for thisselection involveonly
the N-terminalportion
of the enzyme.In summary, correlation of the clone 23
phenotype
withthe
precise
localization of the C terminus of the mutant reversetranscriptase
has allowedustoproposeamapof theMuLVpolgene and to
begin analysis
ofstructure-functionrelationships
within the reverse transcriptase molecule.ACKNOWLEDGMENTS
It is apleasure to thankMichael Seddon, Melody
McClure.
andDorothy Cavanaugh for outstanding technical assistance. We are indebted to Aya Leder for encouragement and adviceon cloning
techniquesduringanearly phaseofthiswork, MarianNauforhelp withfractionationandpackagingof minkcellDNA,AlanSchultzfor adviceonprotein gels,and JohnOwens forassistance with
comput-eranalysis. We arealsogratefulto DolfHatfield and CharlesVan Beveren for valuable
discussion.
Larry Boone, WinshipHerr.
andStephen
Oroszlanforcommunicatingresults beforepublication,and Robert Crouch and Stephen Oroszlan for critical readings of themanuscript. Catherine Miceli provided expert assistance in the
preparation ofthe manuscript.
ADDENDUM IN PROOF
A model for the structure of the
pol
gene with some similaritiestotheoneproposed
herewaspreviously
suggest-edby
Kopchick
et al.(J.
J.Kopchick,
W. L.Karshin,
and R. B.Arlinghaus,
J. Virol.30:610-623, 1979).
LITERATURE CITED
1. Bassin,R.H., B.I.Gerwin,J.G.Levin,G.Duran-Troise,B. M.
Benjers, and A. Rein. 1980. Macromolecular requirements for
abrogation ofFt-] restriction by murine leukemia viruses. J. Virol. 35:287-297.
2. Bassin,R. H., N. Tuttle,and P.J. Fischinger. 1971. Rapidcell culture assay technique for murine leukaemiaviruses. Nature (London)229:564-566.
3. Benton, W. D.,and R. W. Davis. 1977. ScreeningXgt recombi-nant clonesby hybridization to single plaques in situ. Science 196:180-182.
4. Blattner, F. R., B. G. Williams, A. E. Blechl, K. Denniston-Thompson, H. E. Faber,L. A. Furlong,D. J.Grunwald, D.0.
Kiefer, D. D. Moore, J. W. Schumm, E. L. Sheldon, and 0. Smithies. 1977. Charon phages: safer derivatives of
bacterio-phagelambda for DNA cloning. Science 196:161-169. 5. Boone, L. R., F. E. Myer,D. M. Yang,C.-Y. Ou, C. K. Koh,
L. E.Roberson,R. W.Tennant,and W. K.Yang.1983. Reversal of Ft-] host rangebyinvitrorestrictionendonucleasefragment
exchange between molecular clones of N-tropic and B-tropic murine leukemiavirus genomes. J. Virol. 48:110-119.
6. Brewer, L. C., and R.D. Wells. 1974. Mechanisticindependence of avian myeloblastosis virus DNApolymerase and
ribonucle-ase H.J. Virol. 14:1494-1502.
7. Chattopadhyay, S. K., M. R. Lander, E. Rands, and D. R. Lowy. 1980. Structure ofendogenous murine leukemia virus DNAin
mouse genomes. Proc. Natl. Acad.Sci. U.S.A. 77:5774-5778. 8. Chattopadhyay, S. K., A. I. Oliff, D. L. Linemever. NI. R.
Lander, and D. R. Lowv. 1981. Genomes of murine leukemia viruses isolated from wild mice. J. Virol. 39:777-791.
9. Cohen, M., A. Rein, R. M. Stephens, C. O'Connell, R. V. Gilden, M. Shure, M. 0. Nicolson, R. M. McAllister, and N.
Davidson. 1981. Baboon endogenousvirus genome: molecular cloning and structural characterization of nondefective viral genomes from DNA of a baboon cell strain. Proc. Natl. Acad. Sci. U.S.A. 78:5207-5211.
10. Copeland, T. D., D. P. Grandgenett, and S. Oroszlan. 1980. Aminoacid sequence analysis of reversetranscriptasesubunits from avian myeloblastosis virus. J. Virol. 36:115-119. 11. Crouch, R. J., and M. L. Dirksen. 1982. Ribonucleases H. p.
211-241. In S. Linn and R. Roberts (ed.). Nucleases. Cold Spring Harbor Laboratory. Cold Spring Harbor. N.Y. 12. Dickson, C., R. Eisenman, H. Fan, E. Hunter, and N. Teich.
1982. Protein biosynthesis and assembly. p. 513-648. In R. Weiss. N. Teich. H. Varmus. and J. Coffin (ed.). Molecular biology oftumor viruses. RNA tumor viruses. 2nd ed. Cold Spring HarborLaboratory. Cold Spring Harbor, N.Y. 13. Dittmar,K.J., and K. Moelling.1978.Biochemical propertiesof
p15-associated protease in an avian RNA tumor virus. J. Virol. 28:106-118.
14. Eisenman,R. N., W. S. Mason, and M. Linial. 1980. Synthesis and processing ofpolymerase proteins ofwild-type and mutant avianretroviruses. J. Virol. 36:62-78.
15. Gerard, G. F. 1978. MultipleRNase H activities in mammalian typeC retraviruslysates.J. Virol. 26:16-28.
16. Gerwin,B.I.,A.Rein, J. G. Levin, R. H. Bassin, B. M. Benjers, S. V. S.Kashmiri, D. Hopkins, and B. J. O'Neill. 1979. Mutant ofB-tropic murine leukemia virus synthesizing an altered poly-merase molecule. J. Virol. 31:741-751.
17. Gisselbrecht,S.,R. H. Bassin,B. I.Gerwin, and A. Rein. 1974. Dual susceptibility of a 3T3 mouse cell line to infection by N-andB-tropicmurine leukemia virus:apparent lack of expression of the Fv-1gene. Int. J. Cancer 14:106-113.
18. Gorecki, M., and A. Panet. 1978. Discrimination of DNA polymeraseand RNase H activities in reversetranscriptase of avianmyeloblastosisvirus. Biochemistry 17:2438-2442. 19. Graham, F. L., andA.J.vander Eb. 1973.Anewtechnique for
the assay ofinfectivityof human adenovirus 5 DNA. Virology 52:456-467.
20. Grandgenett,D.P.,A.C.Vora,and R. D.Schiff.1978. A 32.000-dalton nucleicacid-binding proteinfrom avian retrovirus cores possesses DNA endonuclease activity. Virology 89:119-132. 21. Grunstein,M., and D. S. Hogness. 1975.Colony hybridization:a
method fortheisolation of clonedDNAsthat containaspecific gene. Proc.Natl. Acad. Sci. U.S.A. 72:3961-3965.
22. Hager, G. L., E. H. Chang, H. W. Chan, C. F. Garon, M. A. Israel, M. A. Martin, E. M. Scolnick, and D. R. Lowy. 1979. Molecularcloningof the Harveysarcoma virus closed circular DNA intermediates: initial structural andbiological character-ization. J. Virol. 31:795-809.
23. Herr, W. 1984. Nucleotide sequence of AKVmurine leukemia virus.J. Virol.49:471-478.
24. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmouse cellcultures. J. Mol. Biol. 26:365-369.
25. Hohn,B. 1979. In vitropackagingof lambda and cosmidDNA.
MethodsEnzymol. 68:299-309.
26. Jamjoom,G., W.L. Karshin,R. B.Naso, L.J. Arcement, and
R. B. Arlinghaus. 1975. Proteins of Rauscher leukemia virus:
resolution of a 70,000-dalton, nonglycosylated polypeptide containing p30 peptide sequences. Virology68:135-145. 27. Jamjoom, G. A., R. B. Naso, and R. B. Arlinghaus. 1977.
Further characterization of intracellular precursorpolyproteins of Rauscher leukemia virus. Virology78:11-34.
28. Kopchick, J. J., J. Harless, B. S. Geisser, R. Killam, R. R.
on November 10, 2019 by guest
http://jvi.asm.org/
478 LEVIN ET AL.
Hewitt, and R. B. Arlinghaus. 1981. Endodeoxyribonuclease activity associated with Rauscher murine leukemia virus. J. Virol. 37:274-283.
29. Lai, M.-H. T., and I. M. Verma. 1978. Reversetranscriptase of RNA tumorviruses.V. Invitroproteolysisof reverse transcrip-tasefrom avianmyeloblastosis virusandisolation ofa polypep-tide manifesting onlyRNase H activity.J. Virol. 25:652-663. 30. Lai,M.-H.T.,I. M. Verma, S. R. Tronick, and S. A. Aaronson.
1978. Mammalian retrovirus-associated RNase H is virus cod-ed.J. Virol.27:823-825.
31. Levin, J. G., B. F. Hughes, J. S. Graeter, A. Rein, E. Rands, and A. B. Mukherjee. 1982. Transfer of murine leukaemia and murinesarcomavirus genetic informationby transfection with isolated metaphasechromosomes. J. Gen. Virol. 62:227-237. 32. Levin, J. G., and M. J. Rosenak. 1976. Synthesis of murine
leukemia virus proteins associated with virions assembled in actinomycin D-treated cells: evidence for persistence of viral messenger RNA. Proc. Natl.Acad. Sci. U.S.A. 73:1154-1158. 33. Levin, J. G., and J. G. Seidman. 1981. Effect ofpolymerase mutations on packaging of primer tRNA""r during murine leukemia virusassembly. J. Virol. 38:403-408.
34. Lowy, D. R., E. Rands, S. K. Chattopadhyay, C. F. Garon, and G. L. Hager. 1980. Molecularcloning of infectious integrated murine leukemia virus DNA from infected mouse cells. Proc. Natl. Acad. Sci. U.S.A. 77:614-618.
35. Maniatis, T., E. F. Fritsch, andJ; Sambrook. 1982. Molecular cloning-a laboratory manual,p. 113-116. ColdSpring Harbor Laboratory,Cold Spring Harbor. N.Y.
36. Mark, G. E., and U. R. Rapp. 1984. Envelope gene sequence of twoinvitro-generated mink cell focus-formingmurineleukemia viruses which contain the entiregp7O sequenceof the endoge-nousnonecotropic parent. J. Virol. 49:530-539.
37. Maxam, A. M., andW. Gilbert. 1980. Sequencingend-labeled DNAwithbase-specific chemical cleavages. Methods Enzymol. 65:499-560.
38. Messing, J., R. Crea, and P. H. Seeburg. 1981. A system for shotgunDNA sequencing. Nucleic AcidsRes.9:309-321. 39. Miller, C. K., and H. M. Tenmin. 1983. High-efficiency ligation
and recombination of DNA fragments by vertebrate cells. Science220:606-609.
40. Modak, M. J. 1976. Pyridoxal 5' phosphate: a selective inhibitorofoncornaviral DNA polymerases. Biochem.Biophys. Res.Commun. 71:180-187.
41. Modak, M. J., and A. Srivastava. 1979. Reverse transcriptase-associatedribonuclease H does notrequire zinc forcatalysis.J. Biol. Chem. 254:4756-4759.
42. Moelling, K. 1974.Characterization of reverse transcriptase and RNase H from Friend-murine leukemia virus. Virology 62:46-59.
43. Nissen-Meyer,J., andI. F. Nes. 1980.Purificationandproperties ofDNA endonuclease associated with Friend leukemia virus. Nucleic Acids Res.8:5043-5055.
44. Ostrowski, M. C., D. Berard, andG. L. Hager. 1981. Specific transcriptional initiation in vitro on murine type C retrovirus promoters. Proc. Natl. Acad. Sci. U.S.A.78:4485-4489. 45. Perucho, M., D. Hanahan, and M. Wigler. 1980. Genetic and
physical linkage ofexogenous sequences intransformed cells. Cell 22:309-317.
46. Peters, G. G., and J. Hu. 1980. Reverse transcriptase as the majordeterminant for selectivepackagingof tRNA'sintoavian
sarcoma virus particles. J. Virol. 36:692-700.
47. Polsky, F., M. H. Edgell, J. G. Seidman, and P. Leder. 1978. High capacitygel preparative electrophoresis forpurificationof fragments ofgenomic DNA. Anal. Biochem. 87:397-410. 48. Rands, E., D.R.Lowy, M. R. Lander, andS. K.Chattopadhyay.
1981. Restriction endonuclease mapping ofecotropic murine
leukemia viral DNAs: size and sequence heterogeneity of the longterminal repeat. Virology 108:445-452.
49. Rao, R. N., and S. G. Rogers. 1979. Plasmid pKCI: a vector containing tenrestriction endonuclease sites suitable for cloning DNAsegments. Gene 7:79-82.
50. Rein, A., E. Athan, B. M. Benjers, R. H. Bassin, B. I.Gerwin, and D. R. Slocum. 1979. Isolation of a replication-defective murine leukaemia virus from cultured AKR leukaemia cells. Nature (London) 282:753-754.
51. Rein, A., and R. H. Bassin. 1978. Replication-defective ecotro-pic murine leukemia viruses: detection and quantitation of infectivity using helper-dependent XC plaque formatioh. J. Virol. 28:656-660.
52. Rein, A., B. M. Benjers, B.I. Gerwin, R. H. Bassin, and D. R. Slocum. 1979. Rescue and transmission of a replication-defec-tive variant ofMoloney murineleukemiavirus. J. Virol. 29:494-500.
53. Rigby, P. W. J., M. Dieckmann, C. Rhodes,and P. Berg. 1977. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase 1. J. Mol. Biol. 113:237-251.
54. Rowe, W. P., W. E.Pugh,and J. W.Hartley.1970.Plaqueassay techniques for murine leukemia viruses. Virology42:1136-1139. 55. Sawyer, R. C., and H. Hanafusa. 1979.Comparison of the small RNAs of polymerase-deficient and polymerase-positive Rous sarcomavirus and anotherspecies of avian retrovirus. J. Virol. 29:863-871.
56. Schwartz, D. E., R. Tizard, and W. Gilbert. 1983. Nucleotide sequenceof Rous sarcoma virus. Cell 32:853-869.
57. Shinnick, T. M., R. A. Lerner, and J. G. Sutcliffe. 1981. Nucleotide sequence ofMoloneymurine leukaemia virus. Na-ture(London) 293:543-548.
58. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separated by gelelectrophoresis. J. Mol. Biol. 98:503-517.
59. Southern, P. J., and P. Berg. 1982.Transformation of
mammali-an cells to antibiotic resistance with a bacterial gene under controlof theSV40 early region promoter. J. Mol. Appl. Genet. 1:327-341.
60. Varmus, H., and R. Swanstrom. 1982. Replication of retrovi-ruses, p. 369-512. In R. Weiss. N. Teich. H. Varmus, and J. Coffin (ed.), Molecular biology oftumor viruses, RNA tumor viruses,2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
61. Verma, 1. M. 1975. Studies on reverse transcriptase of RNA tumor viruses. lII. Properties of purified Moloney murine leukemia virus DNA polymerase and associated RNase H. J. Virol. 15:843-854.
62. Vogelstein, B., and D. Gillespie.1979.Preparativeandanalytical purification of DNA from agarose. Proc. Natl. Acad. Sci. U.S.A. 76:615-619.
63. Vogt, V. M., A. Wight, and R. Eisenman. 1979.Invitrocleavage ofavian retrovirus gagproteinsby viral proteasep15.Virology 98:154-167.
64. vonderHelm,K.1977.Cleavage ofRoussarcomaviral polypep-tide precursorinto internal structuralproteinsin vitro involves viral proteinp15. Proc.Natl. Acad. Sci. U.S.A. 74:911-915.
65. Wahl,G.M.,M.Stern,and G. R. Stark. 1979.Efficiehttransfer oflarge DNAfragments from agarosegelsto diazobenzyloxy-methyl-paper andrapid hybridization byusingdextran sulfate. Proc. Natl. Acad. Sci. U.S.A. 76:3683-3687.
66. Wilbur, W. J., and D. J. Lipman. 1983. Rapid similarity searches of nucleic acid and protein data banks. Proc. Natl. Acad.Sci. U.S.A. 80:726-730.
67. Yoshinaka, Y., and R. B. Luftig. 1977. Properties of a P70 proteolyticfactor of murine leukemia viruses. Cell 12:709-719. J. VIROL.