0022-538X/89/072893-08$02'.00/0
Copyright g 1989. American Society for Microbiology
A
Deletion Mutant of the
Latency-Associated Transcript of Herpes
Simplex
Virus Type
1
Reactivates from the Latent State with
Reduced Frequency
DAVID A. LEIB,'- CONNIE L. BOGARD,' MAGDALENA KOSZ-VNENCHAK2t- KAREN A. HICKS,3
DONALD M. COEN,3 DAVID M. KNIPE,- AND PRISCILLAA.
SCHAFFER'2*
Laboratory ofTumlizor Viruits Genethis, Dana-Farber CancerInstitute, Deparitntiielt ofBiological Chlmistry aci(i Molecul(ar
P11h1arnaologv, (aizdDepaiwittiieitofMicrobiology,aidI Moeculahr
Getie.s2
Htaatlrd Medi(cal School, Bostoni, MisstIsllesttus 02115Received 23 September 1988/Accepted 20 March 1989
We havegenerated andcharacterized a deletion mutant of herpes simplex virus type-1, dlLAT1.8, which lacks the putative promoter region, transcriptional start site, and 1,015 base pairs of the DNA sequences specifying the latency-associated transcripts (LATs). When tested in a CD-1 mouse ocular model, dlLATI.8 wasreplicationcompetent in the eye and in ganglia during acute infection but reactivated from explant cultures of gangliawith reducedefficiency (49%) relative to those of wild-type and marker-rescued viruses (94 and 85%, respectively) despite the fact that levels of mutant viral DNA in gangliaduring latent infection were comparable to wild-type levels. The neurovirulence of KOS was not significantly altered by the removal of sequences specifyingthe LATs, as judged bynumbers of animals dying on or before 30 days postinfection. Examination ofganglialatently infectedwithdlLAT1.8 by in situ hybridization revealed no LAT expression. The genotype of reactivated virus was identical to that of inputdlLAT1.8 virus asjudged by Southern blot analysis. These studiessuggest thatalthough the LATs are not essential for the establishment and reactivation of latency in our model, they may play a role in determining the frequency of reactivation of virus from the latent state.
Herpes simplex virus (HSV) exhibits two modes ofgene expressionwithin theinfected host. During the lytic phase of infection, viral genes are expressed in coordinate fashion. accordingtotheir temporal class: immediate early(IE or c), early (E or 3), orlate (L ory) (6. 7). In contrast, duringthe latent phase of infection, HSV type 1 (HSV-1) gene expres-sionisalmost completelyrepressed.Theonlyabundantviral gene products detected to date in the neuronal nuclei of latently infected sensory ganglia ofmice. rabbits, and hu-mans are the abundant latency-associated transcripts (LATs; 1, 20, 31). Similar patterns of latency-related RNA transcription have been reported during bovine herpesvirus type 1 latency in a rabbit model and pseudorabies virus latencyinswine(18, 19). TheHSV-1 LATs arederivedfrom the DNA strand opposite that which encodes the mRNA
specifying the IE regulatory protein ICPO andoverlaps 30%
of the 3' terminusof theICPOgene(Fig. 1).Northern(RNA) blotanalysis(27, 28, 31, 38) hasdemonstrated thatthe LATs consist of at least three transcripts of 2.0. 1.5. and 1.45 kilobases (kb), which are partially colinear.
Stevensetal. (31)proposedthat LATsfunctiontorepress the expression ofthe immediate-early transactivating gene product ICPO which if active would initiate productive infection. Thus, these investigators hypothesized, LAT expression is the
factor-
responsible forsuppressing produc-tive infection during latency. This theory, if correct, would mean that a virus lacking the sequences coding for LATs would be unable toestablish or maintain alatent infection. Thisisconsistentwiththeobservation thatdeletionmutants lacking sequences that code forICPO can establish latency but failto reactivate efficiently (12).*Correspondingauthor.
t
Permanent
address:Jagiellonian University.InstitLute
ofMolec-LllarBiology, Cracow. Polaind.
In the present study, we have begun to address the question of the function of the LATs by the isolation and characterization of a mutant specifically deleted in the se-quences which encode these transcripts. The mutant was tested in aCD-1 mouse eye model, and its behavior during acute infection was compared with those of the wild-type parent, HSV-1 strain KOS, andFSLAT',a marker-r-escued virus. Latently infected trigeminal ganglia were examined forthe presence and number- of viral genomes by slot-blot DNA hybridization, for LAT expression by in situ RNA hybridization, and forthe ability toyield reactivatable virus by cocultivation techniques. We found that in spite of the replication competence ofthe LATdeletion mutant, it was reactivation impaired when compared with wild-type and marker-rescued viruses. It thus appears that the LATs, although not essential for the establishment or reactivation of
latency.
are required forthe efficient reactivation ofvirus fromthe latent state.(Part ofthis research was presented at the 13th Interna-tional Herpesvirus Workshop, Irvine, Calif., 7-13 August 1988.)
MATERIALS AND METHODS
Cells and viruses. African green monkey kidney (Vero)
cells were propagated as previously described (22). Proce-duresfor thegrowth andassayoftheKOS strain of HSV-1 have been previously described (24), and identical proce-dures were used for growth and assay of dILAT1.8 and
FSLAT' . One-step growth assays were performed
using
standard procedures as described previously (25). The growthand assayofthetemperature-sensitivemutanttsY46, which containsa mutation in the gene specifying the essen-tial immediate-early regulatory
protein
ICP27, has been described elsewhere(22).Plasmids. The locations of HSV-1 DNA sequences in
2893
on November 10, 2019 by guest
http://jvi.asm.org/
a b UL b' a' c' Us c 8
ICPO ICP27ICPO ICP22 ICP47
_~ I_ , _f _4
ICP4 ICP4
E - B S Sm H F H P P H S KX B Sc
I II ,lIII II| | | | II| I
D. IlCP27
B
B
B
ICPO
"-tS Y46 LATsITs'
IppBbl
P B
i g LPB
P_
P B
I IIPA1.8LPB
P P
P H
I I-pPH
F S
I I|pRFS
1kb
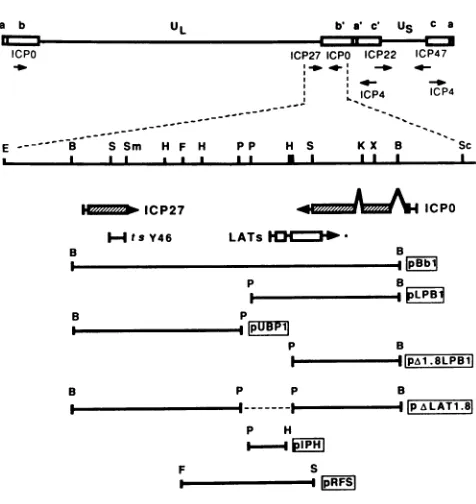
FIG. 1. Physical mapof the HSV-1 genome showing the
loca-tionsof genes andplasmids relevant to this study. Beneath the map
showingthepositions of the immediate-early genes, thepositions of
transcripts encoding ICPO, ICP27, and LAT are shown along with the location of the mutation in the ICP27 temperature-sensitive mutant tsY46, relative to selected restriction sites. . Known protein-coding sequences; W, LAT open reading frames:
deletion in plasmidpAXLAT1.8; *,precise 3' limits of the LATs are not known. Plasmids used in the production of the dILAT1.8 deletion mutant, its rescue, and detection of the LATs are shown below. B,BainHl;E, EcoRI; F, Fspl; H, Hpal; K,Kpntl:P,Pstl; S.
Stil; Sc, Stil; Sm,SinaIil;X, Xlol. Forconvenience, onlyonecopy ofHSV DNA containing ICPO and LAT is shown, although the deletionwasintroduced into bothcopies ofLAT.
plasmids used for the production of the mutant inthis study are shown in Fig. 1. Plasmid pBbl containing the BamHI B fragment in pBR325 was subcloned into pUC8 as two pieces whose limits are the following restriction sites: Bta,nHI to PstI (pUBP1) and PstIl to BaimHI (pLPB1). Plasmid pA1.8LPB1 was derived from pLPB1 by cutting the plasmid with HpaI, adding PstI linkers, and cutting to completion with Pstl, followed by religation.
Plasmid pALAT1.8 was produced by cutting pUPB1 with
Ba,nHI,
filling in the ends with the Klenow fragment of DNApolymerase I, ligating HindIII linkers to the plasmid, and cutting with HindIIl and Pstl. The resultant 5.3-kb fragment wasseparatedby agarose gel electrophoresis, electroeluted, purifiedover anElutip-d column (Schleicher & Schuell, Inc.. Keene, N.H.), and ligated to
pzA1.8LBP1
which had been cut with HindIll and PstI. The deletion in pALAT1.8 removes the putative promoter region (39), transcriptional start site, and 1,015 base pairs of the sequences specifying the LATs. Inaddition, the limits of the deletion were selected so as not to affect ICP0 (15) or the 0.9- and 1.1-kb transcripts de-scribed by Spivack and Fraser (29). Plasmid pIPH was constructed by cutting pLPB1 with Pstl andHpaI.
The resultant 1.4-kb fragment was separated, electroeluted, and purified as described above and ligated to pTZ18R (U.S. Biochemical Corp., Cleveland, Ohio) which had been cutwithPstI and SinaI. All enzymes and linkers utilized in the construction of the plasmids were obtained from New
En-gland BioLabs, Beverly, Mass., and used according to the instructions of the manufacturer.
Nucleic acidisolation.Bacterial plasmid DNAwasisolated and purified as previously described (13). Infectious HSV-1 DNA waspurified as described by Goldin et al. (4).
Generation ofmutants by transfection. Mutant W/LAT1.8 was introduced into the HSV genome by rescuing the ts mutationintsY46. For thispurpose,plasmid pALAT1.8was linearized and mixed with infectious t,sY46 DNA and Vero cells were transfected with the mixture asdescribed
previ-ously (22). Viral DNAwasobtained fromplaqueisolates(2),
cut with Pstl and BanHI, and analyzed by Southern blot hybridization (26) using pLPB1 as the probe for detecting
deleted LAT sequences (Fig. 1). The deletion mutant was plaque purified three times, and a high-titer stock was prepared.
Generation ofmarker-rescuedvirus. The deletion mutation indlLAT1.8wasrepairedby cotransfection of pRFS (Fig. 1)
with infectious dlLAT1.8 DNA into Vero cellsaspreviouLsly described (22). Plasmid pRFS(Fig. 1) does notoverlap with sequences that encode ICP0 (15), ICP27 (14), or the 20-kilodalton transcript described by Spivack and Fraser (29), although there is a small region of overlap with the 22-kilodalton transcript (29). Viral DNA was obtained from plaque isolates (2)andscreenedbyDNAhybridizationusing aslot-blotapparatus(Schleicher & Schuell) accordingtothe instructions of the manufacturer, using pIPHastheprobe for inserted sequences. Plaques which hybridized with pIPH were further tested by Southern blot hybridization (26), usingpLPB1 asthe probe for repaired LATsequences(Fig. 1). The marker-rescued virus was plaque purified three times, and a high-titer stockwas prepared.
Animal procedures. Seven-week-old randomlybred CD-1 mice (Charles River Laboratories, Kingston, N.Y.) were anesthetized with sodium pentobarbitol, corneas were scar-ified, and 20
[l
of virus atthe appropriate titerper eyewasadded as previously described (35). Assays of acute and latent infectionwereperformedaspreviously described(12). To test for non-latent infectious virus on day 30, ganglia were removed and immediately frozen, thawed, homoge-nized, and assayed on Vero cells. Reactivated viruseswere retainedforanalysis by Southern blot hybridization (26) for comparison of theirgenotypes with thoseofinput virus.
Slot-blothybridization. Slot-blot DNAhybridization, per-formed to assess levels of viral DNA within individual latently infected ganglia, was done as previously described (12).
Insituhybridization. The methodsused forin situ hybrid-ization have been previously described (5, 34). For probes, double-stranded DNAs were labeled by nick translation using [3H]deoxynucleoside triphosphates. Purified insert DNAs or whole plasmidsgave similarresults when used as hybridization probes. DNAprobes used for this study were pIPH, containing a 1.4-kb fragment from within the LAT-encoding region (Fig. 1), and pSG1-ES1 (17), containing the largest EcoRI-SaclI fragment from EcoRIJ-K of the HSV-1 genome in the 'L form.
RESULTS
Introduction ofthe LAT deletion into theviral genome. In addition to the 1.8-kb deletion in LAT sequences, the plasmid pALAT1.8 contains the wild-type gene for ICP27 (Fig. 1)and was therefore used to rescuethe ts mutation in
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.66.304.72.324.2].5
X .45~-iA
° * ; 1 2 3 4 5 6 7 8 9 10
_
t0W. 4.a*U_
I
0 2 4 6 8 10 12 14 16 18 20
Time (Hours)
FIG. 3. Growth of KOS, d/LAT1.8. and FSLAT- during a
one-step growth curve experiment in Vero cells infected ait a
multiplicity of10PFU percell. Totalvirus yieldsweremeasuredat
1. 2,and 18 hpostinfection. The 0h timepointsweredetermined by back-titration of virus inocula.
FIG. 2. Southern blot showing a comparison of LAT-coding
sequencesinvirusesresulting fromcotransfection ofVerocellswith pALAT1.8 withtsY46DNA(lanes 1 through 10). Arrowsshow the positions ofwild-type (4.6 kb) anddeleted (2.9 kb)bands in lanes containing marker plasmids. Viral DNAs were cut with Pstl and BaimiiHIandprobed with pLPB1 (Fig.1). d/LAT1.8wasobtained by threeplaque purifications of the viruswhose DNAisshowninlane
10.
tsY46. This procedure was utilized because demonstration
ofts' virus in transfection progeny would likely reflect the
simultaneous transfer of the deletion in LAT-coding se-quences into the viral genome. Plaque isolates were
screened for the ability to grow at the nonpermissive
tem-perature, and
ts'
plaque isolates were selected for furtheranalysis.
The Southern blot in Fig. 2 shows the DNA restriction fragment profiles of the LAT-coding region of KOS, tsY46.
and 10 ts' plaque isolates resulting from cotransfection of Verocellswith infectious tsY46 DNA and pALAT1.8, using
parental plasmids as markers for wild-type and deleted
fragments. Plaque isolates in lanes 7 and 10 show that the deletionwasintroducedinto bothcopiesof LAT. the sizes of
their LAT-encoding fragments being indistinguishable from that of pALAT1.8, but 1.7 kb smaller than the analogous fragments in KOS, tsY46, and pBbl. The plaque isolate
analyzed in lane 10 (I/LAT1.8) was plaque purified an
additional two times and used for further study.
Southern blot analysis of the marker-rescued virus
FSLAT' (produced by cotransfection of Vero cells with pRFS and infectious cl/LAT1.8 DNA) indicated repair of both copies of LAT, thesizeofthe LAT-encoding fragment
being indistinguishable fromthat of the analogous fragment ofthe wild-type virus(data not shown).
Growth properties of dlLATI.8 in vitro. The deletion mutant d/LAT1.8 exhibited growth kinetics and yields of infectiousvirus comparableto thoseof wild-type and mark-er-rescued (FSLAT+) virus on Vero cells in a one-step growth assay (Fig. 3), demonstrating that LATs were not required for growth in Vero cells in culture. When plated
onto Verocell monolayers underamethylcellulose overlay,
dlLAT1.8, FSLAT',andKOS producedplaques of
compa-rable size.
Growth properties of dlLATI.8 in vivo. Having shown previously that a dose of 2 x 106 PFU per eye of KOS
routinely produces reactivatable latency in 100% ofganglia
while killing less than 30% of the animals (12),we selected 2 x 10'PFU pereyeasthestandarddosefor testingcl/LAT1.8
and FSLAT+.
The mutant d/LAT1.8, FSLAT'a and wild-type virus
behavedverysimilarly duringacuteinfectionin theeye(Fig.
4) and in trigeminal ganglia (Fig. 5) over a series of time
points. Similar results were obtained in three separate
ex-periments. The viruses were almost at the lower limit of detection in eyeswabs 3 h following inoculation of 2 x 10"
PFU per eyebut werereadilydetectable ineyeswabs by 24
h (approximately one growth cycle), when peak titers were
reached. Levels of virus in eyes declined during the next 6 days postinfection.
At 18 h postinfection, low levels of c//LAT1.8 and FSLAT weredetected in trigeminal ganglia by directvirus assay. KOS. llLAT1.8, and FSLAT+ were all readily
de-tectableinganglia by48 h postinfection, reaching peaktiters
around 72 hpostinfection. Becausewewishedto testganglia
for the presence of reactivatable virus on day 30, we first
assayed extracts ofhomogenized gangliaatthistime for the
presenceof nonlatent infectious virus. Nonewasdetectedin
six ganglia infected 30 days previously withdILAT1.8, four gangliainfected with
FSLATV,
orsixgangliainfectedatthat time with wild-type virus.46-I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.314.555.79.315.2] [image:3.612.60.289.84.390.2]4*
3-2
0 24 48 72 96 120 144 168
Time(Hours)
FIG. 4. Geometricmeantiters ofKOS,dILAT1.8,andFSLAT' ineyeswabs during acute infection in CD-1mice. Eye swabs were
taken at 3, 24, 48, 72, 96, 120, 144, and 168 hfollowing corneal
infection with2 x 106PFU per eyeof eithervirus. The titersat0 h weredetermined byback-titration of inocula.
Measurementofviral DNA levels ingangliaduring latency. Although no infectious viruswasdetected ingangliaonday
30, viral DNAwas readilydetectablebyslot-blot
hybridiza-tion ingangliafrom mice inoculated 30dayspreviouslywith KOS or dlLAT1.8 (Fig. 6). The levels of viral DNA in mutant- and wild-type-infected ganglia were comparable, containingbetween 0.3 and 3.0copiesof viral DNA per cell. The observation that viral DNA levels in ganglia latently
infected with the two viruses were comparable has been further confirmed by the polymerase chain-reaction
tech-nique (data not shown).
Insituhybridization studiesoflatently infectedganglia. To compare LAT expression in ganglia latently infected with
dILAT1.8,
FSLAT+, andKOS,weperformedinsitu hybrid-izationstudies on 30-dayganglia. Using pSG-ES1 (Fig. 7)or pIPH (data not shown) as probes for LATs, hybridizationstudies ofganglia latently infected with KOS and FSLAT+
revealed the presenceofLATstypicallyasdensegrainsover neuronalnuclei.All 24ganglia from12micelatentlyinfected with KOS and 4 ganglia from 2 mice latently infected with FSLAT+ showed strong nuclear hybridization in neurons. In contrast, noneof 18ganglia from nine mice latently infected withdlLAT1.8or 14 ganglia from seven mock-infected mice demonstrated significant nuclear hybridization with pSG-ES1 (Fig. 7). Thus, no significant level of transcription from thisregion of the viral genome could be detected in ganglia
latently infected with the LAT deletion mutant.
Reactivation andneurovirulencestudies of KOS,dlLAT1.8, andFSLAT+. While the growth kinetics in vitro and in vivo ofKOS,
dlLAT1.8,
andFSLAT+weresimilar(Fig. 3, 4, and5) and the levels of viral DNA in latently infected ganglia were found to be comparable for KOS and dlLAT1.8 (Fig. 6), the latency phenotypes of the viruses were significantly
different (Table 1). Virus reactivated from 17 of 18
(94%)
4
3-2
0 ;6
0 24 48 72 96 120 144 168
[image:4.612.318.564.79.321.2]Time(hour)
FIG. 5. Geometricmean titersofKOS,dILAT1.8,andFSLAT+ intrigeminal ganglia duringacuteinfection inCD-1 mice. Ganglia
wereremoved and assayed directly forthe presence ofinfectious
virus at18, 42, 66, 90, 114, 138, and 162 hfollowingcornealinfection
with 2 x 106PFU per eyeof either virus.
ganglia latentlyinfected with KOSand 17of20(85%) ganglia
latently infected with FSLAT+ after 5 days in explant
culture, whereas reactivation fromganglia latently infected with dlLAT1.8 was reduced: only 21 of 43 (49%) ganglia producedvirusafter 5days in explantculture.
Whitby et al. (40) have reportedthat the addition of200 mM dimethyl sulfoxide (DMSO) to cultures during
coculti-.7 ....
0: ::..
3
MO i_..
.. .. _
KOCS Standar s
(copies percel)
3
03 0-1 0-03
tjj
0-oi
X2
0FIG. 6. Viral DNAintrigeminal gangliaof mice mockinfectedor infected 30dayspreviously with 2 x 106PFU per eyeof KOS or dlLAT1.8.Ganglion DNAwassubjectedtoslot-blot hybridization by usingEcoRI fragments A, D,I, N, and0 as probes (12). The
right-handcolumn showshybridizationofprobe fragmentsto mouse tail DNAcontaining 3, 1, 0.3, 0.1, 0.03, 0.01,andnoviral genomes percellequivalent.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.66.301.81.330.2] [image:4.612.371.512.484.669.2]A
D,
JO'
W, . X st
FIG. 7. In situ hybridization of trigeminal ganglia mock infected (B) or infected with KOS (A) d/LAT1.8(C),orFSLAT+(D). The viral DNA insert in pSG-ES1 (20) was purified and labeled as the hybridization probe for panel C.
vation greatly enhances the reactivation frequency of HSV-1 from latently infected ganglia. In this study, the addition of 200 mM DMSO did not greatly increase the recovery rate of clLAT1.8 (8 of 14 or 57%) from latently infected ganglia compared with the rate from untreated cultures (21 of 43 or
49%)(Table 1). This is in contrast to our latency studies with the ICP0 deletion mutant dlx3.1, in which the recovery of virus following the addition of DMSO to explant ganglion
TABLE 1. BehaviorofKOS. LAT deletion mutant d/LAT1.8. and marker-rescued virus FSLAT+ during acute and
latent infection in a CD-1 mouse eye model No. ofganglia
Peak acute titerin': reiactivated/no. No.of
Virus tested"((4) deaths/no. infected"
Eye Gianglia -DMSO +DMSO
KOS 8 x 1(i 4 x 1(0 17/18(94) 8/8 13/43 d/LAT1.8 2 x 106 3 x 106 21/43 (49) 8/14 15/55 FSLAT+ 7 x 10i 5 x 105 17/20(85) ND 13/38
"Peak eye swab and ganglion titers are derived from Fig. 4 and 5.
Geometricmeantitersareshown foracuteinfection.
" Explant cuIltures were performed on day 330 for the assay of latent
infection. DMSOwasaddedtothe mediumof someexplantcuilturestoafinal concentrattionof200 mM.
`The nuimber of animals which died on or before day 30 postinfection
followingcornealinoculaitionof2 x 10"PFU per-eye.
cultures wasgreatly increased, from0of 46(0%)to 7 of20
(35%)(12).
Theremovalof sequencesspecifying theLATshad littleif any effect on neurovirulence, as judged by the relative numbers of animals which died in the 30 days following inoculation of2 x 10" PFU per eye of KOS (13of43 or30%), FSLAT' (13of 38or 34%),ord/LAT1.8 (15of55 or27%). Examination of thegenotypesofreactivated strains. South-ern blot analysis of the DNAs of five reactivated isolates from ganglia latently infected with dlLAT1.8 demonstrated that the genotype ofthe reactivated virus was identical to thatofthe input (deleted)virus withregardto thesequences whichspecifythe LATs(Fig. 8).Thisexperiment eliminated the remote possibilities of contamination or genetic rever-sion.
Taken together, these studies indicate that LATs are not
requiredfortheestablishmentorreactivation of virallatency
but that they are required for wild-type frequencies of reactivation of virus from the latent state in this model system.
DISCUSSION
The discovery that HSV-1 is transcriptionally active in humans and mice during ganglionic latency (3, 34, 36) and that the mRNAs so produced partially overlap and are complementary to the message encoding ICPO (20, 28, 31)
,t,-.#
:.1.
4
.1*1.
A
;r,if'I <-lo-
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.79.543.87.430.2]t 2 3 4 5 6 7 8
46-P-0 00'
29-FIG. 8. Southern blot analysis of viral DNA from reactivated
viral isolates fromoneKOS(lane 2)andfive(/ILAT1.8 (lanes 4to8)
latently infected ganglia. DNA was cut with PstI andBa,nHI and
probed with plasmid pLPB1 shown in Fig. 1. Arrows show the positions ofwild-type (4.6 kb) and deleted (2.9 kb) bandsofcleaved KOS (lane 1)anddlLAT1.8 (lane 3) viral DNA.
has led to much speculation as to the function of these
so-called LATs. Three possible roles for the LATs were
postulated by Stevens et al. (31). First, the LATs may
encode a protein which is important in triggering the
reacti-vation of virusfrom the latentstate, andthe nuclear restric-tion of the LATs prevents their translation, thereby
main-taining latency. Second, the LATs may code for a protein
which is involved in maintaining the latent state. Third! the
LATs may act by antisense repression to suppress the
expression of ICPO during latency.
Examination of thesequence specifying the LATsreveals
open reading frames but no convincing evidence that they are likelyto codefor aprotein (14). Inaddition, the nuclear
localization and lack ofpolyadenylation of these transcripts
make it unlikely that a protein is specified by the LATs
during latency (38). Wagneretal. (38) used a14-amino-acid
peptide synthesized on the basis of the second HSV-1 LAT
open reading frame to raise serum in rabbits. Despite the
provenreactivity ofthisserumwith thesynthetic peptide,no
antigens were detected by immunohistological examination
ofacutely orlatently infected ganglia. Takentogether, these
data argue against the possibility that the LATs code for a
protein, although furthercharacterization ofminorpoly(A)t
transcripts (16, 38) may reveal otherwise.
Theidea that the LATs functionbyantisenserepression of ICPO is an attractive one which has received support by analogy from other recently described antisense repression
systems (8, 10, 11, 33, 37). To date, however, there is no
direct evidence of such a mechanism for the LATs from
studiesof HSV-1 latency. There is, however,evidence ofan
important role for ICPO in latency. ICPO is a potent
tran-scriptional transactivator of all three classes of HSV genes.
It is one of the first proteins to be synthesized during productive infection and hence is anexcellent candidate for
an initiator protein in reactivation. Consistent with this hypothesis is the observation that certain deletion mutants
that lack ICPO butspecify the LATs areunable toreactivate
fromlatencyinaCD-1mouseocularmodel(12;
unpublished
data). Although it is not
absolutely
essential for virusrepli-cation,
ICPO has been shown to beimportant
for virusgrowth in cell culture at low multiplicities ofinfection
(23,
32). The small amounts of virus likely produced and the consequent low
multiplicities
ofinfection encountered dur-ing reactivation from the latentstate may explain theimpor-tance ofICPO in the reactivation process.
The data
generated
in thisstudy
show that LATs are not essential for theefficientgrowthof the virus in cell culture or in mice and that establishment orreactivation of the latent state can be achieved in the absence of LATs. This is in agreement with other studies(9, 30),although
this is the first mutational study of the LATs to be performed using a defined deletion mutation which completely removes both copies of the LATs and a marker-rescued virus in an isogenic system. It is difficult at this stage to envisage how d1LAT1.8 can become latent in ganglia if ICP0 antisense repression is the sole meansby
which the latent state is established and maintainedfollowing lytic
infection. Thisdifficulty
is reinforced by thefinding
that dILAT1.8 is no more lethal than KOS or FSLAT+ in our mouse model. In the absence of LATs,therefore,
the suppression of gene expression characteristic of the establishment oflatency
must be achieved by an alternative pathway, possibly, as suggested by Roizman and Sears (21), involving ICP4. ICP4 is both a negative and positive regulator of viral gene
expression and may act in these respective capacities to establish and reactivate latency. This alternative
pathway
may actually augment the function ofLAT in the
wild-type
virus, serving tofurther down- andup-regulategene expres-sion during the establishment and reactivation oflatency.
Although theLATs are notessentialfor the establishment or reactivation of
latency,
reactivation appears to be more efficient in the presence of LAT sequences. This is consis-tent with the studies of Steiner et al. (30) but different from the work of Javier et al. (9), in which the lack ofsequencesspecifying the LATs was not associated with any change in reactivation efficiency. The observed reduction in reactiva-tion efficiency of
cllLAT1.8
can be attributed specifically tosequences within pRFS (most likelythose which specify the LATs), since the marker-rescued virus FSLAT reactivates with a frequency comparable to that of the wild-type virus. The removal or restoration of the LATs did not alter the lethality of KOS inour system,asjudged by the numbers of mice dying on or before 30 days postinfection with
d/LATI.8, KOS,orthe marker-rescuedvirus FSLATX This is at variance with the findings of Steiner et al. (30), who found that their LATdeletion mutant (1704) was somewhat less lethal than its wild-type parent strain 17' Although the reason for this difference is not certain at this time, it is
possiblethatthe reduced virulence of 1704is associated with asecondary mutation outside LAT-encoding sequences (30). Although comparable levels ofdILAT1.8 and KOS DNA were found in ganglia during latency, as judged by slot-blot hybridization, this assay fails to address the question ofthe structural configuration or biological activity of the latent genomes. Microscopic examination of tissue sections from ganglia latently infected withdlLAT1.8 byin situ hybridiza-tion revealed more extensive cytopathic changes than in ganglia infected with KOS or FSLAT' (unpublished results). This is consistent with the finding that dlLAT1.8 tends to reachslightly higher titersthan KOS orFSLATt in eyesandin ganglia atearlytimes duringacute infection. One could speculate, therefore, that the repression of gene expression in the absence ofthe LATs during the
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.121.243.82.297.2]ment of latency may have been less efficient, leading to increased virus-specific damage and possibly fewer viable genome-containing neurons available for reactivation. Un-der these circumstances, the levels of viral DNA could appear similar, suggesting similar establishment capability. but the levels of biologically relevant DNA may be quite disparate, possibly accounting for the differingratesof viral reactivation upon explant culture. Further studies are needed todetermine whether the increased histopathologic changes associated with d/LAT1.8 infection are due to
increased spread of the virus during early stages in the establishment of latency.
It is also possible, as suggested by Spivack and Fraser (28), that the LATs may play a more critical role in the maintenance of latency once it has been established. The enhanced cytopathic damage associated with d/LAT1.8 in-fection of ganglia could therefore be a consequence of limited reactivation events which lead to the destruction of some genome-containing neurons prior to explant culture. However, no free infectious virus was found in ganglia 30 days postinfection, suggesting that if such limited reactiva-tion events occur, the amounts of virus produced are quite small or thevirus is rapidly cleared.
From these results it can be stated that the sequences which code for the LATs are not essential for the establish-mentorreactivation of viral latency in this model system. It is clear, however, that deletion of these sequences is asso-ciated withareducedfrequency of reactivation, although the reasonfor this reduction is not clear at this time.
ACKNOWLEDGMENTS
We thank Jack Stevens for providing his in situ hybridization protocol.
Studies in the laboratories of D.K.. D.C. and P.A.S. were supported in part by Public Health Service grantP01 A124010 from
theNational Institute for Allergy and InfectiousDiseasesandinpart
by a National Multiple Sclerosis Society postdoctorill fellowship
(FG766-A-1) awardedtoD.A.L.
LITERATURE CITED
1. Croen,K.D., J.D. Ostrove, L.J. Dragovic, J. E.Smialek,and S. E. Straus. 1987. Latent herpes simplex virus in human
trigeminal ganglia: detection ofanimmediate-early gene "inti-sense' transcriptby in situhybridization. N.Eng. J. Med.317: 1427-1432.
2. DeLuca, N. A., M. A. Courtney, and P. A. Schaffer. 1984.
Temperature-sensitive mutants in herpes simplex virus type 1 ICP4permissive forearly geneexpression.J.Virol. 52:767-776. 3. Galloway, D. A., C. Fenoglio, and J. K. McDougall. 1982. Limited transcription of the herpessimplexvirusgenome when latent inhuman sensory ganglia. J. Virol.41:686-691. 4. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C.
Glorioso. 1981. Cloning of herpes simplex virus type 1 se-quences representingthe whole genome. J. Virol.38:50-58.
5. Haase, A., M. Brahic,L. Stowring, and H. Blum. 1984. Detec-tion of viral nucleic acids by in situ hybridization. Methods Virol. 7:189-226.
6. Honess,R.W.,and B. Roizman. 1974.RegulaItionofherpesvirus macromolecularsynthesis. 1. Cascade regulationof the synthe-sisof three groups of viral proteins. J. Virol. 14:8-19. 7. Honess,R.W.,and B. Roizman. 1975.Regulationofherpesvirus
macromolecularsynthesis: sequential transition ofpolypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA72:1276-1280.
8. Izant, J. G., and H. Weintraub. 1984. Inhibition ofthymidtine kinase gene expression by anti-sense RNA: a moleculcar
ap-proachtogeneticanalysis. Cell36:1007-1015.
9. Javier, R.T., .I. G. Stevens, V. B. Dissette,and E. K.Wagner.
1988. A herpes simplex virus transcript abundant in latently
infected neurons is dispensablefor establishment of the latent
state. Virology 166:254-257.
10. Kim, S., and B. A. Wold. 1985. Stable reduction ofthymidine kinaseactivityincellsexpressinghighlevels ofanti-senseRNA. Cell42:129-138.
11. Knecht, D. A., and W. F. Loomis. 1987. Anti-sense RNA inactivation of myosin heavychain gene expressionin Dictyo-steliifin discoi(lem. Science 236:1081-1086.
12. Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe, K. L. Tyler, and P. A. Schaffer. 1989. Immediate-earlyregulatorygenemutantsdefine different stages
in the establishment and reactivation ofherpes simplex virus
latency. J. Virol. 63:759-768.
13. Maniatis, T., E. F. Fritsch, andJ. Sambrook. 1982. Molecular cloning: a laboratory manual. p. 90-91. Cold Spring Harbor Laboratory. Cold SpringHarbor. N.Y.
14. McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M.C.Frame,D.McNab,L.J.Perry, J.E.Scott,and P.Taylor.
1988. ThecompleteDNA sequence ofthelonguniqueregionin the genome ofherpes simplex virus type 1. J. Gen. Virol. 69: 1531-1574.
15. Perry,L.J.,F.J.Rixon,R. D.Everett,M.C.Frame,and D.J. McGeogh. 1986. Characterization of the IE110 gene ofherpes simplexvirustype 1.J.Gen. Virol. 67:2365-2380.
16. Puga, A.,and A. L. Notkins. 1987. Continued expression ofa
poly(A)'transcript ofherpes simplexvirus type1intrigeminal gangliaoflatentlyinfected mice. J. Virol. 61:1700-1703. 17. Quinlan,M.P., and D.M.Knipe. 1985. Stimulation of
expres-sionofaherpes simplexvirusDNA-binding protein bytwoviral functions. Mol. Cell. Biol. 5:957-963.
18. Rock, D. L., S. L. Beam, andJ. E. Mayfield. 1987. Mapping bovine herpesvirus type 1 latency-related RNA in trigeminal ganglia oflatentlyinfected rabbits. J. Virol. 61:3827-3831. 19. Rock, D. L., W. A. Hagemoser, F. A. Osorio, and H. A.
McAllister. 1988. Transcription from the pseudorabies virus
genomeduringlatent infection. Arch. Virol. 98:99-106.
2'0. Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L. Lewis, J.R.Lokensgard,andS.L.Wechsler. 1987.Detectionoflatency related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol. 61:3820-3826.
21. Roizman, B., and A. E. Sears. 1987. An inquiry into the mechanisms of herpes simplex virus latency. Annu. Rev. Mi-crobiol. 41:543-571.
22. Sacks,W.R.,C. C.Greene,D. P.Aschman,and P. A.Schaffer. 1985. Herpessimplexvirustype 1 ICP27isanessential regula-tory protein.J. Virol. 55:796-805.
23. Sacks,W. R.,and P. A.Schaffer. 1987. Deletionmutantsinthe geneencodingthe herpessimplexvirustype 1immediate-early protein ICPO exhibit impaired growth in cell culture. J. Virol. 61:829-839.
24. Schaffer, P. A., V. C. Carter, and M. C. Timburv. 1978.
Collaborativecomplementation study oftemperature sensitive mutantsofherpessimplexvirustypes1and2.Virology 27:490-504.
25. Schaffer, P. A., R. J. Courtney, R. M. McCombs, and M. Benyesh-Melnick. 1971. A temperatur-e-sensitivemutantof her-pessimplexvirus defectiveinglycoproteinsynthesis. Virology
46:356-368.
26. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separated by gelelectrophoresis. J. Mol. Biol. 98:5()3-517.
27. Spivack, J. G., and N. W. Fraser. 1987. Detection ofherpes
simplexvirustype 1 transcriptsduringlatentinfection in mice. J. Virol. 61:3841-3847.
28. Spivack, J. G.,and N. W. Fraser. 1988. Expression ofherpes
simplexvirustype1latency-associatedtranscriptsinthe
trigem-inal ganglia of miceduring acute infection and reactivation of latent infection. J. Virol. 62:1479-1485.
29. Spivack, J. G., and N. W. Fraser. 1988.
Expression
ofherpes simplex virListype 1(HSV-1) latency-associated transcriptsandon November 10, 2019 by guest
http://jvi.asm.org/
transcriptsaffected by thedeletion inavirulentmutatnt HFEM: evidence for a new class of HSV-1 genes. J. Virol.
62:3281-3287.
30. Steiner, 1., J. G. Spivack, R. P. Lirette, S. M. Brown, A. R. MacLean,j.H.Subak-Sharpe, and N. W. Fraser.1989. Herpes simplex virustype1latency-associatedtranscriptsareevidently
notessentialfor latent infection. EMBOJ. 8:505-511. 31. Stevens, J.G., E. K. Wagner,G.Devi-Rao, M. L.Cook, and L.
Feldman. 1987. RNA complementar-y to herpesvirus a(-gene mRNA ispredominant in latently infectedneurons.Science235:
1056-1059.
32. Stow,N. D., and E. C.Stow. 1986.Isolation and characterisation ofa herpes simplextype 1 mutant containingadeletion within
thegeneencoding the immediate-early polypeptide Vmw110.J.
Gen. Virol. 67:2571-2585.
33. Strickland, S.,J.Huarte, D. Belin, A. Vassalli, R. J. Rickles, and J. D. Vassali. 1988. Anti-sense RNA directed against the 3' noncodingregionpreventsdormant mRNAactivation inmouse
oocytes. Science 241:680-684.
34. Stroop,W. G., D. L. Rock, and N. W. Fraser.1984. Localization ofherpes simplex virus in the trigeminal and olfactorysystems
of the mouse central nervous system during acute and latent
infections by in situ hybridization. Lab. Invest. 51:27-38. 35. Tenser, R. B., and M. E. Dunstan. 1979. Herpes simplex virus
thymidine kinase expression ininfection of the trigeminal
gan-glion. Virology99:417-422.
36. Tenser, R. B., M.Lawson, S. J. Ressel, and M. E. Dunstan. 1982. Detection ofherpes simplex virus mRNA in latently infected
trigeminal ganglion neurons by in situ hybridization. Ann. Neurol. 11:285-291.
37. vanderKrol, A. R., P. E. Lenting, J. Veenstra, I. M. vander Meer, R. E. Koes, A. G. M. Gerats, J. N. M. Mol, and A. R. Stuitje. 1988. An anti-sense chalcone synthase gene in
trans-genic plants inhibits flower pigmentation. Nature (London) 333: 866-869.
38. Wagner, E. K.,G.Devi-Rao, L.T.Feldman,A.T.Dobson, Y. F.
Zhang, W. M. Flanagan, and J. G. Stevens. 1988. Physical characterization of theherpessimplex virus latency-associated
transcript inneurons. J. Virol. 62:1194-1202.
39. WVechsler, S. L., A. B. Nesburn, R. Watson, S. M. Slanina, and H. Ghiasi. 1988. Fine mapping of the latency relatedgene of
herpes simplex virus type 1: alternative splicing produces
dis-tinct latency-related RNAs containing open reading frames. J.
Virol. 62:4051-4058.
40. Whitby, A. J., W. A.Blyth, and T. J. Hill. 1987.The effect of DNA hypomethylating agents on the reactivation of latent herpes simplex virus from latently infected mouse ganglia in
vitro. Arch. Virol. 97:137-144.