Rochester Institute of Technology
RIT Scholar Works
Theses
Thesis/Dissertation Collections
1-2006
LigEvolver: A Tool for ligand formulation using
genetic algorithm
Priyadarsini Soundararajan
Follow this and additional works at:
http://scholarworks.rit.edu/theses
This Thesis is brought to you for free and open access by the Thesis/Dissertation Collections at RIT Scholar Works. It has been accepted for inclusion in Theses by an authorized administrator of RIT Scholar Works. For more information, please [email protected].
Recommended Citation
LigEvolver: A Tool for Ligand Formulation Using Genetic
Algorithm
Approved: _ _
P_a_u_I_A_._C_r
a---=i
9=----__
Thesis AdvisorG. R. Skuse
Director of Bioinformatics orHead, Department of Biological Sciences
Submitted in partial fulfillment of the requirements for the Master of Science
degree in Bioinformatics at the Rochester Institute of Technology.
Thesis/Dissertation Author Permission Statement
Ti tIe of thesis or dissertation:
_--"
l===luf:.zJ.'2Z~e~\J~O~L<JlV--bf:Rd::>..~
~
---I
A
~---,
'
fc!..10~O:!..!L
""--_
EI--..\.<Oh.R
..----J
L""'-..lI.I.J('!I[JJA~NuD
L---Name of author: &1L(ADBRsI N\
~\
\f\I'DI\B.f\Rft TBN Degree:N"smR..
Of
XIf:.J\lC.E:.Program: l?lbII\lFDR.\-j t\IICS
College:
Sc/f
=-
NCfe
I understand that I must submit a print copy of my thesis or dissertation to the RIT Archives, per current RIT guidelines for the completion of my degree. I hereby grant to the Rochester Institute of Technology and its agents the non-exclusive license to archive and make accessible my thesis or dissertation in whole or in part in all forms of media in perpetuity. I retain all other ownership rights to the copyright of the thesis or dissertation. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation.
Print Reproduction Permission Granted:
I, fBNfiDAfS5lNI
c
~
O
N'D8f<tlRf\rAN
,hereby grant permission to the Rochester Institute Technology to reproduce my print thesis or dissertation in whole or in part. Any reproduction will not be for commercial use or profit.Signature of Author: _ _ _
P_r_iy~a_d_a_r_s_i_n_i
___
Date:1
i
/&4/0
b
Print Reproduction Permission Denied:
I, , hereby deny permission to the RIT Library of the Rochester Institute of Technology to reproduce my print thesis or dissertation in whole or in part.
Signature of Author: _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ Date: _ _ _ _ _
Inclusion in the
RITDigital Media Library Electronic Thesis
&Dissertation (ETD) Archive
I, ' additionally grant to the Rochester Institute of Technology Digital Media Library (RIT DML) the non-exclusive license to archive and provide electronic access to my thesis or dissertation in whole or in part in all forms of media in perpetuity.
I understand that my work, in addition to its bibliographic record and abstract, will be available to the world-wide community of scholars and researchers through the RIT DML. I retain all other ownership rights to the copyright of the thesis or dissertation. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation. I am aware that the Rochester Institute of Technology does not require registration of copyright for ETDs.
Thesis
Advisory
CommitteeCommittee Chair
Dr. PaulCraig
ProfessorofChemistry
DepartmentofChemistry
CollegeofScience
Rochester InstituteofTechnology
Committee Member
Professor Al Biles
ProfessorandUndergraduate Program Coordinator
InformationTechnologyDepartment
Golisano CollegeofComputingandInformation Sciences
Rochester InstituteofTechnology
Committee Member
Dr. Anne Haake
Associate Professor
InformationTechnologyDepartment
Golisano CollegeofComputingandInformationSciences
Abstract
Human
Immunodeficiency
Virustype 1 (HIV-1)has been foundtobe thecause of
Acquired Immune Deficiency Syndrome (AIDS). This virus caused uproar during the
1980's and started a major drug discovery race among the pharmaceutical companies. It
also proved to be a milestonein theestablishmentoftheuse of computers inrational drug
design.
Evolutionary computation is a commonly used computational approach that has
been successfully applied to a variety of fields ranging from engineering to life science.
The main reason for its effectiveness is that it is driven by the principles of evolution. A
genetic algorithm is an approach to evolutionary computation that allows random
combination of data to occur in a series of generations and enables the identification of
novel systemsthatmight otherwisehavegone undetected.
This work explores the use of genetic algorithms to generate new ligand structures
thatmay be effectivein inhibitingHIV- 1
Protease, one ofthemajordrug targets in HIV.
One of the computational challenges associated with drug discovery is the conversion of
chemical and biological entities into formats that the computer can use. Chemical
structures can be represented by linear character strings called SMILES strings. SMILES
(Simplified Molecular Input Line Entry Specification) strings are taken as the genetic
representation for our approach to drug discovery, and a genetic algorithm has been
the ligands are evaluated and either kept or removed from the gene pool following the
Acknowledgements
I would like to thank Dr. Paul Craig, committee chairman and advisor for hisunrelenting
guidance and encouragement. His patience and enthusiastic supervision has been the
driving force behind the successful completion of the project. I am grateful to Prof. Al
Biles for his zeal andsupport. Thenumerous technicaldiscussions withhimenabled me to
approach the problem in various angles and his advices towards the computational aspect
oftheresearch were useful. I would like to offer my sincere gratitude to Dr. Anne Haake
for herassistance andinterest.Dr. Haake had been a source of supportand encouragement
at all times. Iwould like to thankallthe committee members fortrusting me and being a
part ofthis thesis.
I would like to extend my thankfulness to Mr. Srinivas Naidu, my uncle and a constant
Table ofContents
1. Introduction 1
1.1 Fundamental Concepts 1
1.2 PurposeofInvestigation 2
1.3Problem Description 3
1.4Rationale 4
2. MethodsandMaterials 5
2.1 DiseaseandEnzyme Selection 5
2.1.1 HIVGenome 5
2.1.2Lifecycle ofHIV 6
2.1.3 HIVProtease 8
2.1.4ConservedRegion 9
2.1.5 EnzymaticReaction 10
2.2 Genetic RepresentationinLigEvolver 12
2.2.1 Chromosome Representation 12
2.2.2Sample SMILES string and2D structure ofligand 13
2.2.3 PharmacophoricFeaturesConsidered 14
2.3Input 15
2.3.1 XML/Perl Parser 16
2.3.2 Protein Data Bank 17
2.4 Output 17
2.6.1 Genetic Algorithm 19
2.6.2 Pharmacophore Elucidator 27
2.6.3 Molecular Weight Calculator 32
2.6.4 Van Der Waals Volume 33
2.6.5 Polar Surface Area 35
2.6.6 Motif Finder 35
2.6.7 Fitness Computor 36
3. Results 41
3.1 Simulation 1 41
3.1.1 Significant Results 41
3.2 SimulationII 46
3.2.1 Significant Results 46
3.3 Simulation HI 52
3.3.1 Significant Results 52
3.4 Simulation IV 58
3.4.1 Significant Results 58
3.5Simulation V 61
3.5.1 Significant Results 61
3.6SimulationVI 64
3.6.1 Significant Results 65
3.7 FitnessBehavior 66
5. Conclusion 72
Bibliography 73
Appendix 1 78
ListofFigures
Figure 2.1.1.1 HIV Genome 5
Figure 2.1.2.1 HIV Lifecycle 7
Figure 2. 1.3.1 HIV Protease and activesite 8
Figure 2.1.3.2HIV Protease inhibited
by 1D4Y 8
Figure 2.1.4.1 Asparatic Acid 9
Figure 2.1.4.2Threonine 9
Figure 2.1.4.3 Glycine 9
Figure 2. 1.4.4Catalytictriad 1 0
Figure2.1.5.1 HIV Proteasecleavingreaction 11
Figure 2.2.1.1 Transformationfrom3D structuretoSMILES string 12
Figure2.2.2.2TPVStructure 14
Figure2.2.3.1 TPV Structurewithlabeledpharmacophoricfeatures 15
Figure2.3.1 Input Construction 17
Figure 2.6.1 ComponentsofLigEvolver 19
Figure 2.6.1.1 Flowchart ofGA 20
Figure 2.6.1.1.1.1 ExampleofGenFrag 22
Figure 2.6.1.1.2.1 ExampleofCrossover 23
Figure2.6.1.1.3 Mutationrates and geneticadaptability 24
Figure 2.6.1.2.1 Tournament Selection 26
Figure 2.6.2.1.1 Aromatic StructureI
- ABenzene
ring 27
Figure2.6.2.2. 1 Hydrogen Bond AcceptorStructure I 28
Figure 2.6.2.2.3 HydrogenBond Acceptor StructureIII 28
Figure 2.6.2.2.4 Hydrogen Bond Acceptor StructureIV 29
Figure 2.6.2.3. 1 Hydrogen Bond A/D Structure I 29
Figure 2.6.2.3.2 Hydrogen Bond A/D Structure II 29
Figure 2.6.2.3.3 Hydrogen BondA/D StructureIII 30
Figure 2.6.2.4. 1 HydrophobicStructure I 30
Figure 2.6.2.4.2 Hydrophobic StructureII 30
Figure 2.6.2.4.3 Hydrophobic StructureIII 3 1
Figure 2.6.2.4.4 Hydrophobic StructureIV 3 1
Figure 2.6.2.4.5 Hydrophobic Structure V 3 1
Figure 2.6.2.4.6 Hydrophobic Structure VI 32
Figure 2.6.2.4.7 Hydrophobic StructureVII 32
Figure3.1.1.1.1.1 Simulation I- Generation 100- CompoundI
42
Figure 3.1.1.1.1.2 SimulationI- Generation 100- Compound II
43
Figure 3.1.1.2.1.1 SimulationI- Generation 85
-CompoundI 44
Figure3.1.1.2.2.1 Simulation I
- Generation 85- CompoundII
45
Figure 3.2. 1.1.1.1 Simulation II
- Generation40
-CompoundI 47
Figure 3.2.1.1.2.1 Simulation II
- Generation
40- Compound II
48
Figure3.2.1.1.3.1 SimulationII
- Generation 40- Compound HI
49
Figure 3.2. 1.1.4.1 SimulationII
- Generation 40- Compound IV
50
Figure3.2. 1.2.1.1 SimulationII
- Generation41 - Compound I
5 1
Figure3.3.1.1.1.1 SimulationHI- Generation 100- Compound I
(a) 53
Figure 3.3.1.1.1.2SimulationIII- Generation 100- Compound I
Figure 3.3.1.1.1.3 Simulation III
-Generation 100 - CompoundI
(c) 55
Figure 3.3.1.1.2.1 SimulationHI
-Generation 100 - CompoundII
(a) 56
Figure 3.3.1.1.2.2 SimulationIII- Generation 100- CompoundII
(b) 57
Figure 3.3.1.1.3.1 Simulation III- Generation 100 - CompoundIII 57
Figure 3.4.1.1.1.1 SimulationTV- Generation 100
-Compound I 59
Figure 3.4.1.1.2.1 SimulationIV
-Generation 100- CompoundII 60
Figure 3.5.1.1.1.1 SimulationV
-Generation 12- Compound I 62
Figure 3.5.1.2.1.1 SimulationV
- Generation 10- CompoundI 63
Figure 3.5.1.3.1.1 SimulationV- Generation 9- Compound
I 64
Figure 3.6.1.1.1.1 SimulationVI
-Generation 199- CompoundI 65
Figure 3.7.1 Fitness Behavior 66
Figure 4.1 LigEvolver CompoundI 67
Figure4.2Similaritiesof compoundIwithknown inhibitors 68
Figure 4.3 LigEvolverCompoundII 68
Figure4.4 Similarities of compoundIIwithknown inhibitors 69
Figure4.5 LigEvolverCompound III 69
Figure4.6Similarities of compoundIIIwithknown inhibitors 70
List ofTables
Table 2.2.2. 1 Ligand
Summary
1 3Table 2.3.1 ListofDatabases 16
Table2.6.1.1.3.1 Amino AcidsandtheirSMILES string 25
Table 2.6.3.1 Molecularweights 32
Table 2.6.4.1 AtomsandtheirVolumes 34
Table 2.6.6.1 FDA
-Approved Protease Inhibitors 35
Table 2.6.7.1.1 Aromatic
RingCount ScoringScheme 37
Table 2.6.7.2.1 Hydrogen Bond Donor CountScoringScheme 38
Table 2.6.7.3.1 Hydrogen Bond Acceptors CountScoringScheme 38
Table 2.6.7.4.1 Hydrophobic Regions CountScoringScheme 38
Table 2.6.7.5.1 Molecular WeightScoringScheme 39
Table 2.6.7.6.1 Polar Surface AreaScoring Scheme 39
Table 2.6.7.7.1 VanDer Waals VolumeScoringScheme 40
Table 3.1.1 SimulationIProperties 41
Table 3.2.1 Simulation II Properties 46
Table 3.3. 1 Simulation HI Properties 52
Table 3.4.1 SimulationIVProperties 58
Table 3.5.1 SimulationVProperties 61
Table 3.6.1 Simulation VI Properties 64
1.
Introduction
LigEvolver, the tool that produces novel ligand structures, has been implemented for
specific reasons. These intentions and the waythis tool is different from previous genetic
algorithm based tools is presented in this section. The necessary background and the
problem are alsodescribed.
1. 1 Fundamental Concepts
Enzymes.
Enzymes are a specialized class of proteins that act as catalysts to increase the rate of
chemicalreactions.
Inhibitor
Theinhibitorsarechemical compoundsthatservetoblockenzymesfrom functioning.
Ligands
The substrate that is bound to by a protein is also called a ligand. These can be used as
effectiveinhibitors. Theterms ligandandinhibitorareusedinterchangeablyinthis thesis.
GeneticAlgorithm
Genetic Algorithms employ concepts of evolution to search for solutions in domain
problem spaces. The individualsin apopulation are made to crossover and mutateto give
apre-defined quality criterion,they areeither allowed toexist or eliminated fromthe next
generation.
Pharmacophore
This is a common template that depicts the desired spatial arrangement of chemical
propertiesofa potential candidate. It istheminimal structure requiredforinhibition.
SMILES
This isan acronym for SimplifiedMolecularInputLineEntry Specification. Itisa method
ofrepresenting amoleculeinalinear form.
1.2 Purpose ofInvestigation
Inhibitordesign isa major part ofthe drug discoveryprocess. Structure-baseddrug design
has been shown to produce effective results and has led to the discovery of a number of
inhibitors. There are plentyoffeatures thatlead to thedesignof a reliable drug such as its
bioavailability,bindingaffinityandinteractionspecificity, amongotherfeatures.
In the case of enzymes with known inhibitors, it would be worthwhile to explore the
possibility of generating ligands based on scalar properties without other added
complexities. The focus of this study is directed towards determining if new ligand
structures could be derived by using a minimal set of physiochemical properties and
Previous studiessuch as thosebyGlenetal., (1994) andVenkatasubramanianetal., (1995)
have shownthe useofevolutionarytechniques fordrugdesign. Douguetet al made use of
SMILES line notation for their program LEA (Ligand by Evolutionary Algorithm).
LigEvolver differs from the past research as it makes use of the smallest number of
constraints and genetic operators for evolving ligands. Its main objective is to see if this
limited approach canleadtopowerful structures.
1.3 Problem Description
There are many undiscovered therapeutic structures that have the ability to function as
inhibitors. The goal of this study is to uncover such structures by constructing a genetic
algorithm based computer tool, LigEvolver. The task is to identify new inhibitors for the
HIV- 1 Protease
enzyme and LigEvolver aides this task byevolving solutions that seem
more likely to performthe task. The level of analysis of the chemical structures has been
restrictedto twodimensions becauseoftheminimalisthypothesis.
Given a set of chemical structures in the SMILES notation, LigEvolver evolves potential
inhibitors for the target enzyme. The algorithm conserves the fittest individuals in each
generation and iterates until fairly competent candidates have been produced. Each
1.4 Rationale
Discovering new drugs is an extremely tedious and expensive task. Computational
derivation ofpotential ligand structures wouldhelp toreducethe time andthe costofdrug
development. The use of a genetic algorithm to generate candidate structures could give
riseto avariety ofcombinations that wouldenable the scientist topursue lead compounds
thathavenotbeen identified usingtraditionalapproaches.
The intention is to develop a program using a well-characterized system of an enzyme
(HIV protease) that has been crystallized in the presence of a large number on inhibitors.
In this case, the 3D structures of the drugs and the target protein are known. Once an
approachtopredictingnew ligandsbasedon existing ligands has beendeveloped,itcanbe
extendedto open cases, where a number ofdrugs havebeen identified,but thestructure of
2.
Methods
andMaterials
2. 1 Disease andEnzyme Selection
2.1.1 HIV Genome
The HIV genome consists of three main genes, namely gag, pol and env. Gag codes for
structural proteins that form the viral core. Pol genes code for Reverse Transcriptase, Integrase and Protease, which arethreeessential enzymes thatplay a significant roleinthe
formation and maturation of the virus. The env gene codes the envelope for the viral
[image:20.509.90.408.339.550.2]proteins [39]. Figure 2.1.1.1 showstheHIVgenomeencompassingthe threegenes.
2.1.2 Lifecycle of HIV
Humanimmunodeficiencyvirus(HIV)is alethalretrovirus thatattackstheimmune
system.Thisretrovirus followsaspecificlife
-cycletoinfectthehostcells.Theviral
BINDING The HIVbinds itselftotheCD4
receptorinthecell membrane
FUSION
TRANSCRIPTION
INTEGRATION
Theviruspenetratesthe
membrane and entersthecell with
thehelpoftheco-receptor
Reverse Transcriptasetranscribes
theviralRNA into DNA
Integrase integratestheviralDNA
withthehostcell'sDNA
CLEAVING Proteasecleavestheviral
polyproteinsinto functional
proteins andthevirus exitsthecell
2.1.3 HIV Protease
Ascan beseen fromthe lifecycle, HIV Protease is a crucial enzyme thathydrolyzes viral
polyproteins into functional protein products. These products then contribute to the
further propagate the virus. This work concentrates on identifying new ligands for HIV
Protease. Theflexibilityandmutabilityofthisenzymemakesit achallengingdrugdesign
target. HIVprotease acts as adimerwithtwo identicalpolypeptidechains. It has onlyone
active site thatis depicted intheboxedregionoffig. 2.1.3.1 [40].
[image:23.509.169.341.230.359.2]Figure2.1.3.1 HIVProtease andactive site
*Figuregeneratedusing DeepView
2.1.4 Conserved Region
During the transcription of RNA to DNA, Reverse Transcriptase has an error rate of about 1 in 2000 bases. Dueto this the HIV strain undergoes mutation in each generation and the nucleotide sequence ofHIV Protease changes from strain to strain. However, a conserved region has been detected. It isthe residues, Asp - Thr
-Gly, ofthe catalytic
triad.
Figure 2.1.4.1 Aspartic Acid(Asp)*
Figure 2.1.4.2 Threonine(Thr)*
)H H2N'
Figure 2.1.4.3 Glysine (Gly)*
O
"^p
H O H H1
H O ^
H3CT
1 II
L H O
OH
-R'
Figure 2.1.4.4 Catalytictriad
2.1.5 Enzymatic Reaction
The HIV protease enzyme cleaves the protein at the carbonyl group ofthe peptide bond.
The entire process is shown with respect to the active site (Asp - Thr
-Gly) in figure
2.1.5.1 [20]. Step 1 shows the binding ofHIV
-1 protease to a peptide substrate; step 2
demonstratesthe attack ofthecarbonyl ofthescissile amidebyHIV - 1
proteaseactivated
water; step 3 illustrates the cleavage of the scissile bond while step 4 depicts the final
H
\
I
K
CatalyticHTV-1 ProteaseMechanismof/ / C=^ ff Asp25 .nr'% o^c/ (-) Asp25'
\
3" C^
Pi \^^-x^Vpi*X7->c.
AsP^25 A -,r) Asp25'
C (-)
Xd
pr x-'"N< 1
H / -,
(-) H
9 / H
Asp25 Asp25'
^
4.
V
P'A
/ 9" h
\
H
A/
VH
c \
Asp25
[image:26.509.89.423.59.539.2]Asp25'
2.2 Genetic Representation in LigEvolver
2.2.1 Chromosome Representation
"Chromosome"
is the general term used to address the genotype of the individuals in a
population. The SMILES string serves as the input to the genetic program. Its simplified
representation supplies thecomputer with an accuratelinearrepresentation ofcomplicated
two dimensional structures, which can be readily manipulated during evolutionary
programming [4]. It is also a standard and universal form of representation used by
chemists all over the world [1]. These reasons make it a good choice for the depiction of
the chemical compounds and a means to represent the chromosome for the genetic
program. The mapping of the chemical compound from its 3D structure to the SMILES
notationis shownin figure 2.2.1.1.
3D
2D
ID
CC(C)(C)NC(=0)ClCC2CCCCC2CNlCC(0)C(Cc3ccccc3)NC(-0)C(CC(N)=0)NC(=0)c4ccc5ccccc5n4
Figure 2.2.1.1 Transformation from 3D structuretoSMILES string
2.2.2 Sample SMILES string and 2D structure of ligand
An example of the ligand structure is shown below along with the SMILES string. The
common nameisTipranavir,whichis abbreviatedTPV.
N-(3-{(lR)-l-[(6R)-4-HYDROXY-2-OXO-6-PHENETHYL-6-PROPYL-5,6-DIHYDRO-2H-Name
PYRAN-3-YL]PROPYL}PHENYL)-5-(TRIFLUOROMETHYL)-2-PYRIDINESULFONAMIDE
TPV
HETID
Formula C3, H33 N2 05 F3S SMILES
CCCC2(cCclcccccl)CC(0)=C(C(CC)c3cccc(NS(=0)(=0)c4ccc(cn4)C(F)(F)F)c3)C(=0)02
String
T^\
/%
Figure 2.2.2.2TPVStructure
2.2.3 Pharmacophoric Features Considered
For each of the ligand structures the following are the important features considered to
form the basic pharmacophore template. These features are thought to be important
interactionpointsbetweentheenzyme andtheligand.
1. AromaticRings [Ar]
2. HydrogenBond Acceptors [A]
3. HydrogenBondAcceptors andDonors [A/D]
4. Hydrophobic center [F]
A O
Figure 2.2.3.1 TPVStructurewithlabeledpharmacophoricfeatures
Key
Hydrophobic Center
A Hydrogen Bond Acceptor
A/D Hydrogen Bond Acceptor/Donor
Ar- Aromatic Ring
2.3Input
Before beginningthe geneticalgorithm, certain pre
-processingstepshadto be completed
for the construction of the input files. SMILES databases, consisting of various chemical
compounds represented as SMILES strings, were assembled. The data for these databases
were extracted from various public databases. The public databases are listed in the
ListofDatabases
Kyoto EncyclopediaofGenes andGenomes (KEGG)
ChemPDB
ChemBank
Macromolecular Structure Database (MSD)
Anti - HIV National Cancer Institute
(NCI)
Protein Data Bank (PDB)
Table2.3.1 ListofDatabases
The above mentioned databases have been downloaded from Ligand.Info which is a
collectionof various small-moleculedatabases [27].
2.3.1 XML/Perl Parser
An XMLparser was constructedtoelucidatethedata fromthedatabases andtoproduce the
output in a format easily transferable to the database. In other words, the files from the
public databases contain information regarding a ligand such as its molecular weight,
geometrical coordinates, etc. Theparser scans thedump files forthetoken "SMILES" and
extracts only the SMILES strings forthe compilation of the various input files. The lava
Parserworksthe same way.Dependingonthe type ofinputdata fileeither oftheparsersis
2.3.2 Protein Data Bank
A HIV database was constructed using the ligand data of the various HIV
-1 Protease
inhibitors. These data were extracted from the Protein Data Bank (PDB) and had to be
compiled manually since the dump files from the PDB did not contain SMILES string
information of the ligands. This file contained the identifier, molecular formula and the
SMILES stringinformation.
Public DBs r PDB Manual Input SMILES .txt HIVPDB .txt
$%
LigEvolver<*
Figure2.3.1 Input Construction
2.4 Output
Two files are produced as aresult ofrunning LigEvolver. Onegives the details regarding
the number of children produced, maximum, minimum and average fitness for each
generation. The otherfile gives the final set ofSMILES strings along withthe generation
2.5 ExternalPackages
Certain calculations required the use of PerlMol, an external package comprised of
modules for performing basic computational chemistry related functions. This is freely
distributed software and a detailed description of PerlMol can be found at
(www.perlmol.org). PerlMolprovidedthefollowingfunctionalitiestoLigEvolver:
1. CalculationofChemical formula fromtheSMILES string
2. Calculationofbonds fromtheSMILES string
3. PolarSurfaceArea(PSA)
A perl script was written to combine the PSA script (available at
http://www.perlmol.org/examples/polar_surface_area/)withthechemicalformulaandbond
calculation.
2.6Design ofLigEvolver
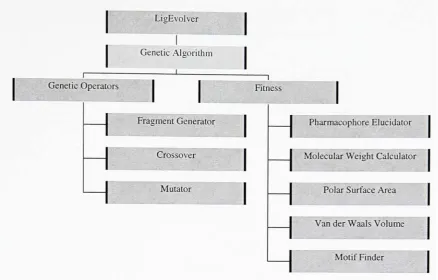
This section gives adetaileddescription oftheindividual components ofLigEvolver. Each
LigEvolver
Genetic Algorithm
GeneticOperators
1
Fitness
Fragment Generator
Crossover
Mutator
Pharmacophore Elucidator
Molecular Weieht Calculator
Polar SurfaceArea
Van der Waals Volume
Motif Finder
Figure2.6.1 ComponentsofLigEvolver
2.6.1 Genetic Algorithm
A genetic algorithm (GA) is used to evolve the population in a random manner to
generatereasonablygoodindividuals. It is driven bytheconceptofDarwinianevolution.
Theuse of genetic algorithmsinthegeneration ofligands has been successfullyexplored
[3,4,5]. The main parts of the algorithm fall into two broad categories namely, Genetic
operators and Fitness function, which will be discussed in the following sections. The
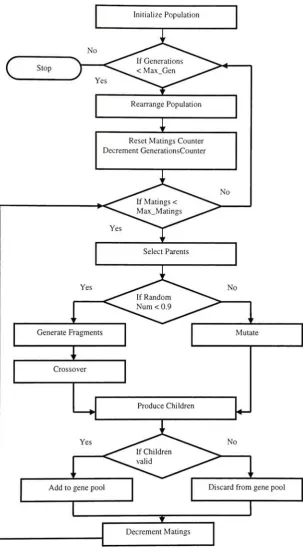
genetic algorithm is the main module. It comprises of functions to initialize the
population, mate individuals and generate new population. This is illustrated in fig.
[image:34.509.37.476.30.310.2]Reset Matings Counter Decrement GenerationsCounter
Select Parents
Yes No
Generate Fragments
T
Mutate
Crossover
ProduceChildren
Yes No
Addtogene pool Discard fromgene pool
[image:35.509.104.408.35.591.2]DecrementMatings
Theinitialpopulationisseeded in differentstyles.Theways ofinitialization are asfollows:
1. Initializewithknown HIVinhibitors
2. Initializewithrandominhibitors
3. Initializewitha mixtureofknownand randominhibitors
For each generation, the population is rearranged according to the fitness function and
only afixed number of individuals is retainedto contribute towards evolution while the
restarediscarded.
2.6.1.1 Genetic Operators
The genetic operators are themechanisms through which theindividuals in a population
are manipulated and changed. If evolution were viewed as a reaction, then genetic
operators can be considered catalysts. Two dominant operators observed in many
applications are the Crossover and Mutation operator. Depending on the need of the
problem customized operators may also be included as is the case in this study. The
operators usedin LigEvolver are, GenFrag,CrossoverandMutator.
2.6.1.1.1 GenFrag
The functionality of this operator is to divide the input SMILES string into meaningful
fragments. This operator was introduced because random segmentation ofthe SMILES
string maygive riseto chemically impossibleor meaninglessindividuals. Thenumbersin
Original String
CC(C)CN(CC(0)C(Cclcccccl)
NC(=0)OC2CCOC2) (=0)c3ccc(N)cc3Fragmentsafter applicationofGenFrag
The SMILES string isinitiallycutinto fragments.
<> <
CC(C)CN(CC(0)C(Cclcccccl)
NC(=0)OC2CCOC2) (=0)c3ccc(N)cc3< > < ?
Arandom pointofconcatenationischosentogive rise to twostringstoserveashead and
tail. Inthis casethe concatenation pointis taken tobetwo. So thestart ofthefragment is
combined with the second fragment for the head and the rest of the fragments are
combinedtogether toformthe tail.
Fragment One (Serves asthehead fortheCrossoveroperator)
CC(C)CN(CC(0)C(Cclcccccl) NC(=0)OC2CCOC2)
Fragment Two (Servesasthe tailfortheCrossoveroperator)
(=0)c3ccc(N)cc3
Figure 2.6.1.1.1.1 ExampleofGenFrag
Overlapand duplicity checks areperformed withinthe GenFrag,to avoidredundancy in
the gene pool. The fragments to be combined for the head and the tail portion are
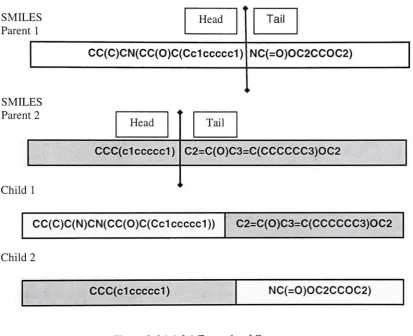
2.6.1.1.2 Crossover
The crossover operator is used to mate the parents. The GenFrag operator returns two
fragmentsthataretheheadandtailofanindividual. Crossoverstakeplace90%ofthe time.
Thecrossover operator works as shownin figure 2.6.1.1.2.1.
SMILES
Parent 1
Head
CC(C)CN(CC(0)C(Cc1ccccd)
Tail NC(=0)OC2CCOC2) SMILES Parent 2 Head CCC(c1 ccccd) Child 1 Tail C2=C(0)C3=C(CCCCCC3)OC2
CC(C)C(N)CN(CC(0)C(Cc1ccccd)) C2=C(0)C3=C(CCCCCC3)OC2
Child2
[image:38.509.48.462.145.481.2]CCC(d ccccd) NC(=0)OC2CCOC2)
Figure 2.6. 1.1.2.1ExampleofCrossover
2.6.1.1.3 Mutator
Mutations have played an important role in the evolution of living organisms. These
operators cause variations in the gene pool. They can be both beneficial and harmful,
leads tonoadaptabilityin thepopulation whereas levelsof mutationsthataretoohigh lead
togeneticbreakdown as shownin fig. 2.6.1. 1.3[5].
Off*- DNA
myiases -*On
Mutation rate
Evolutionary Immediate
death death
[image:39.509.115.379.99.388.2]{no adaptability) (genetic breakdown)
Figure 2.6.1.1.3 Mutationrates and geneticadaptability [5]
InLigEvolver,mutationstakeplace 10%ofthe time.
2.6.1.1.3.1 Amino Acid Insertion
Random places of insertion are chosen and an amino acid is included. The choice ofthe
Amino Acid SMILES String
Alanine NC(C)C(0)=0
Arginine NC(=N)NCCCC(N)C(=0)0
Asparagine NC(=0)CC(N)C(=0)0
Asparatic Acid OC(=0)CC(N)C(=0)0
Cysteine NC(CS)C(=0)0
Glutamic Acid C(CC(=0)N)C(C(=0)0)N
Glutamine NC(CCC(=0)N)C(=0)0
Glycine C(C(=0)0)N
Histidine NC(Cc 1 cnc[nH]1)C(=0)0
Isoleucine CCC(C)C(N)C(=0)0
Leucine CC(C)CC(N)C(=0)0
Lysine NCCCCC(N)C(=0)0
Methionine C(N)(C(0)0)CCSC
Phenylalanine NC(Cclcccccl)C(=0)0
Proline C1CCNC1C(=0)0
Serine NC(CO)C(=0)0
Threonine CC(0)C(N)C(=0)0
Tryptophan NC(Cc1cc2ccccc2[nH]1 )C(=0)0
Tyrosine NC(Cc1ccc(0)cc1 )C(=0)0
Valine CC(C)C(N)C(=0)0
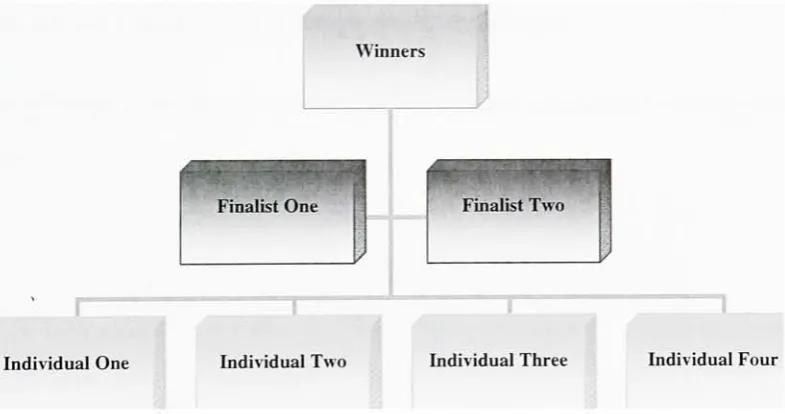
2.6.1.2 Parent Selection
Parent selection is another important design factor for a genetic algorithm. There are
various selectionalgorithms and theone employed inthisstudy istournament selection. In
thismethodtwogroupsoffour individuals areselectedrandomly.The one withthehighest
fitness score is selected fromeach group. These two individuals then serve as the parents.
In case of atie with respect to fitness score, the parentischosen randomly. Thisselection
approachfollowstheconcept of atournamentwithteamscompetingtoget selected. Finally
thefitness scoreisthedeterminantofthewinners.
FinalistOne Finalist Two
[image:41.509.54.447.284.491.2]Individual One IndividualThree Individual Four
2.6.2 Pharmacophore Elucidator
This module is used to extract the pharmacophore from a SMILES string. The
pharmacophoric features include aromatic rings, hydrogen bond acceptor, hydrogen bond
acceptors/donors,hydrophobicregions,positivechargecenters and negative charge centers.
2.6.2.1 Aromatic Rings
Aromaticrings are a circulararrangement ofatoms thatcanberepresentedwith alternating
single and double
-bonds. It is aconjugated system oflone pairs, unsaturatedbonds and
empty orbitals that put together form a strong stabilization [36]. In chemical terms,
aromatic rings contain(4n+2)IIelectrons,where nisaninteger.
The following arethe chemical compounds thatwouldbe detectedby the tool as aromatic
rings.
o
Figure2.6.2.1.1 Aromatic Structure I
- ABenzenering*
*
Drawinghas beengeneratedusingSmi2Depict developedby UniversityofCalifornia,
2.6.2.2HydrogenBondAcceptor
Hydrogenatomsthatarebound toelectronegative atoms such as oxygen or nitrogen can
interactwith lonepairstoformadditionalbonding [39]. The lonepairsthatinteractwith
thehydrogenatomaretermed ashydrogen bondacceptors.
Thefollowing arethe chemical compoundsthatwouldbedetected bythe toolashydrogen
bondacceptors.
Figure 2.6.2.2.1 Hydrogen Bond Acceptor Structure I
=
0
Figure 2.6.2.2.2 Hydrogen Bond Acceptor Structure II
O
N
Figure 2.6.2.2.4 Hydrogen Bond Acceptor StructureIV
2.6.2.3 Hydrogen Bond Acceptor/Donor
A groupthatcaneither acceptordonate hydrogen bonds is identifiedas hydrogenbond
acceptor/donor.
Thefollowingarethechemical compounds thatwould be detectedbythe tool as hydrogen
bond acceptors/donor. This is not an exhaustive list, but contains a few of the common
bondsthatoccurinHIV inhibitors.
-OH
Figure 2.6.2.3.1 Hydrogen bond A/D Structure I
-NH2
Figure 2.6.2.3.3 Hydrogen bondA/D StructureIII
2.6.2.4 Hydrophobic Regions
Theregions thatrepel water aretermedhydrophobicregions.
The following are the chemical compounds that would be detected by the tool as
hydrophobicregions.
HaC ^ CHa
-N H
Figure 2.6.2.4.1 Hydrophobic StructureI*
-CHa
HaC
Figure2.6.2.4.2 Hydrophobic StructureIP
/s.
o
o
Figure 2.6.2.4.3 Hydrophobic Structure IIP
CH,
HaC-
//
o
Figure 2.6.2.4 .4Hydrophobic Structure
TV*
c
CH-,
CHn
c
Figure 2.6.2.4.6HydrophobicStructureVI*
A
O
Figure 2.6.2.4.6 Hydrophobic StructureVII*
*
Drawings have beengeneratedusing Smi2Depict developedby UniversityofCalifornia,
Irvine [44]
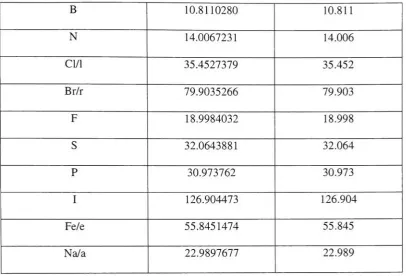
2.6.3 Molecular Weight Calculator
As the name indicates, the molecular weight calculator computes the molecular weight
giventhe chemicalformula. The molecular weight values (round
-offweight) usedbythe
toolis shown inthe tablebelow.
Molecule Weight Round- off weight
C 12.0110369 12.011
H 1.0079759 1.007
B 10.8110280 10.811
N 14.0067231 14.006
Cl/1 35.4527379 35.452
Br/r 79.9035266 79.903
F 18.9984032 18.998
S 32.0643881 32.064
P 30.973762 30.973
I 126.904473 126.904
Fe/e 55.8451474 55.845
[image:48.509.47.454.40.316.2]Na/a 22.9897677 22.989
Table 2.6.3.1 Molecularweights
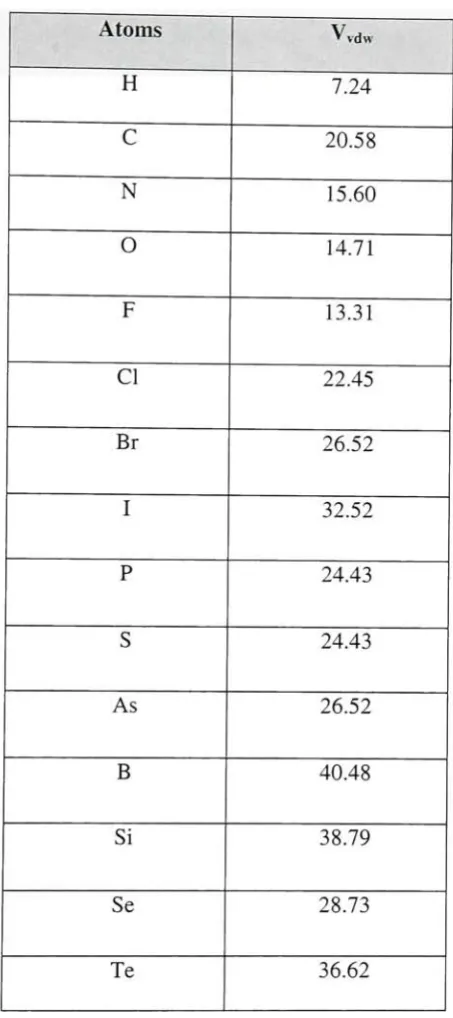
2.6.4 Van DerWaalsVolume
Van derWaalssurfacesdefinetheclosest contactthatatomsina molecule could make with
one another. Hence it acts as anindicatorof atompacking in a molecular structure. Italso
has animportantrolein establishingthe specificityofinteractions betweenproteinbinding
pockets andligands [36].
Zhao et alhave described a methodforthecalculation of vanderWaals volumeusing the
formula, V(vdW) = summation operator all atom contributions 5.92N(B) 14.7R(A)
3.8R(NR) (N(B) isthe numberofbonds,R(A) is thenumber of aromaticrings, andR(NA)
Table 2.6.4.1 givesthevalues oftheatoms andtheirvolumes.
Atoms vdw
H 7.24
C 20.58
N 15.60
O 14.71
F 13.31
CI 22.45
Br 26.52
I 32.52
P 24.43
S 24.43
As 26.52
B 40.48
Si 38.79
Se 28.73
Te 36.62
2.6.5 Polar Surface Area
Polar Surface Area(PSA) is a descriptorusedto indicate the bioavailability ofdrugs. The
surface belonging to polar atoms correlates with the transport of molecules through
membranes. Ertl et al proposed an approach for the calculation of PSA based on the
summation of surface contributions of polar fragments. PerlMol
uses this method and
provides aperl scripttocalculatethePSA.
2.6.6 Motif Finder
Themotiffinder isresponsiblefor observingcommon patternsbetweena set ofstrings.It is
used to derive structural similarities between compounds through the longest common
substringpresentintheirSMILES string.
The structures ofthe various FDA (U.S. Food and Drug Administration) approved drugs
forHIVprotease were compiledintheformofSMILES strings. These werethen matched
withtherecentlygeneratedligands todetermine iftheyhave acommon substringoflength
greaterthan 9.Table2.5.6.1 isthelistofdrugsthathas beentakenintoconsideration.
Brand Name GenericName Manufacturer Name
Agenerase Amprenavir GlaxoSmithKline
Aptivus Tipranavir BoehringerIngelheim
Crixivan indinavir, IDV, MK-639 Merck
Fortovase Saquinavir Hoffmann-LaRoche
Kaletra lopinavirand ritonavir Abbott Laboratories
Lexiva FosamprenavirCalcium GlaxoSmithKline
Norvir ritonavir,ABT-538 Abbott Laboratories
Reyataz Atazanavirsulfate Bristol-Myers Squibb
Viracept nelfinavirmesylate, NFV Agouron Pharmaceuticals
Table 2.6.6.1 FDA
-Approved ProteaseInhibitors*
*Datatakenfrom FDAsite [42]
2.6.7 Fitness Computor
The fitness function isusedtoevaluatethestrength and robustness of anindividualelement
in the population. The fitness is calculated for every entity and it determines if the
individual needs to beretained or eliminated.In a way, thefitness function biases thepath
of evolution.Thefollowingisthefitness function.
F=Ar+Hd+Ha+Hy+Mw +Ps+Vd+Sm
Where,
Ar- Aromatic
RingCountScore
Hd- Hydrogen Bond DonorScore
Ha- Hydrogen Bond Acceptor Score
Hy- Hydrophobic Region CountScore
Mw- MolecularWeight Score
Ps- PolarSurface AreaScore
Sm
-Structural Motif Score
For each of the above mentioned features scores were assigned according to their
resemblance to the existing known inhibitors with the lowest score
being the minimum
matching score and the highest being the maximum match. Ifthere are no matches to the
known inhibitor feature count then no points are assigned. A list of the known existing
inhibitors was made and theirfeature pattern was analyzed manually to obtain the scoring
scheme.This list has beenincludedas appendixII.
2.6.7.1 Aromatic
Ring
CountThe Aromatic ring is one of the pharmacophoric features that are vital components in
determining the way the inhibitors interact with the enzyme. Table 2.6.7.1.1 gives the
scoringschemeforthearomaticringcount.
Aromatic RingCount Score
>4and <7 1
>1 and<4 2
==4 3
Table 2.6.7.1.1 AromaticRingCountScoringScheme
2.6.7.2 Hydrogen Bond Donor Count
Acount ofthehydrogen bond donors istaken andscoresareassignedaccordingto table
Hydrogen Bond Donor Count Score
>1 and <5 1
>7 2
>4 and <8 3
Table 2.6.7.2.1 Hydrogen Bond Donor CountScoringScheme
2.6.7.3 HydrogenBond Acceptor Count
Acount ofthehydrogen bond acceptorsistakenandscoresare assignedaccordingto table
2.6.7.3.1.
Hydrogen BondAcceptorCount Score
>1 and<5 1
>7 2
>4and<8 3
Table 2.6.7.3.1 Hydrogen Bond Acceptors CountScoring Scheme
2.6.7.4 HydrophobicRegion Count
A countofthehydrophobicregionsistaken and scoresareassignedaccordingto table
2.6.7.4.1.
Hydrophobic RegionCount
>1 and<5
>7
>4 and <8 3
Table 2.6.7.4.1 Hydrophobic Regions CountScoringScheme
2.6.7.5 Molecular Weight
Scores are assigned according to therange of molecular weights for each compound. The
range andtheircorrespondingscorearesummarizedintable2.6.7.5.1.
Molecular Weight (Daltons) Score
<500 1
>600and<806 2
>500and<600 3
Table 2.6.7.5.1 Molecular WeightScoringScheme
2.6.7.6 Polar Surface Area
The ideal polar surface range has been takenfromthe list ofknown inhibitors and scores
are attributedto anewlygeneratedindividualif itspolarsurface areafalls withintherange.
The detailsregardingtherange andthe scores are givenintable2.6.7.6.1.
PolarSurface Area(Angstroms)
>150and<200
>100and<150
Score
2.6.7.7 Van DerWaals Volume
Van derwaals volume is another descriptorthat isused to assign scores to thepopulation
units.Thevolume range andtherespective scorehavebeenshownintable2.6.7.7.1.
Van Der Waals Volume(Angstroms3) Score
>400and<750 1
>800and<900 2
>900 and<1000 3
Table2.6.7.7.1 Van DerWaalsVolumeScoring Scheme
2.6.7.8 Structural Motif
Thestructuresofthegovernment approveddrugswere comparedto thenew ligandtoseeif
they include similarities. The presence of a significant resemblance gives a score of one
3.
Results
LigEvolver was run using the data from various public databases. The results obtained
are discussed inthis section. Unless otherwise specified theentire chemical figures have
been generatedusing CS ChemDraw Pro.
3.1 Simulation I
Theinhibitorgenerationwas done usingtheparameters shownintable3.1.1.
Property Value
Database ChemPDB
InitialPopulation Size 4009
NumberofMatingsperGeneration 150
NumberofGenerations 100
Populationsize perGeneration 500
Table 3.1.1 Simulation I Properties
3.1.1 Significant Results
A fewnoteworthy compoundsthatwere generatedduringthissimulationarediscussed in
3.1.1.1Generation#100
3.1.1.1.1 Compound1
SMILES string
C(=0)NC(Cc3ccccc3)C(NC(=0)C(N)c3ccccc3)C(=0)N2C
1 CC 1=CN(C2)ccccCC(C)CNc2cccc3CC(C)CNc23
[image:57.509.157.356.182.463.2]HoC
Figure 3.1.1.1.1.1 SimulationI- Generation 100- CompoundI*
3.1.1.1.1.2 Compound II
SMILES String
NC(=0)clc[n+](cccl)C(NC(=0)C(N)c3ccccc3)C(=0)N2ClCCl=CN(C2)cccc3CC(C)C
Nc2cccc3CC(C)CNc23
Reconstruction
Elements underlined,eitherin the original SMILES stringor in thereconstructed string,
signify the portion ofthe SMILES string that is modified. This convention is followed
throughout theremainingsections.
NC(=0)c1c[n+](ccc1)C(NC(=0)C(N)c3ccccc3)C(=0)N2C1CCl=CN(C2)ccccCC(C)CN
c2cccc3CC(C)CNc23
^ /Nfl /N^
[image:58.509.153.358.323.621.2]HoC
*Figuregeneratedusing ChemSketch [45].
3.1.1.2Generation#85
3.1.1.2.1 CompoundI
SMILESString
COC(=0)C(Cc1ccccc1)CCC1=C(C)C(=0)NC1
C(0)CN(CCC4CCC50COC5C4)C(=0)
[image:59.509.128.380.248.613.2]CCN6C(=0)c7ccccc7C6
Figure 3.1.1.2.1.1 SimulationI-Generation 85
3.1.1.2.2 CompoundII
SMILES String
CC(C)C(NC(C)=0)C(=0)NC(Cc1ccccc1 )C(0)CN(CCC4CCC50COC5C4)C(=0)CCN6
C(=0)c7ccccc7C6
Figure3.1.1.2.2.1 SimulationI
[image:60.509.97.411.191.549.2]3.2 Simulation II
Property Value
Database KEGG
Initial Population Size 10005
NumberofMatingsperGeneration 100
NumberofGenerations 45
Populationsize perGeneration 500
Table 3.2.1 Simulation II Properties
3.2.1 Significant Results
A fewnoteworthycompounds thatwere generatedduringthis simulation arediscussedin
3.2.1.1 Generation #40
3.2.1.1.1 Compound 1
SMILES String
CCC(C)C(N(C)C)C(-0)NClC(Oc2ccc(cc2)C(0)CNC(=0)C(Cc3ccccc3)NCl)CC(=0)N
[image:62.509.113.401.220.598.2]C1C(0)CC(0C1)
Figure 3.2.1.1.1.1 Simulation II- Generation40
3.2.1.1.2Compound II
SMILES String
CC(C)CC(NC(=0)C(NC(=0)NC(Cc1ccccc1)C(Oc4ccc(C=CNC(=0)C(Cc5ccccc5)NC3
=0)cc4)c6ccccc6
Reconstruction
CC(C)CC(NC(=0)C(NC(=0)NC(Cclcccccl))C(Oc4ccc(C=CNC(=0)C(Cc5ccccc5)NC=
0)cc4)c6ccccc6)
3.2.1.1.3 Compound III
SMILES String
NC(=0)OC2CCOC2cccc3CC(C)CNc2cccc3CC(C)CNc2cccc3CC(C)CNc2cccc3CC(C)C
Nc23
Reconstruction
NC(=0)OC2CCOC2cccc3CC(C)CNc2cccc3CC(C)CNc2cccc3CC(C)CNc2cccc3CC(C)C
[image:64.509.151.373.239.551.2]Nc2
Figure 3.2.1.1.3.1 SimulationII- Generation40- CompoundIIP
3.2.1.1.4 Compound IV
SMILESString
CC(C)(C)NC(=0)C1 CN(CCN 1 )COC(=0)CC(0)(CCC(C)(C)0)C(=0)OC 1C2c3cc40C
[image:65.509.58.409.166.376.2]Oc4cc3CCN5CCCC25C=C1
3.2.1.2 Generation #41
3.2.1.2.1 Compound I
SMILES String
[nH]c(C=C4NC(=0)C(C)=C4)C(=0)N2CCCC20c2ccc(C=CNC(=0)C(Cc3ccccc3)NC(=
0)ClNC(=0)C4CCCN4C)cc2
Reconstruction
[nH]c(C=C4NC(=0)C(C)=C4)C(=0)N2CCCC20c2ccc(C=CNC(=0)C(Cc3ccccc3)NC(=
[image:66.509.146.376.285.582.2]0)CNC(=0)C4CCCN4C)cc2
Figure 3.2.1.2.1.1 SimulationII- Generation41
-CompoundP
3.3 Simulation III
Property Value
Database MSD
Initial Population Size 6433
NumberofMatingsperGeneration 100
NumberofGenerations 100
Population sizeperGeneration 500
Table 3.3.1 SimulationmProperties
3.3.1 Significant Results
A fewnoteworthycompoundsthatwere generatedduringthis simulation arediscussedin
3.3.1.1 Generation #100
3.3.1.1.1 Compound1
SMILES String
CN(C)C(C 1 CCNC1C(=0)Oc1ccccc1)C(=0)N2CCCC2C(=0)NC3C(Oc4ccc(C=CNC(= 0)C(Cc5ccccc5)NC3
Reconstruction
CN(C)C(C1CCNC1C(=0)0c1ccccc1)C(=0)N2CCCC2C(=0)NC3COcccc(C=CNC(=0)
C(Cc5ccccc5)NC3c4))
[image:68.509.192.324.256.534.2]^^
Reconstruction
[image:69.509.144.374.140.347.2]CN(C)C(C 1 CCNC1C(=0)Oc1ccccc1)C(=0)N2CCCC2C(=0)NC3C(Oc4ccc(C=CNC(= 0)C(Cc5ccccc5)N)C34)
Reconstruction
CN(C)C(ClCCNClC(=0)Oclcccccl)C(=0)N2CCCC2C(=0)NC3C(Oc4cccC=C4iNC(= 0)C(Cc5ccccc5)NC3
Figure3.3.1.1.1.3 Simulation HI- Generation 100- Compound I(c)*
*"
[image:70.509.208.304.146.453.2]3.3.1.1.2 Compound II
SMILES String
NC(CS)C(=0)OC2(0)C3C(OC)C1 (COC(=0)c6ccccc6)C45C(CCC8(C0C(=0)c6)C(=0)
NCC(=0)N(C)C(Cc2ccccc2)
Reconstruction
NC(CS)C(=0)OC2(0)C3C(OC)Cl(COC(=0)c6ccccc6)C23CCCC(COC(=0)cl)C(=0)N
[image:71.509.108.410.201.555.2]CC(=0)N(C)C(Cc2ccccc2)
Figure 3.3.1.1.2.1 SimulationIII- Generation 100- Compound II
Reconstruction
NC(CS)C(=0)OC2(0)C3C(OC)Cl(COC(=0)c6ccccc6)C23C(CCClCOC(=0)c)C(=0)N
CC(=0)N(C)C(Cc2ccccc2)
\^<*
[image:72.509.206.309.130.285.2]o
Figure 3.3.1.1.2.2 SimulationIII
-Generation 100- Compound II(b)*
*Figure has beengeneratedusing Smi2Depict developedby UniversityofCalifornia,
Irvine [44].
3.3.1.1.3 Compound III
SMILES String
CCCl=C(C)CN(C(=0)NCCc2ccc(cc2)S(=0)(=0)NC(=0)NC3CCC(C)CC3)C 1OCC1=C NC(=0)NClCc5cccnc5
3.4Simulation IV
Property Value
Database ChemBank
Initial Population Size 2344
NumberofMatingsperGeneration 100
NumberofGenerations 100
Populationsize perGeneration 300
Table 3.4.1 SimulationIV Properties
3.4.1 Significant Results
A fewnoteworthycompoundsthatwere generatedduringthis simulation arediscussedin
3.4.1.1 Generation #100
3.4.1.1.1 Compound I
SMILES String
NC(=0)C2CCCCN2C(=0)C(Cc3ccccc3)NC(=0)C(Cc4ccccc4)NC1 CN1CCN(CC 1)CC(
C)ClNC(=0)C(Cc2)
Reconstruction
NC(=0)C2CCCCN2C(=0)C(Cc3ccccc3)NC(=0)C(Cc4ccccc4)NC 1 CN1CCN(CC1)CC( C)C1NC(=0)C(C)
3.4.1.1.2Compound II
SMILES String
CN 1 CCN(CC1)CC(C)C 1NC(=0)C(Cc2ccccc2)NC(=0)C(CCCCCC(=0)NO)NC(=0)C3 CCCN3Clc5ccccc35
Reconstruction
CN1CCN(CC1)CC(C)C1NC(=0)C(Cc2ccccc2)NC(=0)C(CCCCCC(=0)NO)NC(=0)C3 CCCN3Clc5ccccc5
[image:75.509.51.463.218.449.2]3.5 Simulation V
Property Value
Database HIV_PDB
Initial Population Size 71
NumberofMatingsperGeneration 100
NumberofGenerations 100
Population size perGeneration 150
Table 3.5.1 Simulation V Properties
3.5.1 Significant Results
Afewnoteworthycompoundsthatwere generatedduringthis simulationare discussedin
3.5.1.1 Generation #12
3.5.1.1.1 Compound I
SMILESString
CC(C)(C)NC(=0)C1CC2CCCCC2CN 1 3=CN[C@](C [C @H] (O)[C@H](Cc4ccccc4)NC(
=0)0[C@H]5CCOC5)(Cc6ccccc6)C3
Reconstruction
CC(C)(C)NC(=0)ClCC2CCCCC2CN13=CN[C](C[C@H](0)[C@H](Cc4ccccc4)NC(=
0)0[C@H]5CCOC5)(Cc6ccccc6)C3
[image:77.509.111.405.233.505.2]
3.5.1.2 Generation #10
3.5.1.2.1 Compound I
SMILESString
CC 1CCC(0)C 1 CC(C)(C)NC(=0)C1CC2CCCCC2CN1C(0)C(0)C(OCc4ccccc4)C(=0)
[image:78.509.119.401.172.492.2]NCc5c(F)cccc5
3.5.1.3Generation #9
3.5.1.3.1CompoundI
SMILES String
(CNC(=0)c4ccccc3)CN(Cc2ccccc2)C(=0)COc3c(C)cccc3NC(=0)C(CC(N)=0)NC(=0)
c4ccc5ccccc5n4
Reconstruction
(CNC(=0)c4ccccc4)CN(Cc2ccccc2)C(=0)COc3c(C)cccc3NC(=0)C(CC(N)=0)NC(=0)
c4ccc5ccccc5n4
Figure 3.5.1.3.1.1 SimulationV
-Generation 9 - CompoundI*
*Figure has been generatedusing Smi2Depict developedbyUniversityofCalifornia,
Irvine [44],
3.6 Simulation VI
Property Value
Database HIVNCI
Initial PopulationSize 42689
NumberofMatingsperGeneration 100
PopulationsizeperGeneration 1000
Table 3.6.1 SimulationVI Properties
3.6.1 Significant Results
3.6.1.1 Generation #199
3.6.1.1.1 Compound I
SMILES String
COC(=0)C(0)C(0)(CCC(C)C)C(=0)OClC2c3cc40COc4cc3CCN5CCCC25C=ClCCN
5CCCC25C=ClN=Nc5ccccc5
Reconstruction
COC(=0)C(0)C(0)(CCC(C)C)C(=0)OC1C2c3cc40COc4cc3CCN5CCCC25C=C1 CCN
[image:80.509.53.396.367.574.2]5CCCC5C=CN=Nc5ccccc5
3.7Fitness Behavior
It was noted that the fitness steadily increased with the number of generations. After a
certain number ofgenerations the fitness values approached an upper limit and this value
remained constant through the subsequent generations. Figure 3.7.1 shows the fitness
behaviorchart across twosimulations.
AverageFitness
4. Discussion
The approachusedinthisstudy does nottakeintoaccounttheentirecomplexitiesinvolved
in ligand design. It is limited due to the non - incorporation ofthe 3D features. The main
objective of this research was to investigate if scalar properties such as those used in the
fitness function would derive promising ligand structures. Another aspect was to
experiment withtheapplicationof geneticalgorithmsto thedomain ofligandgeneration.
From the results it can be inferred that the tool generated ligands that were hopeful and
novel by nature. It also proved that genetic algorithm is an efficient computational
approach towards solvingproblems ofthis nature. Certain compounds that were produced
by LigEvolver had striking similarities to existing known inhibitors. A few of those are
[image:82.509.124.435.357.633.2]presentedhere.
(a) 1NPW (b) 1C6S
-CO
[image:83.509.126.381.307.629.2](c) 1W5X
Figure 4.2 SimilaritiesofCompoundIwithknown inhibitors[43]
\
o
l
UN'
>- HO-M.
y^,.
r
i.-.UH
\
V^
'y
-rjH
c^-/Sr\.
Figure4.4Similaritiesof compoundIIwithknowninhibitor 1D4K[43]
[image:84.509.60.459.285.516.2]?-,
(a)2A4F (b) 1HWR
Figure 4.6 SimilaritiesofcompoundIIItoknown inhibitors [43]
Afew compounds were generatedthathadatotallynew structurethan theonesalready
provedtobe inhibitors. Onesuch compoundfromthis classof resultis shownin Figure
4.7.
Figure 4.7LigEvolver CompoundIV*
LigEvolverCompound # InputDatabase
I Protein Data Bank(onlyHIVinhibitors)
II KEGG
III ChemBank
TV KEGG
5. Conclusion
LigEvolversucceededin generatingnew ligands usingtheminimalist approach. Theresults
have beenpromising, but given thenature oftheproblemthere is nocomputational wayof
verifyingthesolutions.Aworthwhilefutureenhancement wouldbetoimprovisethefitness
function by incorporating the 3D properties. Despite some unresolved issues, this tool
provides an extensible first step towards generation ofinhibitorsthatpossess thepotential
Bibliography
Paper References
1. David Weininger. (1987) SMILES, a chemical language and information system.
1. Introduction to Methodologyand EncodingRules. J. Chem. Inf. Comput. Sci.,
28,31-36.
2. Schneider and Fechner. (2005) Computer - based de
novo design ofDrug - like
molecules. Nature, 4,649- 663.
3. Glen and Payne. (1994) A genetic algorithm for the automated generation of
molecules within constraints. Journal ofComputer - Aided
molecular design, 9,
181 202.
4. Douguet, Thoreau and Grassy. (2000) A genetic algorithm for the automated
generation of small organic molecules: Drug design using an evolutionary
algorithm. JournalofComputer- AidedMolecular
Design, 14,449- 466.
5. Genetic algorithm for the design of molecules withdesired properties. Journal of
Computer- Aided
moleculardesign, 16(2002).
6. Miroslav Radman. (1999) Enzymes of evolutionary change. Nature, 401, 866
-869.
7. Zhao, Abraham and Zissimos. (2003) FastCalculation of van der Waals volume
as asum of atomic andbondcontributions anditsapplicationto drugcompounds.
JournalofOrganicChemistry, 68,7368- 7373.
8. Wlodawer and Vondrasek. (1998) Inhibitors of HIV - 1 Protease: A Major
Success of Structure - Assisted
Drug Design. Annual Review of Biophysical
9. Draghici and Potter. (2003)
Predicting
HIV drugresistance with neural networks.Bioinformatics, 19,98- 107.
10. Tossi et al. (2000) Aspartic protease inhibitors: An integrated approach for the
design and synthesis of diaminodiol - based
peptidomimetics. Eur. J. Biochem,
267, 1715-1722.
11. Gohand Foster.EvolvingMolecules for
DrugDesign using Genetic Algorithms.
12. You, Garwicz and Rognvaldsson. (2005) Comprehensive Bioinformatic Analysis
ofthe SpecificityofHuman ImmunodeficiencyVirus Type 1 Protease. Journal of
Virology,79, 12477- 12486.
13. Fernandez et al. (2004) Inhibitor designby wrapping packing defects in HIV - 1
proteins. PNAS, 101, 11640- 11645.
14. Akaho et al. (2001) A Study on Docking Mode of HIV Protease and Their
Inhibitors. J. Chem.Software, 7, 103- 1 14.
15. Jones et al. (1997) Development and Validation of a Genetic Algorithm for
FlexibleDocking.J. Mol. Biol.267, 727- 748.
16. Head et al. (1995) VALIDATE: A New Method for the Receptor
-Based
PredictionofBindingAffinitesofNovel Ligands. J. Am. Chem. Soc, 118, 3959
-3969.
17. Griffith et al. (2004) Combining structure
-based drug design and
pharmacophores.Journal ofMolecularGraphics andModelling, 23,439- 446.
18. Jones,WillettandGlen. (1995) Ageneticalgorithmfor flexiblemolecularoverlay
and pharmacophore elucidation. Journal ofComputer
-Aided Molecular Design,
19. Xue and Bajorath. (2000) Molecular Descriptors in Cheminformatics,
Computational Combinatorial Chemistry, and Virtual Screening. Combinatorial
ChemistryandHighThroughputScreening,3, 363- 372.
20. Becketal.(2002) DefiningHIV
-1 Protease Substrate Selectivity. CurrentDrug
Targets
-InfectiousDisorders, 2, 37- 50.
Books
21. Baker, B.R. (1967). Design of Active - Site
-Directed Irreversible Enzyme
Inhibitors. New York: JohnWileyandSons.
22. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P. (2002).
MolecularBiologyoftheCell.(FourthEdition).New York: Garland.
23. Gasteiger, J., & Engel, T., (Eds.). (2003). Chemoinformatics. New York: John
WileyandSons.
24. Mitchell,M. (1996).AnIntroduction toGenetic Algorithms. Cambridge,MA: The
MIT Press.
25. Copeland, R. (2000).Enzymes:APractical IntroductiontoStructure, Mechanism,
andData Analysis. (SecondEdition). NewYork: Wiley-VCH.
26. Kubinyi, H., & Muller, G. (2004) Chemogenomics in Drug Discovery: A
MedicinalChemistryPerspective. New York: JohnWileyandSons.
Web References
27. Ligand.Info: Small-Molecule Meta-Database, http://ligand.info/, accessed as on
17th
28. Pharmacophore Identification and the Unknown Receptor Problem,
http://cnx.rice.edu/content/mll538/latest/, accessed as on
7th
Dec2005.
29. 3DPharmacophoreSearching,
http://www.netsci.org/Science/Cheminform/feature02.html, accessed as on
7th
Dec2005.
30. Survivalofthefittest inDrugDesign,
http://pubs.acs.org/subscribe/iournals/mdd/v03/i09/html/felton.html,accessedas
on7thDec 2005.
31. Pharmacophore Perception by Molecule Alignment,
http://www2.chemie.uni-erlangen.de/projects/sol/drugdesign.html,accessedason7thDec 2005.
32. GeneticAlgorithmsandEvolutionaryComputation,
http://www.talkorigins.org/faqs/genalg/genalg.html, accessed as on
7l
Dec 2005.
33. SMILES Home Page, http://www.daylight.com/smiles/, accessed as on
7th Dec
2005.
34. California LutheranUniversity,
http://www.clunet.edu/BioDev/omm/hivrt/hivrt.htm,accessedas on28 Jan 2006.
35. California LutheranUniversity,
http://www.clunet.edu/BioDev/omm/hiv_protease/molmast.htm,accessedas on
28th
Jan 2006.
36. Rutgers,DepartmentofChemistryandChemicalBiology,
http://rutchem.rutgers.edu/content dynamic/faculty/edward arnold.shtml,
accessedason
28th
37. Research ofJoannaTrylska, http://www.trvlska.com/research.html.accessedas on
28th
Jan 2006.
38. Wikipedia, http://en.wikipedia.org/wiki/. accessed ason28th
Jan 2006.
39. ThemoleculesofHIV,http://www.mcld.co.uk/hiv/?q=HIV%20genome,accessed
as on 16thMay 2006.
40. BiologyofHIV ProteaseInhibitors,
www.unc.edu/~brybar/evolutionmodule/Biologv%20of%20HIV%20Protease%20
Inhibitors.doc. accessed ason
16th
May2006.
41. UniversityofCalgary,
http://www.chem.ucalgary.ca/courses/351/Carey5th/Ch27/ch27-4-4.html
accessed on
10th
March 2006.
42. FDAApprovedDrugs,http://www.fda.gov/oashi/aids/virals.html,accessedas on
10th
March 2006.
43. Protein DataBank,http://www.rcsb.org/pdb/home/home.do,accessed ason 12th
October 2005.
44. ChemDB /Smi2Depict:Generate 2D Images fromMoleculeFiles,
http://cdb.ics.uci.edu/CHEM/Web/cgibin/Smi2DepictWeb.py, accessed as on 15th
December2005.
45. ACD/ChemSketch 10.0Freeware,
http://www.acdlabs.com/download/chemsk.html, accessedason 18th
November,
Appendix I
SmilesEncodingRules
1. Non
-hydrogen atoms are specified by their atomic symbol enclosed in square
brackets. The second letter of the two - letter
symbol must be entered in lower
case.
2. Elements inthe "organicsubset",B, C, N, O, P, S, F, CI,BrandI,may be written
without brackets if the number of attached hydrogens conforms to the lowest
normal valence consistentwith explicitbonds.
3. Elements notintheorganic subset mustbe described in brackets.
4. Attached hydrogens andformal charges are always specifiedinside brackets. The
numberof attachedhydrogens is shownbythe symbolHfollowed byan optional
digit(e.g., [NH4+]
-ammoniumcation).
5. Similarly, aformal chargeis shownbyone ofthe symbols +or-, followedby an
optional digit (e.g., [Fe+2] - iron
(II) cation). The form [Fe+++] is considered
synonymous withtheform[Fe+3].
6. Ifunspecified, the number of attached hydrogens and charges is assumed to be
zeroforan atominsidethebracket.
7. Atoms inaromatic rings are specifiedbylowercaseletters; e.g.,normal carbon is
representedbytheletterC,aromatic carbonbyc.
8. Singlebond isrepresentedbythesymbol
-9. Double bondisrepresentedbythe symbol=
10. Triplebond isrepresentedbythesymbol#
12. Singleand aromaticbonds maybe, andusuallyare, omitted.
13. Hydrogenscan beomittedfrom theSMILESnotation
Structure
CH2=CH-CH2-CH=CH-CH2-OH
Valid SMILES
c=ccc=cco
c=c-c-c=c-c-o
occ=ccc=c
14. Branchesare enclosed withinparentheses
Triothylamine
CH3
I
CH2
I
H2C CH2 N CH2 CH3
SMILES CCN(CC)CC
Isobutyricacid
CH3 O
I
II
H3C CH C OH
SMILES CC(C)C(=0)0
3
-propyl- 4
-isopropyl- 1
-heptene CH3 CH2
I
CH2 CH3 CH-CH3 H2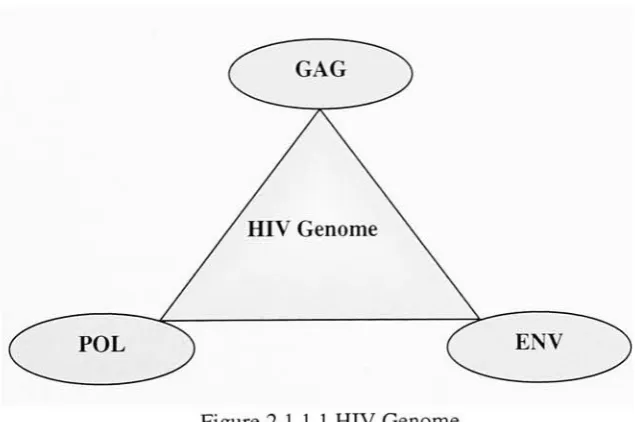


![Figure 2.6.1.1.3 Mutation rates and genetic adaptability [5]](https://thumb-us.123doks.com/thumbv2/123dok_us/120734.11628/39.509.115.379.99.388/figure-mutation-rates-and-genetic-adaptability.webp)