0022-538X/07/$08.00⫹0 doi:10.1128/JVI.02135-06
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Relative Fitness and Replication Capacity of a Multinucleoside
Analogue-Resistant Clinical Human Immunodeficiency Virus
Type 1 Isolate with a Deletion of Codon 69 in the
Reverse Transcriptase Coding Region
䌤
Cristina Villena,
1Julia G. Prado,
1Maria Carmen Puertas,
1Miguel A
´ ngel Martı´nez,
1Bonaventura Clotet,
1Lidia Ruiz,
1Neil T. Parkin,
2Luis Mene
´ndez-Arias,
3and Javier Martinez-Picado
1,4*
irsiCaixa Foundation, Hospital Germans Trias i Pujol, Universitat Auto`noma de Barcelona, Badalona, Spain1; Monogram Biosciences,
South San Francisco, California2; Centro de Biologı´a Molecular Severo Ochoa, Consejo Superior de
Investigaciones Cientı´ficas—Universidad Auto´noma de Madrid, Madrid, Spain3; and
Institucio´ Catalana de Recerca i Estudis Avanc¸ats, Barcelona, Spain4
Received 29 September 2006/Accepted 14 February 2007
Deletions, insertions, and amino acid substitutions in the3-4 hairpin loop-coding region of human
immuno-deficiency virus type 1 (HIV-1) reverse transcriptase (RT) have been associated with resistance to nucleoside RT inhibitors when appearing in combination with other mutations in the RT-coding region. In this work, we have
measured the in vivo fitness of HIV-1 variants containing a deletion of 3 nucleotides affecting codon 69 (⌬69) of the
viral RT as well as the replication capacity (RC) ex vivo of a series of recombinant HIV-1 variants carrying an RT
bearing the⌬69 deletion or the T69A mutation in a multidrug-resistant (MDR) sequence background, including the
Q151M complex and substitutions M184V, K103N, Y181C, and G190A. Patient-derived viral clones having RTs with
⌬69 together with S163I showed increased RCs under drug pressure. These data were consistent with the viral
population dynamics observed in a long-term-treated HIV-1-infected patient. In the absence of drugs, viral clones
containing T69A replicated more efficiently than those having ⌬69, but only when patient-derived sequences
corresponding to RT residues 248 to 527 were present. These effects could be attributed to a functional interaction between the C-terminal domain of the p66 subunit (RNase H domain) and the DNA polymerase domain of the RT. Finally, recombinant HIV-1 clones bearing RTs with MDR-associated mutations, including deletions at codon 69, showed increased susceptibilities to protease inhibitors in phenotypic assays. These effects correlated with impaired Gag cleavage and could be attributed to delayed maturation and decreased production of active protease in those variants.
Antiretroviral therapy including nucleoside and nonnucleo-side reverse transcriptase (RT) inhibitors, protease (PR) in-hibitors, and entry inhibitors as part of combination drug reg-imens has contributed to a decrease in mortality and morbidity among human immunodeficiency virus type 1 (HIV-1)-infected patients (26, 31). However, drug-resistant HIV-1 variants, which are a major factor contributing to treatment failure (11, 37), often emerge during the course of antiretroviral treatment as a result of impotent regimens, suboptimal adherence, phar-macological hurdles, or ineffectively treated compartments. Long-term HIV chemotherapy with repetitive treatment fail-ure and frequent antiretroviral drug changes is often associ-ated with the accumulation of drug resistance mutations that confer increased phenotypic resistance and lead to the clini-cally undesirable selection of multidrug-resistant (MDR) HIV-1 strains.
Resistance to multiple nucleoside RT inhibitors has been associated with an amino acid substitution at the nucleoside
binding site of the enzyme (e.g., Q151M) and with insertions or deletions in the3-4 hairpin loop in the finger subdomain (amino acid residues 56 to 77) of HIV-1 RT. The acquisition of resistance through the Q151M pathway was first observed in virus isolated from patients receiving zidovudine and di-danosine (40). Viral clones harboring this amino acid substi-tution displayed moderate resistance to zidovudine and zalcit-abine and low-level resistance to other nucleoside analogues (17, 40). Further acquisition of additional mutations, such as A62V, V75I, F77L, and F116Y, rendered viruses that were highly resistant to zidovudine, didanosine, zalcitabine, and stavudine. Another group of MDR viruses are those having insertions or deletions in HIV-1 RT (reviewed in reference 25). Viruses with a dipeptide insertion (usually Ser, Ser-Gly, or Ser-Ala) between RT codons 69 and 70 and additional mutations, such as M41L, A62V, K70R, and T215Y, display high-level resistance to zidovudine and moderate levels of re-sistance to other nucleoside analogues (3, 6, 7, 19, 23, 39, 41, 43, 46). Similarly, deletions around positions 67 to 70 of the RT are associated with resistance to RT inhibitors, in some cases through complex interactions with other mutations in the RT-coding region (14, 15, 42, 45). However, deletions are less frequently observed than insertions, accounting for approxi-mately 0.2% of the HIV-infected patients treated with
nucle-* Corresponding author. Mailing address: irsiCaixa Foundation, Hospital Germans Trias i Pujol, Ctra. de Canyet s/n, 08916 Badalona, Spain. Phone: 34 93 4656374. Fax: 34 93 4653968. E-mail: jmpicado @irsicaixa.es.
䌤Published ahead of print on 21 February 2007.
4713
on November 8, 2019 by guest
http://jvi.asm.org/
oside RT inhibitors (24), and are usually accompanied by other drug resistance mutations. The mechanisms and evolutionary pathways by which these deletions develop are not known.
The aims of this work were to elucidate mutational pathways leading to the emergence of HIV-1 variants carrying a deletion of 3 nucleotides affecting codon 69 (⌬69) of the viral RT in the presence of a relatively complex array of mutations associated with resistance to multiple RT inhibitors and to determine the role of accessory mutations in viral fitness. Viral isolates con-taining the deletion were obtained from an HIV-1-infected patient who had been treated for more than 15 years with frequent changes of antiretroviral drug regimen. Population-based sequencing as well as clonal genotype analyses of the viral RT-coding region was used to track the evolution and population dynamics of HIV-1 variants carrying the⌬69 dele-tion. We further evaluated the impact of⌬69 on phenotypic resistance, replication capacity (RC), and Gag polyprotein pro-cessing in different sequence contexts, including the presence of MDR-associated mutations in the viral RT-coding region.
MATERIALS AND METHODS
Patient sample.Plasma and peripheral blood mononuclear cells (PBMCs) were obtained from a heavily treated HIV-1-infected patient. The donor was an HIV-1-infected 40-year-old man who had been diagnosed with HIV-1 infection in 1989 and extensively treated with several RT and PR inhibitors since 1991. An initial treatment with zidovudine monotherapy (1991 to 1994) was followed by a series of drug regimens that included combinations of antiretroviral drugs, such as zidovudine, didanosine, zalcitabine, stavudine, lamivudine, abacavir, nevirap-ine, efavirenz, saquinavir, ritonavir, nelfinavir, amprenavir, lopinavir, and enfu-virtide. Plasma HIV-1 RNA was quantified using the Amplicor HIV Monitor test, version 1.5 (Roche Diagnostics), with a limit of detection of 50 copies/ml. Lymphocyte CD4⫹T-cell counts were determined in whole blood by flow cy-tometry. Population-based sequencing of the HIV-1 RT-coding region showed that it contained the deletion (⌬69) as well as 25 additional mutations scattered throughout the entire RT-coding sequence, including 9 amino acid substitutions associated with drug resistance.
Length polymorphisms detection.The proportion of the viral population con-taining⌬69 in the RT from longitudinal viral isolates was determined by analysis of length polymorphisms, as previously described (34).
Generation of RT recombinant viruses.Viral RNA was extracted from 140l of a plasma sample obtained in April 2001 (viral RNA kit; QIAGEN). Combined cDNA synthesis and PCR (one-step RT-PCR; QIAGEN) was performed using primers 1633U23 (5⬘-ATT CTG GAC ATA ARA CAR GGA CC-3⬘; positions 1633 to 1655 in the HXB2 numbering system) and 4461L25 (5⬘-CTT CTA TAT ATC CAC TGG CTA CAT G-3⬘; positions 4461 to 4485) to amplify the RT-coding region. One microliter of the amplified product was submitted to a second amplification round (PlatinumTaqDNA polymerase, high fidelity; Invitrogen) with inner cloning primers 2574U29 and 4120L35 (21), which included restriction sites for XmaI and PacI, respectively. The product was ligated into pJM14⌬RT
(21), which had been previously digested with the same restriction enzymes. After transformation ofEscherichia coli DH5␣competent cells (Invitrogen), individual recombinant clones were obtained and their genotypes verified by DNA sequencing.
Although the reconstructed pJM14 (containing the 5⬘ half of HIV-1NL4-3)
could be cotransfected with p83-10 (containing the 3⬘half of HIV-1NL4-3) (9)
through ligation of their unique EcoRI restriction sites (position 5743 in HIV-1NL4-3) to produce infectious virus, the presence of an additional EcoRI restriction
site in the RT of the viral isolate forced us to modify the original protocol (21). Infectious HIV-1 molecular clones were generated after cotransfection of MT-4 cells with 2g of a PCR-amplified fragment containing the entire RT-coding sequence (amino acids 15 to 527) of the reconstructed pJM14 with primers 1811U24 and 4335L25 (21) and 5g of pJM31⌬GPRT linearized with XbaI. Homologous re-combination in MT-4 cells between overlapping ends of the reconstructed PCR-amplified fragment and pJM31⌬GPRT regenerated a complete HIV-1 genome. Culture supernatants were tested for p24 antigen production using an enzyme-linked immunosorbent assay (Innotest HIV antigen monoclonal antibody; Innogenetics) every 3 to 4 days to monitor viral production.
In order to evaluate the specific phenotypic contribution of the first 248 amino acid residues of the RT, new viral clones containing the wild-type (WT) region of HIV-1NL4-3, from residue 249 to residue 527, were generated by megaprimer
mutagenesis (12) (http://www.irsicaixa.com/downloads/external/VillenaJV07.pdf).
Mutagenesis.Site-directed mutagenesis was used to introduce either a 3-nucleotide deletion or a single-3-nucleotide change at codon 69 of HIV-1NL4-3to
generate mutation⌬69 or T69A, respectively (http://www.irsicaixa.com/downloads /external/VillenaJV07.pdf). Site-directed mutagenesis was also used to revert the Ile-163 in MDR clone MDRc7 back to the WT residue, Ser-163 (MDRc7b), and the Arg-20 in MDRc3 back to its WT residue, Lys-20 (MDRc3b) (Table 1).
Phenotypic analysis. Drug susceptibility assays were performed using the PhenoSense HIV system (Monogram Biosciences) (32). This assay is based on the use of a modified HIV-1 vector derived from the NL4-3 molecular clone, which contains an insert derived from amplification of patient plasma samples that includes the viral p7-p1-p6 PR cleavage sites ingag, the entire PR-coding region, and the first 915 nucleotides of the RT-coding region. Relative fitness was determined for each drug concentration as the ratio between the luciferase activity (relative light units [RLU]) obtained for each virus and the activity of the WT reference HIV-1NL4-3strain. Values were then normalized according to the
transfection efficiencies.
RC analysis.Three different assays were used. The first was a single-cycle-infectivity assay using a GHOST CCR5/CXCR4 cell line which was stably trans-fected with a construct containing an HIV-2 long terminal repeat driving the expression of green fluorescent protein (35). In these assays, a total of 5⫻104
cells/well were infected in duplicate with 50 ng of p24 antigen equivalents of virus in the presence of 20g of Polybrene/ml by spinoculation (3 h at 1,500⫻gand 22°C). The proportion of green fluorescent protein-positive cells was measured by fluorescence-activated cell sorting analysis 24 h after infection. Second, viral replication rates were measured by determining the slope of the p24 antigen production curve during the exponential phase in culture supernatants of in-fected PBMCs obtained from a single donor with a multiplicity of infection of 0.001. Third, the RCs of the recombinant viral clones were also determined using a modification of the PhenoSense drug susceptibility assay (Monogram Bio-sciences), as previously described (4).
[image:2.585.43.548.80.185.2]Gag processing analysis.Time course infections of MT-4 cultures were carried out using 0.0001 50% tissue culture infective dose per cell with either WT HIV-1 TABLE 1. Amino acid substitutions within the RT of recombinant HIV-1 clones used in this studya
aAmino acid differences in the RT between the viral clones obtained from the plasma of the patient in April 2001. Mutations associated with resistance to nucleoside
RT inhibitors are shown in red. Mutations associated with resistance to nonnucleoside RT inhibitors are shown in green.
on November 8, 2019 by guest
http://jvi.asm.org/
or patient-derived clones. Aliquots of culture supernatants and cell lysates were collected daily to perform an immunoblot, as previously described (30). Briefly, aliquots of 450-l cell culture supernatants were overlaid onto 300l of a 20% sucrose cushion and centrifuged at 25,000⫻gfor 2 h at 4°C. Virus pellets or 105
MT-4 cells collected at different times after infection were suspended in 80 mM Tris buffer, pH 6.8, with 2% sodium dodecyl sulfate (SDS) and 1% glycerol and subjected to electrophoresis in a 4 to 12% SDS-polyacrylamide gel. Separated proteins were electrotransferred to a nitrocellulose membrane and probed with specific rabbit antiserum against HIV-1 p24 (ARP432; Medical Research Coun-cil AIDS Directed Programme) or-actin (Sigma). Detection of membrane-bound antibodies was performed using goat anti-rabbit immunoglobulin G or anti-mouse immunoglobulin G, respectively, conjugated with horseradish perox-idase (Pierce), and the reaction was developed with enhanced chemilumines-cence detection reagents and Hyperfilm-ECL (Amersham).
Statistical analysis.Statistical analyses were performed using the R language (http://www.r-project.org) and GraphPad Prism 4 software, version 4 (GraphPad Software Inc., San Diego, CA).
Nucleotide sequence accession numbers.The sequences obtained from the population-based sequencing of the HIV-1 RT-coding region were deposited in GenBank under accession numbers EF154391, EF154392, and EF154395. Clonal sequences were deposited in GenBank under accession numbers EF154393 and EF154394.
RESULTS
Clinical evolution. The multinucleoside analogue-resistant
[image:3.585.138.449.65.487.2]HIV-1 variant bearing a deletion of 3 nucleotides at codon 69
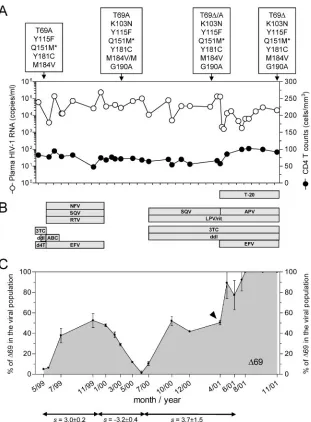
FIG. 1. Longitudinal clinical evolution of the study patient. (A) Determination of plasma HIV-1 RNA copies and CD4 T lymphocytes counts along the period of the study. Boxes show the mutations in the RT associated with resistance to RT inhibitors. In all cases, the amino acid substitution Q151M was accompanied by A62V, V75I, F77L, and F116Y (indicated as Q151M*). The⌬69 deletion was always found together with S163I. The substitution Y181C was most likely selected by a previous nevirapine-containing regimen. (B) Antiretroviral therapy during the follow-up study, grouped by drug family, is indicated in boxes. Drug abbreviations: 3TC, lamivudine; ddI, didanosine; d4T, stavudine; ABC, abacavir; EFV, efavirenz; NFV, nelfinavir; SQV, saquinavir; RTV, ritonavir; APV, amprenavir; LPV/rit, lopinavir boosted with ritonavir; and T-20, enfuvirtide. (C) In vivo selection of⌬69 based on length polymorphism analysis. The selection coefficients (s) for the⌬69-containing viral population are indicated below the graph for each different treatment period. The arrow indicates the clinical isolate used to obtain representative HIV-1 variant clones used in the construction of patient-derived recombinant viruses.
on November 8, 2019 by guest
http://jvi.asm.org/
in the RT-coding region was isolated from a patient that was known to be seropositive for 12 years and who had received antiretroviral treatment since 1991. During the follow-up of this study (May 1999 to November 2001), the patient’s plasma viral loads fluctuated between 103and 2 ⫻ 105 HIV-1 RNA
copies/ml (Fig. 1A), despite antiretroviral therapy regimens containing up to six drugs representing all currently FDA-approved drug families (Fig. 1B). CD4 T-cell counts never rose above 120 cells/l. A 6-month antiretroviral treatment inter-ruption (first half of 2000) due to alpha interferon therapy for hepatitis C did not change the clinical course of the HIV-1 infection. Population-based genotypic analysis of HIV-1 RNA from multiple longitudinal samples showed the accumulation of at least nine amino acid substitutions in the RT-coding region associated with nucleoside and nonnucleoside RT in-hibitor resistance.
In vivo selection of⌬69.The RT-coding regions of 18 viral
isolates derived from plasma samples collected during the fol-low-up period were amplified by RT-PCR for length polymor-phism analysis to follow the viral population dynamics of HIV-1 variants containing ⌬69. A significant proportion of deletion-containing HIV-1 variants were already detected in May 1999, although the relative amounts of virus carrying the deletion fluctuated over time (Fig. 1C). No association be-tween the appearance of the deletion and the effect in vivo on plasma viremia was observed.
Although the presence of⌬69 was already detected during the antiretroviral treatment with lamivudine, didanosine, and stavudine, its relative amount increased from 5 to 50% after a treatment switch to a regimen including abacavir and efa-virenz. A subsequent 6-month-long treatment interruption
re-sulted in a sharp decrease, down to 1%, of the viral population containing⌬69, without changes in plasma viral load. Reiniti-ating a four-drug antiretroviral treatment including lamivudine and didanosine induced the resurgence, up to 50%, of the viral population containing ⌬69. Upon treatment intensification with efavirenz and enfuvirtide,⌬69 became dominant. During this time period, a transient reduction in plasma viral load and an increase in CD4 cell counts were observed, most likely due to the intensification with enfuvirtide (Fig. 1A).
The relative fitness of the⌬69 viral population in each treat-ment period was estimated using a selection coefficient as previously described (27). Under the two independent antiret-roviral treatment periods (end of 1999 and 2000 to 2001), the calculated selection coefficients (s) for HIV variants containing ⌬69 were 3.0⫾0.2 and 3.7⫾1.5, respectively. Conversely, in the absence of drug pressure, HIV variants containing ⌬69 disappeared at similar rates (s ⫽ ⫺3.2 ⫾ 0.4). These data suggest that, in a background containing other drug resistance-associated mutations and in the absence of RT inhibitors, viral populations containing⌬69 are less fit than those without the deletion.
Variations in RC.In order to study the in vitro RC impact of
⌬69, we selected replication-competent viral clones obtained from a patient’s plasma sample containing approximately 50% of the⌬69 viral population (April 2001) (arrow in Fig. 1C). As a result, we obtained three representative recombinant HIV-1 clones containing RT residues 15 to 248 derived from the clinical isolate within an HIV-1NL4-3background. These MDR
clones were MDRc3 (which contained the amino acid substi-tution T69A), MDRc7 (which contained⌬69 and the substi-tution S163I), and MDRc7b (which contained⌬69 and a WT
FIG. 2. Relative RCs in the presence of increasing concentrations of different RT inhibitors. The ratio between the RLU for each recombinant virus and the RLU for the reference HIV-1NL4-3strain is plotted versus the drug concentration. RLU values are derived from the PhenoSense assay
and have been normalized by transfection efficiency. The horizontal dotted line represents the RC of HIV-1NL4-3. Abbreviations: 3TC, lamivudine;
ddI, didanosine; ABC, abacavir; EFV, efavirenz; ZDV, zidovudine; FTC, emtricitabine.
on November 8, 2019 by guest
http://jvi.asm.org/
Ser at position 163) (sequence differences are given in Table 1). These three variants were further characterized and com-pared with mutant HIV-1NL4-3clones differing from the WT in
having either⌬69 or T69A in their RT-coding regions. Because the emergence of T69A preceded the appearance of the dele-tion within the same sequence context, we considered that comparing the effects of T69A and⌬69 was interesting in the context of our fitness and drug susceptibility studies.
The relative RCs of the viral clones selected were deter-mined in the presence of increasing drug concentrations (Fig. 2). All patient-derived recombinant clones consistently showed increasing relative RC values as the drug concentrations in-creased. MDRc7 (⌬69/S163I) showed the highest relative RC under antiretroviral drug pressure, followed by MDRc7b (⌬69) and MDRc3 (T69A). An additional recombinant clone (MDRc2) that differed from MDRc3 in having the amino acid substitution T200A showed the same relative RCs as MDRc3 in the presence of all tested drugs (data not shown). Taken together, these results suggest that in the presence of RT inhibitors, MDR-recombinant viruses containing the substitu-tion T69A are less fit than their homologous counterparts containing⌬69.
Recombinant HIV-1NL4-3 clones containing either ⌬69 or
T69A showed higher relative RCs than the parental WT virus in the presence of high concentrations of lamivudine and emtricitabine. However, differences in the relative RCs were relatively small in the presence of different concentrations of didanosine, stavudine, and abacavir. In contrast, both mutants had impaired relative RCs in the presence of intermediate concentrations of efavirenz and zidovudine (Fig. 2) as well as nevirapine (data not shown). In these cases, the mutant con-taining the deletion had a more deleterious effect on viral replication.
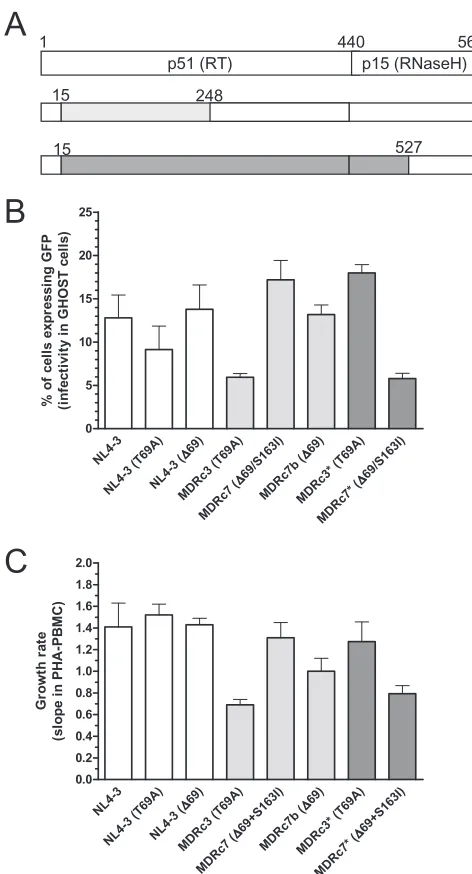
Relative RCs were determined ex vivo for each viral clone in the absence of drugs. The measurements obtained in assays carried out with GHOST cells and PBMCs are given in Fig. 3. The results were also broadly consistent with the relative RCs obtained with the modified PhenoSense assay (Table 2).
Both ⌬69 and T69A had small effects on viral RC when introduced into a WT HIV-1NL4-3clone. However, in the
ab-sence of drugs, MDRc7 (⌬69/S163I) showed the highest RC among the recombinant HIV-1 clones containing residues 15 to 248 of the patient-derived RT. When Ile-163 was replaced by WT Ser in those viruses (MDRc7b), its RC was significantly reduced, suggesting that the substitution S163I contributes to further increases in the RCs of viral clones containing⌬69. In the absence of drugs, recombinant viral clones with Ala at position 69 (MDRc3) showed lower RCs ex vivo than those containing the⌬69 deletion. Reversion of Arg-20 to its WT residue, Lys (MDRc3b), did not have a significant effect on viral growth (data not shown).
These results were somewhat contradictory to evidence showing that antiretroviral drug therapy withdrawal favors the selection of MDR virus without⌬69 (Fig. 1C). Therefore, we obtained two additional HIV clones containing residues 15 to 527 of the RT derived from the clinical isolate. In this sequence background, the clone MDRc3, having the substitution T69A, had a higher RC than MDRc7, in agreement with the evidence obtained in vivo when the antiretroviral therapy was inter-rupted. Therefore, amino acid substitutions within the RT’s
connection subdomain or RNase H domain have a significant effect on viral fitness.
Susceptibility to RT inhibitors. Recombinant viruses
de-rived from HIV-1 RNA obtained from the plasma of the in-fected patient were all highly resistant to all of the RT inhib-itors tested. Only tenofovir retained some activity against the deletion-containing viral clones (Table 2). However, the virus containing the⌬69 deletion within the HIV-1NL4-3backbone
[image:5.585.299.535.64.501.2]showed slightly reduced susceptibility to lamivudine and emtricitabine, while showing hypersusceptibility to zidovudine, an effect that was not observed with mutation T69A. Both viruses containing T69A or⌬69 were fully susceptible to the
FIG. 3. Relative RC in the absence of drugs of RT-recombinant HIV-1. (A) Recombinant virus containing RT residues 15 to 248 (light gray) or 15 to 527 (dark gray) derived from a heavily treated patient’s HIV-1 RNA. (B) RC was measured by a single-cycle-infectivity assay with the GHOST CCR5/CXCR4 cell line. (C) Viral growth rate based on p24 antigen production during the exponential phase in supernatant cultures of PHA-stimulated PBMCs.
on November 8, 2019 by guest
http://jvi.asm.org/
other drugs tested, suggesting that resistance to RT inhibitors was mainly determined by the accompanying mutations (i.e., the 151 MDR complex).
Susceptibility to PR inhibitors.All of the recombinant virus
tested contained RT residues 15 to 248, derived from the patient’s plasma HIV-1 RNA within an isogenic HIV-1NL4-3
backbone. Therefore, they shared a common PR-coding re-gion. Unexpectedly, our data showed that in general, most of the viruses had increased susceptibilities to PR inhibitors, which in some cases represented a⬎2.5-fold reduction in the 50% inhibitory concentration (IC50) for the inhibitor in
com-parison with that for the reference WT virus. We tested whether this effect was related to viral maturation by
measur-ing the amounts of Gag precursor and mature p24 durmeasur-ing a time course involving the infection of MT-4 cells with the viral clones HIV-1NL4-3 and MDRc3 (Fig. 4). Relatively large
amounts of matured p24 were detected in virions obtained from cell culture supernatants infected with HIV-1NL4-3after 6
[image:6.585.40.543.81.193.2]days of infection, while the amounts of Gag precursor were very small (Fig. 4A). However, the virions obtained from cells infected with the recombinant virus MDRc3 showed large amounts of unprocessed Gag precursor and significant amounts of the intermediate p41 protein. These observations were also consistent with data obtained from cell lysates showing that the amounts of p55 and p41 precursors relative to those for p24 were significantly larger in MDRc3-infected cells than in cells infected
FIG. 4. Western blot detection of HIV-1 p24 in MT-4 cells infected with WT (HIV-1NL4-3) and MDRc3 viruses. MT-4 cells were infected with
0.0001 50% tissue culture infective dose per cell, and aliquots of culture supernatants and cells were collected daily. Three micrograms of total protein from the pellets obtained after processing 450 l supernatant (A) or 105cells (B) were applied in SDS-polyacrylamide gels. After
[image:6.585.136.445.423.646.2]electrotransfer, blots were developed with specific rabbit antiserum against HIV-1 p24. Western blots show the Gag processing time course for cultures infected with WT and MDRc3 viruses. Blots shown in panel A were derived from virions found in the culture supernatants, whereas those in panel B were obtained from cell lysates. A loading control for the cell lysate, obtained using a-actin-specific antibody, is shown below each panel. m stands for mock-infected cells. The electrophoretic mobilities of p24 and the 41- and 55-kDa precursors of p24 (p41 and p55, respectively) are indicated.
TABLE 2. In vitro RC and antiretroviral drug susceptibility of recombinant HIV-1 clones
Virus
Fold increase of IC50relative to that for wild-type virus control
RC (% relative to WT value)
NRTI NNRTI PI
ABC ddl FTC 3TC d4T TFV ddC ZDV DLV EFV NVP ATV APV IDV LPV NFV RTV SQV
HIV-1NL4-3 0.87 1.02 0.80 0.88 0.97 0.93 0.96 0.93 1.04 0.79 1.07 0.89 0.88 0.97 0.89 0.86 0.85 0.93 95
HIV-1NL4-3(T69A) 1.00 1.09 1.15 1.14 1.14 0.81 1.13 0.73 0.79 0.66 0.79 0.86 1.01 0.86 0.94 0.86 0.93 0.75 84
HIV-1NL4-3(⌬69) 1.25 1.02 5.30 3.51 1.00 0.67 1.34 0.19 0.61 0.54 0.54 0.86 0.90 0.90 0.92 0.96 0.88 0.86 73
MDRc3 (T69A) >>> 10.88 >>> >>> 8.75 1.79 20.39 392.36 >>> >>> >>> 0.32 0.34 0.35 0.34 0.36 0.31 0.32 10 MDRc7 (⌬69/S163l) >>> 12.03 >>> >>> 8.56 1.60 26.27 570.91 >>> >>> >>> 0.64 0.72 0.72 0.69 0.73 0.70 0.65 45 MDRc7b (⌬69) >>> 19.35 >>> >>> 13.68 2.03 32.89 686.60 >>> >>> >>> 0.49 0.55 0.48 0.45 0.50 0.44 0.49 33
a⬎⬎⬎, high-level resistance. Values (n-fold) for change over the lower cutoff for each drug are highlighted in bold type. RC was determined using a modification
of the PhenoSense drug susceptibility assay. NRTI, nucleoside RT inhibitor; NNRTI, nonnucleoside RT inhibitor; PI, PR inhibitor; ABC, abacavir; ddI, didanosine; FTC, emtricitabine; 3TC, lamivudine; d4T, stavudine; TFV, tenofovir; ddC, zalcitabine; ZDV, zidovudine; DLV, delavirdine; EFV, efavirenz; NVP, nevirapine; ATV, atazanavir; APV, amprenavir; IDV, indinavir; LPV, lopinavir; NFV, nelfinavir; RTV, ritonavir; SQV, saquinavir.
on November 8, 2019 by guest
http://jvi.asm.org/
with the WT virus (Fig. 4B). This result suggests that there are genetic determinants in the recombined RT fragment from the plasma patient’s HIV-1 RNA that contribute to the efficiency of Gag processing and increased PR inhibitor susceptibility. Never-theless, our results are not conclusive and further experiments will be required to address this issue.
DISCUSSION
In this study, we report on the impact on drug susceptibility, in vivo viral fitness, and ex vivo RC of a 3-nucleotide deletion (designated⌬69) in the3-4 hairpin loop-coding region of the HIV-1 RT in the context of a MDR genotype. Nucleotide sequences around RT codons 64 to 71 show remarkable vari-ability, thereby causing some uncertainty regarding the loca-tions of deleloca-tions within the3-4 hairpin loop (25). Align-ment of RT sequences longitudinally obtained during the study revealed that the deletion occurred at codon 69, since we found no additional changes in its vicinity when sequences with and without the deletion were compared. Usually, ⌬69/⌬70 deletions have been associated with the Q151M complex (16, 24, 38, 44), as observed in our study, while there is a related deletion in the3-4 hairpin loop (⌬67) in isolates contain-ing thymidine analogue resistance mutations (TAMs) (16, 24, 38, 44).
Our results show that under drug pressure, the⌬69 deletion increases the RCs of HIV-1 variants with an MDR background (specifically, in the presence of the Q151M complex and mu-tations M184V, K103N, Y181C, and G190A). These observa-tions are consistent with viral population dynamics observed in a long-term-treated HIV-1-infected patient, thereby providing a molecular-based explanation for the emergence of the dele-tion. Enhanced RC and relative infectivity in the presence of abacavir have been previously observed with recombinant HIV-1 clones expressing a patient-derived RT carrying a de-letion affecting codon 70 found in combination with mutations L74V and Q151M (13). The introduction of⌬69 alone within the HIV-1NL4-3sequence background increased viral
suscepti-bility to zidovudine, while producing a slight increase in the IC50for lamivudine and emtricitabine. The influence of
1-ami-no-acid deletions in the 3-4 region of the HIV-1 RT on lamivudine susceptibility in the absence of the M184V muta-tion or the Q151M complex has been controversial (38, 45). However, in combination with other drug resistance mutations (i.e., the Q151M complex, M184V, K103N, Y181C, and G190A),⌬69 confers increased resistance to all RT inhibitors. The molecular mechanism underlying resistance to RT inhib-itors mediated by⌬69 remains to be elucidated. In the pres-ence of accompanying TAMs, recombinant HIV-1 RT contain-ing ⌬67 showed significant nucleoside analogue excision activity on primers terminated with zidovudine, stavudine, and tenofovir (2). However, the contribution of nucleotide selectivity mechanisms affecting discrimination against triphosphorylated derivatives of the inhibitor cannot be ruled out.
Nucleotide sequence analysis of HIV clones found in the patient revealed that the amino acid substitution S163I was found only in clones containing the⌬69 deletion. Interestingly, Ser-163 is well conserved among natural HIV-1 isolates, al-though in the presence of drugs, the lower RCs of viral clones obtained by site-directed mutagenesis and bearing the deletion
plus S163 suggest that this mutation contributes toward the increasing viral fitness of⌬69-containing HIV-1 variants. The identification of the S163N substitution as a second-site amino acid change in the viral RT that restores the viral RCs of compromised HIV-1 mutants bearing TAMs at positions 41 and 70 supports the role of Ser-163 in viral fitness (18). Ser-163 is located in the palm subdomain, near the DNA polymerase active site of the RT, and could affect interactions involving residues that contact the template strand.
In the absence of drugs, viral clones containing T69A repli-cated more efficiently than those having ⌬69, but only when patient-derived sequences containing RT residues 248 to 527 were included. This observation suggests a functional interac-tion between the C-terminal region of the p66 subunit, includ-ing the connection subdomain and the RNase H domain and the DNA polymerase domain of the RT. This interaction would be relevant to an increase in the viral RC ex vivo and consistent with the fluctuations of the viral populations ob-served in vivo.
Changes in drug susceptibility and RC are regularly assessed on viral isolates in order to characterize the pathogenicity and potential transmissibility of HIV-1 variants with specific drug resistance-associated mutations. However, the genotypic con-text in which the mutation has been selected might significantly alter the viral phenotype (34). RT is a multifunctional enzyme that has RNA- and DNA-dependent DNA polymerase activity in addition to endonuclease (RNase H) activity residing within the C-terminal domain of the p66 subunit. RNase H degrades the viral RNA found in RNA-DNA intermediates, which are formed during proviral DNA synthesis. Although currently available drug susceptibility assays for RT inhibitors do not take into account the effects of regions other than the RT polymerase domain, RNase H mutations can significantly con-tribute to nucleoside RT inhibitor resistance when present either alone or in combination with classical mutations in-volved in resistance to RT inhibitors (28, 29). Recently pub-lished reports revealed that mutations within the connection subdomain and the RNase H domain could modulate RT inhibitor resistance, potentially affecting p66/p51 dimerization (10). The comparison of the RT sequence derived from the patient’s viral isolate and the WT HIV-1NL4-3strain showed
several amino acid changes within residues 330 and 560 (com-prising the connection subdomain and the RNase H domain of the RT, respectively) (Table 1).
An unexpected result was the increased susceptibility to PR inhibitors of the patient’s isolate-derived recombinant virus since all tested viruses shared the WT PR-coding region. Ab-normal processing of PR-RT has been associated with lower infectivity in some HIV-1 constructs but not with drug suscep-tibility changes (5). Moreover, the functional interplay be-tween RT and PR has been previously proposed to be relevant to the therapeutic control of HIV-1 infection (1, 8). Viral constructs containing the RT polymerase domain and derived from the patient’s April 2001 isolate showed increased suscep-tibilities to all PR inhibitors, while clone MDRc3 (containing a T69A mutation) showed partially impaired Gag processing. These results were consistent with a reduction in the amount of functional PR, which could be attributed either to Gag-Pol instability or to a defect in Gag-Pol dimerization. Since PR is active only as a dimer, Gag-Pol dimerization is required for PR
on November 8, 2019 by guest
http://jvi.asm.org/
activation and therefore viral maturation (33, 36). Therefore, we speculate about the possibility that RT mutational patterns found in the clinical isolate could either affect Gag-Pol dimer-ization or reduce the stability of the viral polyprotein, causing reductions in the levels of active PR. These reductions could be responsible for PR inhibitor hypersusceptibility, a phenome-non that has been previously observed but whose underlying mechanism remains elusive (20, 22).
In summary, the 3-nucleotide deletion (⌬69) along with S163I in the context of an MDR RT genotype favored the ex vivo RC under drug pressure, in agreement with its in vivo emer-gence and evolution in a long-term-treated HIV-1-infected pa-tient. The C-terminal domain of the p66 subunit, including RNase H, might affect the drug susceptibilities of the virus to RT inhibitors and viral fitness. Finally, MDR-associated mu-tations might affect the final amount of active PR, leading to PR inhibitor hypersusceptibility.
ACKNOWLEDGMENTS
This study was supported by the Fundacio´n para la Investigacio´n y Prevencio´n del Sida en Espan˜a (FIPSE) through grant 36523/05; the Fondo de Investigaciones Sanitarias (FIS) through grants PI050022 and PI051456, the Spanish AIDS network Red Tema´tica Cooperativa de Investigacio´n en SIDA (RD06/0006), and contract 99/3132 (to J.M.-P.); and the Spanish Ministry of Education and Science through grants BMC2003-02148 and BIO2003-01175.
REFERENCES
1.Bleiber, G., M. Munoz, A. Ciuffi, P. Meylan, and A. Telenti.2001. Individual contributions of mutant protease and reverse transcriptase to viral infectiv-ity, replication, and protein maturation of antiretroviral drug-resistant hu-man immunodeficiency virus type 1. J. Virol.75:3291–3300.
2.Boyer, P. L., T. Imamichi, S. G. Sarafianos, E. Arnold, and S. H. Hughes.
2004. Effects of the⌬67 complex of mutations in human immunodeficiency virus type 1 reverse transcriptase on nucleoside analog excision. J. Virol.
78:9987–9997.
3.Boyer, P. L., J. Lisziewicz, F. Lori, and S. H. Hughes.1999. Analysis of amino insertion mutations in the fingers subdomain of HIV-1 reverse transcriptase. J. Mol. Biol.286:995–1008.
4.Campbell, T. B., K. Schneider, T. Wrin, C. J. Petropoulos, and E. Connick.
2003. Relationship between in vitro human immunodeficiency virus type 1 replication rate and virus load in plasma. J. Virol.77:12105–12112. 5.Cherry, E., N. Morin, and M. A. Wainberg.1998. Effect of HIV constructs
containing protease-reverse transcriptase fusion proteins on viral replication. AIDS12:967–975.
6.De Antoni, A., A. Foli, J. Lisziewicz, and F. Lori.1997. Mutations in the pol gene of human immunodeficiency virus type 1 in infected patients receiving didanosine and hydroxyurea combination therapy. J. Infect. Dis.176:899– 903.
7.de Jong, J. J., J. Goudsmit, V. V. Lukashov, M. E. Hillebrand, E. Baan, R. Huismans, S. A. Danner, J. H. ten Veen, F. de Wolf, and S. Jurriaans.1999. Insertion of two amino acids combined with changes in reverse transcriptase containing tyrosine-215 of HIV-1 resistant to multiple nucleoside analogs. AIDS13:75–80.
8.de la Carriere, L. C., S. Paulous, F. Clavel, and F. Mammano.1999. Effects of human immunodeficiency virus type 1 resistance to protease inhibitors on reverse transcriptase processing, activity, and drug sensitivity. J. Virol.73:
3455–3459.
9.Gibbs, J. S., D. A. Regier, and R. C. Desrosiers.1994. Construction and in vitro properties of HIV-1 mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retrovir.10:343–350.
10.Gupta, S., S. Fransen, E. E. Paxinos, W. Huang, E. Stawiski, C. J. Petro-poulos, and N. T. Parkin.2006. Infrequent occurrence of mutations in the C-terminal region of reverse transcriptase modulates susceptibility to RT inhibitors. Antivir. Ther.11:S143.
11.Hirsch, M. S., F. Brun-Vezinet, B. Clotet, B. Conway, D. R. Kuritzkes, R. T. D’Aquila, L. M. Demeter, S. M. Hammer, V. A. Johnson, C. Loveday, J. W. Mellors, D. M. Jacobsen, and D. D. Richman. 2003. Antiretroviral drug resistance testing in adults infected with human immunodeficiency virus type 1: 2003 recommendations of an International AIDS Society-USA panel. Clin. Infect. Dis.37:113–128.
12.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease.1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene77:51–59.
13.Hu, Z. X., H. Hatano, M. Wild, R. Kalayjian, B. Gripshover, and D. R. Kuritzkes.2005. Relative fitness and infectivity of a clinical HIV-1 isolate with a deletion of codon 70 in reverse transcriptase. Antivir. Ther.10:S178. 14.Imamichi, T., S. C. Berg, H. Imamichi, J. C. Lopez, J. A. Metcalf, J. Falloon, and H. C. Lane.2000. Relative replication fitness of a high-level 3⬘ -azido-3⬘-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr3Gly) at codon 69. J. Virol.74:10958–10964.
15.Imamichi, T., M. A. Murphy, H. Imamichi, and H. C. Lane.2001. Amino acid deletion at codon 67 and Thr-to-Gly change at codon 69 of human immunodeficiency virus type 1 reverse transcriptase confer novel drug resis-tance profiles. J. Virol.75:3988–3992.
16.Imamichi, T., T. Sinha, H. Imamichi, Y. M. Zhang, J. A. Metcalf, J. Falloon, and H. C. Lane.2000. High-level resistance to 3⬘-azido-3⬘-deoxythimidine due to a deletion in the reverse transcriptase gene of human immunodefi-ciency virus type 1. J. Virol.74:1023–1028.
17.Iversen, A. K., R. W. Shafer, K. Wehrly, M. A. Winters, J. I. Mullins, B. Chesebro, and T. C. Merigan.1996. Multidrug-resistant human immunode-ficiency virus type 1 strains resulting from combination antiretroviral ther-apy. J. Virol.70:1086–1090.
18.Jeeninga, R. E., W. Keulen, C. Boucher, R. W. Sanders, and B. Berkhout.
2001. Evolution of AZT resistance in HIV-1: the 41-70 intermediate that is not observed in vivo has a replication defect. Virology283:294–305. 19.Larder, B. A., S. Bloor, S. D. Kemp, K. Hertogs, R. L. Desmet, V. Miller,
M. Sturmer, S. Staszewski, J. Ren, D. K. Stammers, D. I. Stuart, and R. Pauwels.1999. A family of insertion mutations between codons 67 and 70 of human immunodeficiency virus type 1 reverse transcriptase confer multinucleoside analog resistance. Antimicrob. Agents Chemother.43:
1961–1967.
20.Leigh Brown, A. J., S. D. Frost, B. Good, E. S. Daar, V. Simon, M. Markow-itz, A. C. Collier, E. Connick, B. Conway, J. B. Margolick, J. P. Routy, J. Corbeil, N. S. Hellmann, D. D. Richman, and S. J. Little.2004. Genetic basis of hypersusceptibility to protease inhibitors and low replicative capacity of human immunodeficiency virus type 1 strains in primary infection. J. Virol.
78:2242–2246.
21.Martinez-Picado, J., L. Sutton, M. P. De Pasquale, A. V. Savara, and R. T. D’Aquila.1999. Human immunodeficiency virus type 1 cloning vectors for antiretroviral resistance testing. J. Clin. Microbiol.37:2943–2951. 22.Martinez-Picado, J., T. Wrin, S. D. Frost, B. Clotet, L. Ruiz, A. J. Brown,
C. J. Petropoulos, and N. T. Parkin.2005. Phenotypic hypersusceptibility to multiple protease inhibitors and low replicative capacity in patients who are chronically infected with human immunodeficiency virus type 1. J. Virol.
79:5907–5913.
23.Mas, A., M. Parera, C. Briones, V. Soriano, M. A. Martı´nez, E. Domingo, and L. Mene´ndez-Arias.2000. Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resis-tance. EMBO J.19:5752–5761.
24.Masquelier, B., E. Race, C. Tamalet, D. Descamps, J. Izopet, C. Buffet-Janvresse, A. Ruffault, A. S. Mohammed, J. Cottalorda, A. Schmuck, V. Calvez, E. Dam, H. Fleury, and F. Brun-Ve´zinet.2001. Genotypic and phe-notypic resistance patterns of human immunodeficiency virus type 1 variants with insertions or deletions in the reverse transcriptase (RT): multicenter study of patients treated with RT inhibitors. Antimicrob. Agents Chemother.
45:1836–1842.
25.Menendez-Arias, L., T. Matamoros, and C. E. Cases-Gonzalez.2006. Inser-tions and deleInser-tions in HIV-1 reverse transcriptase: consequences for drug resistance and viral fitness. Curr. Pharm. Des.12:1811–1825.
26.Mocroft, A., S. Vella, T. L. Benfield, A. Chiesi, V. Miller, P. Gargalianos, A. d’Arminio Monforte, I. Yust, J. N. Bruun, A. N. Phillips, J. D. Lundgren, et al.1998. Changing patterns of mortality across Europe in patients infected with HIV-1. Lancet352:1725–1730.
27.Nagylaki, T.1992. Introduction to theoretical population genetics. Springer-Verlag KG, Berlin, Germany.
28.Nikolenko, G. N., S. Palmer, F. Maldarelli, J. W. Mellors, J. M. Coffin, and V. K. Pathak.2005. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. USA102:2093–2098.
29.Nikolenko, G. N., E. S. Svarovskaia, K. A. Delviks, and V. K. Pathak.2004. Antiretroviral drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase increase template-switching frequency. J. Virol.
78:8761–8770.
30.Olivares, I., M. Gutierrez-Rivas, C. Lopez-Galindez, and L. Menendez-Arias.2004. Tryptophan scanning mutagenesis of aromatic residues within the polymerase domain of HIV-1 reverse transcriptase: critical role of Phe-130 for p51 function and second-site revertant restoring viral replication capacity. Virology324:400–411.
31.Palella, F. J., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, S. D. Holmberg, et al.1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med.338:853–860.
32.Petropoulos, C. J., N. T. Parkin, K. L. Limoli, Y. S. Lie, T. Wrin, W. Huang, H. Tian, D. Smith, G. A. Winslow, D. J. Capon, and J. M. Whitcomb.2000.
on November 8, 2019 by guest
http://jvi.asm.org/
A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother.44:920–928.
33.Pettit, S. C., L. E. Everitt, S. Choudhury, B. M. Dunn, and A. H. Kaplan.
2004. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol.78:8477–8485.
34.Prado, J. G., S. Franco, T. Matamoros, L. Ruiz, B. Clotet, L. Menendez-Arias, M. A. Martinez, and J. Martinez-Picado.2004. Relative replication fitness of multi-nucleoside analogue-resistant HIV-1 strains bearing a dipep-tide insertion in the fingers subdomain of the reverse transcriptase and mutations at codons 67 and 215. Virology326:103–112.
35.Prado, J. G., N. T. Parkin, B. Clotet, L. Ruiz, and J. Martinez-Picado.2005. HIV type 1 fitness evolution in antiretroviral-experienced patients with sus-tained CD4⫹T cell counts but persistent virologic failure. Clin. Infect. Dis.
41:729–737.
36.Quillent, C., A. M. Borman, S. Paulous, C. Dauguet, and F. Clavel.1996. Extensive regions of pol are required for efficient human immunodeficiency virus polyprotein processing and particle maturation. Virology219:29–36. 37.Richman, D. D.2001. HIV chemotherapy. Nature410:995–1001. 38.Ross, L., M. Johnson, R. G. Ferris, S. A. Short, L. R. Boone, T. E. Melby, R.
Lanier, M. Shaefer, and M. St Clair.2000. Deletions in the3-4 hairpin loop of HIV-1 reverse transcriptase are observed in HIV-1 isolated from subjects during long-term antiretroviral therapy. J. Hum. Virol.3:144–149. 39.Ross, L., M. Johnson, N. Graham, M. Shaefer, and M. St. Clair.1999. The
reverse transcriptase codon 69 insertion is observed in nucleoside reverse transcriptase inhibitor-experienced HIV-1-infected individuals, including those without prior or concurrent zidovudine therapy. J. Hum. Virol.2:290– 295.
40.Shirasaka, T., M. F. Kavlick, T. Ueno, W. Y. Gao, E. Kojima, M. L. Alcaide, S. Chokekijchai, B. M. Roy, E. Arnold, R. Yarchoan, and H. Mitsuya.1995. Emergence of human immunodeficiency virus type 1 variants with resistance
to multiple dideoxynucleosides in patients receiving therapy with dideoxy-nucleosides. Proc. Natl. Acad. Sci. USA92:2398–2402.
41.Sugiura, W., M. Matsuda, Z. Matsuda, H. Abumi, A. Okano, T. Oishi, K. Moriya, Y. Yamamoto, K. Fukutake, J. Mimaya, A. Ajisawa, M. Taki, K. Yamada, and Y. Nagai.1999. Identification of insertion mutations in HIV-1 reverse transcriptase causing multiple drug resistance to nucleoside analogue reverse transcriptase inhibitors. J. Hum. Virol.2:146–153.
42.Suzuki, K., G. R. Kaufmann, M. Mukaide, P. Cunningham, C. Harris, L. Leas, M. Kondo, M. Imai, S. L. Pett, R. Finlayson, J. Zaunders, A. Kelleher, and D. A. Cooper.2001. Novel deletion of HIV type 1 reverse transcriptase residue 69 conferring selective high-level resistance to nevirapine. AIDS Res. Hum. Retrovir.17:1293–1296.
43.Tamalet, C., J. Izopet, N. Koch, J. Fantini, and N. Yahi.1998. Stable rear-rangements of the3-4 hairpin loop of HIV-1 reverse transcriptase in plasma viruses from patients receiving combination therapy. AIDS12:F161– F166.
44.Tamalet, C., N. Yahi, C. Tourres, P. Colson, A. M. Quinson, I. Poizot-Martin, C. Dhiver, and J. Fantini.2000. Multidrug resistance genotypes (insertions in the3-4 finger subdomain and MDR mutations) of HIV-1 reverse transcriptase from extensively treated patients: incidence and asso-ciation with other resistance mutations. Virology270:310–316.
45.Winters, M. A., K. L. Coolley, P. Cheng, Y. A. Girard, H. Hamdan, L. C. Kovari, and T. C. Merigan.2000. Genotypic, phenotypic, and modeling studies of a deletion in the3-4 region of the human immunodeficiency virus type 1 reverse transcriptase gene that is associated with resistance to nucleoside reverse transcriptase inhibitors. J. Virol.74:10707–10713. 46.Winters, M. A., K. L. Coolley, Y. A. Girard, D. J. Levee, H. Hamdan, R. W.
Shafer, D. A. Katzenstein, and T. C. Merigan.1998. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J. Clin. Investig.102:
1769–1775.