Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Sequences in the 5
⬘
and 3
⬘
R Elements of Human Immunodeficiency
Virus Type 1 Critical for Efficient Reverse Transcription
YUKI OHIANDJARED L. CLEVER*Department of Microbiology, University of Texas Health Science Center at San Antonio, San Antonio, Texas 78229-3900
Received 31 March 2000/Accepted 18 June 2000
The genome of human immunodeficiency virus type 1 (HIV-1) contains two direct repeats (R) of 97 nucleo-tides at each end. These elements are of critical importance during the first-strand transfer of reverse
tran-scription, during which the minus-strand strong-stop DNA (ⴚsssDNA) is transferred from the 5ⴕend to the 3ⴕ
end of the genomic RNA. This transfer is critical for the synthesis of the full-length minus-strand cDNA. These repeats also contain a variety of other functional domains involved in many aspects of the viral life cycle. In
this study, we have introduced a series of mutations into the 5ⴕ, the 3ⴕ, or both R sequences designed to avoid
these other functional domains. Using a single-round infectivity assay, we determined the ability of these mu-tants to undergo the various steps of reverse transcription utilizing a semiquantitative PCR analysis. We find
that mutations within the first 10 nucleotides of either the 5ⴕor the 3ⴕR sequence resulted in virions that were
markedly defective for reverse transcription in infected cells. These mutations potentially introduce mismatches
between the full-lengthⴚsssDNA and 3ⴕacceptor R. Even mutations that would create relatively small
mis-matches, as little as 3 bp, resulted in inefficient reverse transcription. In contrast, virions containing identically mutated R elements were not defective for reverse transcription or infectivity. Using an endogenous reverse
transcription assay with disrupted virus, we show that virions harboring the 5ⴕor the 3ⴕR mutations were not
intrinsically defective for DNA synthesis. Similarly sized mismatches slightly further downstream in either the
5ⴕ, the 3ⴕ, or both R sequences were not detrimental to continued reverse transcription in infected cells. These
data are consistent with the idea that certain mismatches within 10 nucleotides downstream of the U3-R junc-tion in HIV-1 cause defects in the stability of the cDNA before or during the first-strand transfer of reverse
transcription leading to the rapid disappearance of theⴚsssDNA in infected cells. These data also suggest that
the great majority of first-strand transfers in HIV-1 occur after the copying of virtually the entire 5ⴕR.
Retroviruses harbor two direct repeat sequences (R) at the 5⬘and 3⬘ends of their genomic RNA (for a review, see refer-ence 13). These repeats are necessary for directing the minus-strand strong-stop cDNA (⫺sssDNA) from the 5⬘end of the viral genome, close to where reverse transcription initiates, to the 3⬘ R during the synthesis of the full-length minus-strand cDNA copy (for a review, see reference 48). This is known as the first-strand transfer of reverse transcription. It has been shown that the complementarity of the⫺sssDNA and the 3⬘R is important for directing the first-strand jump (12, 13, 26, 47, 48). It is not entirely clear if this complementarity is the only factor which directs the first-strand transfer. It has re-cently been reported that both complementarity-dependent and complementarity-independent mechanisms guide the first-strand transfer during reverse transcription in Moloney murine leukemia virus (MMLV) (49). It is also unclear whether the terminal complementarity between the growing 3⬘end of the cDNA and the acceptor template or complementarity behind this region is more important for the first-strand switch or if both contribute (49).
The length of the R sequence varies considerably between different retroviruses (for a review, see reference 13). The R is as short as 16 nucleotides in mouse mammary tumor virus and is up to about 250 nucleotides long in the human T-cell leu-kemia and bovine leuleu-kemia virus retroviruses (13). In the hu-man immunodeficiency virus type 1 (HIV-1), the R elements
are each 97 nucleotides long. These facts suggest that very short R sequences can efficiently direct the first-strand jump. In fact, it has been reported that R sequences much shorter than wild-type sequences function efficiently during the first-strand transfer of HIV-1 reverse transcription. This is based on observing the replication characteristics of HIV-1 virions har-boring deletions in the 3⬘acceptor R (5). The R sequences of HIV-1 contain many overlapping functional domains. This fact has made it difficult to functionally dissect the importance of R sequences during the first-strand switch using an infectious viral system.
In addition to being important during the first-strand trans-fer, the R sequences of HIV-1 fold into two important RNA stem-loop structures termed the poly(A) and TAR hairpins (Fig. 1A) (4). The poly(A) stem-loop contains the polyadenyl-ation signal which is exclusively utilized in only the 3⬘R. It has recently been reported that the structure and stability of this hairpin constitute one factor that directs poly(A) site selection to the 3⬘R (19, 31, 32). In addition, it has been shown that the 5⬘copy of the poly(A) hairpin is an integral part of the HIV-1 packaging signal (9, 20, 39). Mutations that disrupt the folding of this element are detrimental to proper RNA encapsidation, while compensatory mutations restore packaging to wild-type levels (9, 20). The other RNA element located in the R, the TAR hairpin, performs several critical functions during the viral life cycle (13). The 5⬘TAR element is the well-established binding site for the viral transcriptional-transactivator protein, Tat. Tat binds to a bulge near the top of this hairpin and recruits several host proteins that in turn lead to the hyper-phosphorylation of RNA polymerase II causing it to become more processive and to synthesize full-length primary tran-* Corresponding author. Mailing address: Department of
Microbiol-ogy, University of Texas Health Science Center, San Antonio, TX 78229-3900. Phone: (210) 567-3935. Fax: (210) 567-6612. E-mail: cleverj @uthscsa.edu.
8324
on November 9, 2019 by guest
http://jvi.asm.org/
scripts. The binding sites for these factors are located near the top of the TAR element (for a review, see reference 15). The 5⬘TAR stem-loop has also been shown to be part of the HIV-1 packaging signal (9, 29, 39). Mutations that disrupt the lower portion of the 5⬘ TAR stem cause severe defects in proper genomic RNA encapsidation, while compensatory mutations restore packaging to wild-type levels (9, 29). These data indi-cate that the structure of the 5⬘TAR element is necessary for the fidelity of genomic RNA encapsidation, while the primary sequence is not. Mutations in the 5⬘ TAR hairpin have also been reported to cause defects in the initiation of reverse
transcription (9, 28). These defects in initiation were not at-tributable to defects in RNA packaging. However, it could be argued that these apparent defects in the initiation of reverse transcription resulted from mismatches between the mutant ⫺sssDNA and the wild-type 3⬘R elements. These mismatches could result in inefficiencies in the first-strand transfer and thus could indirectly cause the ⫺sssDNA to be rapidly degraded leading to the conclusion that initiation of reverse transcription was reduced.
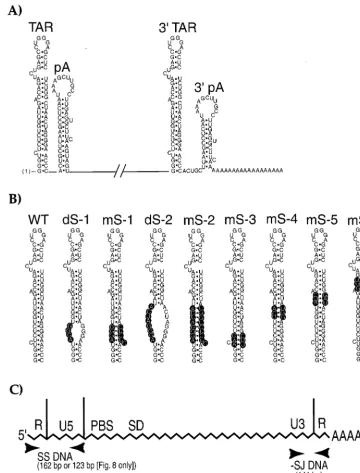
In order to address some of these questions, we have intro-duced a series of mutations into the 5⬘, the 3⬘, or both R FIG. 1. (A) Diagram of the RNA secondary structures located at the 5⬘and 3⬘termini of the HIV-1 genome in the R sequences. The TAR and the poly(A) hairpins (pA) are indicated. The poly(A) tail is shown at the 3⬘end. (B) Diagram of the various TAR mutants used in this study and comparison to wild-type TAR (WT). Mutated nucleotides are highlighted in black. The dS-1, mS-1, dS-2, mS-2, and mS-3 mutations have been described before (9). Previously, they were introduced into only the 5⬘TAR element (9). (C) Relative positions of the primer pairs in the HIV-1 genome used in the semiquantitative PCR analysis of newly synthesized viral DNA. Abbreviations: SS, strong stop;⫺SJ, minus-strand jump; FL, full length; PBS, primer binding site; SD, major 5⬘splice donor. The diagram is not drawn to scale.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.122.483.73.546.2]sequences in an HIV-1 viral clone. We have designed these mutations to avoid the other functional domains, described above, that lie within these regions. The ability of the mutants to undergo the various steps of reverse transcription during one round of viral replication was determined using a semi-quantitative PCR assay. We found that certain mutations in either the 5⬘or the 3⬘R sequence caused marked defects in viral infectivity that were attributable to inefficient reverse transcription. These mutations would cause mismatches be-tween the full-length ⫺sssDNA and the 3⬘ acceptor R. In contrast, these viruses were able to undergo reverse transcrip-tion in detergent-disrupted virions during endogenous reverse transcription assays as efficiently as wild-type virus. This indi-cates that these virions were not intrinsically defective for DNA synthesis. Other R mutations, farther downstream, did not affect viral infectivity or interfere with reverse transcription efficiency in infected cells. Our results suggest that, in HIV-1, the terminal complementarity between the ⫺sssDNA and 3⬘ acceptor R sequences is critically important for the stability of the minus-strand cDNA and thus profoundly affects the effi-ciency of reverse transcription.
MATERIALS AND METHODS
Cell culture.Human osteosarcoma (HOS), 293T, and COS-7 cells were cul-tured in Dulbecco’s modified Eagle medium containing 4.5 g of glucose/liter, 100 U of penicillin G/ml, 0.1 mg of streptomycin sulfate/ml, and 10% fetal calf serum at 37°C in 5% CO2.
Plasmid construction.All mutations were introduced into the previously de-scribed HIV-gpt vector (gift from N. Landau and D. Littman) (35, 40). The amphotropic murine leukemia virus (AMLV) Env expression vector has also been previously described (35, 40). The 5⬘TAR hairpin mutations mS-4, -5, and -6 were created by oligonucleotide-directed mutagenesis within theBspEI-KasI (nucleotides 309 to 637) fragment of HIV-1 subcloned into pBluescript II KS⫹ (pBS/KS⫹; Stratagene), after which DNAs were sequenced in order to confirm the mutations. This fragment was then subcloned back into the HIV-gpt vector through a multistep subcloning process. The other 5⬘TAR hairpin mutations have been previously described (9). HIV-gpt constructs containing mutations in only the 3⬘TAR element were created by swapping theBspEI-HindIII (nucle-otides 309 to 531) fragment from the 5⬘repeat containing the desired mutation with the 3⬘repeat sequence. HIV-gpt constructs harboring mutations in both the 5⬘and the 3⬘repeat elements were constructed by swapping a unique 2.7-kb
BamHI-XbaI fragment containing the 3⬘TAR mutation into the HIV-gpt con-struct harboring the desired 5⬘TAR mutation. Constructs for in vitro transcrip-tion of antisense riboprobes used in the RNase protectranscrip-tion assays were made by subcloning theKpnI-ClaI fragment of wild-type or mutant HIV-gpt into pBS/ KS⫹cut with the same enzymes, as described before (9). Prior to in vitro transcription with T7 RNA polymerase, plasmids were linearized withBspEI. Radiolabeled transcripts were prepared exactly as described previously (8, 10).
Virus production and infectivity assays. All virions used in these studies consisted of HIV-1 core particles (strain HXB2) pseudotyped with the AMLV Env protein. Viral stocks were prepared from transient calcium phosphate co-transfection of 293T cells, exactly as before (9–11). Infectivity assays with HOS cells were performed in duplicate by using serial dilutions of the viral superna-tants as previously described (9–11).
Virus quantitation and exogenous reverse transcriptase assays.The concen-tration of viral antigen (p24) in the stocks was determined using an enzyme immunoassay as recommended by the manufacturer (Coulter-Immunotech) and as previously described (9–11). Reverse transcriptase assays were performed in duplicate on virions pelleted from 0.5 ml of viral stocks at 25,000⫻gfor 2 h at 4°C, as described before (9–11).
RNase protection assays.Viral stocks (12 ml) were layered onto a 5-ml, 20% sucrose cushion (in phosphate-buffered saline [PBS]) and centrifuged at 57,771⫻g in an S-20/20 rotor (Sorvall) for 1.5 h at 4°C. Viral pellets were resuspended in 0.1 ml of PBS, and an aliquot was removed in order to determine the p24 concentration as described above. Virion and cytoplasmic RNAs were extracted exactly as described before (9–11). Viral and cytoplasmic RNA prep-arations were treated with 1.0 U of RQ1 RNase-free DNase (Promega) and 10 U of RNase inhibitor in 0.1 ml for 30 min at 37°C followed by treatment with phenol-chloroform and ethanol precipitation to remove any plasmid DNA con-tamination. Amounts of viral RNAs were quantitated using an RNase protection assay as recommended by the manufacturer (RPA III; Ambion). For virion-derived RNAs, the amount of RNA equivalent to 100 ng of pelleted p24 was annealed to an excess of32P-labeled riboprobe (105cpm,⬃200 pg). For
cyto-plasmic RNAs, approximately 1/20 of the RNA isolated from one T75 flask of 293T cells was used. The protected fragments were electrophoresed on denatur-ing 5% polyacrylamide–8 M urea sequencdenatur-ing gels and subjected to
autoradiog-raphy. Radioactivity in the various bands was quantitated using a Molecular Dynamics PhosphorImager.
Semiquantitative PCR analysis.Viral supernatants containing either 50 or 500 ng of p24 were brought to a final volume of 4 ml using fresh media. After addition of MgCl2(5 mM final concentration) and 100 U of RNase-free DNase
I, supernatants were incubated at 24°C for 30 min. After addition of 8g of Polybrene per ml, the DNase-treated supernatants were split into two samples. The reverse transcriptase inhibitor AZT (zidovudine) was added to one-half of the supernatants, to a final concentration of 10M. COS-7 cell monolayers grown to about 50% confluence on 10-cm2plates were infected with 2 ml of
DNase-treated viral supernatants containing either 25 or 250 ng of p24. Those plates of cells infected with virus in the presence of 10M AZT had been pretreated with the same drug concentration for 3 h prior to infection. After a 90-min infection at 37°C, cell monolayers were extensively washed with PBS. An additional 10 ml of medium was added (with or without AZT [10m]), and cells were cultured for about 20 more hours. After extensive washing with PBS, cells were briefly trypsinized and pelleted. Total-cell lysates were prepared by a previously published procedure (9, 14). Briefly, cells were disrupted by the addition of lysis buffer (100 mM KCl, 20 mM Tris-HCl [pH 8.4], 0.2% Nonidet P-40, 500g of proteinase K per ml) and then incubated at 60°C for 2 h, followed by 15 min at 95°C. Serial dilutions of the lysates were then assayed for the presence of the cellular CC chemokine receptor-5 gene (CCR5) to assure that approximately equal amounts of nucleic acids were present in each sample. A previously described “hot” PCR-based procedure was used (9, 28, 52). Lysates were diluted in 10-fold increments, and 5l of each was used in the PCRs. The reaction contents were essentially as previously described, except that 50 ng of the unlabeled oligonucleotide (5⬘-ATGGATTATCAAGTGTCAAGT-3⬘; sense) and 25 ng of the32P-labeled oligonucleotide (5⬘-GCAGGAGGCGGGCTGCA
ATTT-3⬘; antisense), which hybridized to the CCR5 gene, were added to each reaction mixture. Thirty amplification cycles consisting of 93°C for 1 min and 65°C for 2 min were used, and reaction products were separated on 5% poly-acrylamide gels in 1⫻Tris-borate-EDTA buffer. The CCR5 PCR product was 100 bp in length. Gels were visualized by autoradiography and quantitated using a Molecular Dynamics PhosphorImager. Identical reaction conditions were used for hot PCR of viral DNAs. The 103-fold dilutions of the cellular lysates were
used in the PCRs for cells infected with 25 ng of p24 (104-fold for cells infected
with 250 ng of p24), because it was found that the viral DNA products fell within the linear range of the standard curves. These oligonucleotide pairs, which hybridized to HIV-1 (HXB2), were used to amplify strong-stop (5⬘-ATCTG
AGCCTGGGAGCTCTCT-3⬘ [sense]; 5⬘-ACTGCTAGAGATTTTCCACACT
GA-3⬘[antisense]), minus-strand jump (5⬘ -CTTTCCGCTGGGGACTTTCCA-3⬘[sense]; 5⬘-GAGAGCTCCCAGGCTCAGATCTGG-3⬘[antisense]), and full-length DNAs (5⬘-TGTGCCCGTCTGTTGTGTGACTCT-3⬘[sense]; 5⬘-TCCTG CGTCGAGAGAGCTCCTCTGG-3⬘[antisense]). Reaction products were visu-alized and quantitated as described above. The sizes of the PCR products were 162 bp for strong-stop DNA, 141 bp for minus-strand jump DNA, and 138 bp for full-length DNA. In order to amplify strong-stop DNA produced from HIV-gpt harboring the TAR mS-4, -5, and -6 mutations, a different sense primer, 5⬘-CT GCTTAAGCCTCAATAAAGC-3⬘, that did not overlap with the point muta-tions was used. This produced a PCR product 123 bp in length.
Endogenous reverse transcriptase assays.The endogenous reverse transcrip-tase reactions were performed essentially by a previously described procedure (38). Viral stocks (12 ml) were brought to 5 mM MgCl2and treated with 400 U
of RNase-free DNase I for 30 min at 37°C. DNase I-treated supernatants were layered onto 5-ml, 20% sucrose cushions in TEN buffer (100 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl [pH 8.0]) and centrifuged at 57,771⫻gin an S-20/20 rotor (Sorvall) for 2 h at 4°C. The viral pellet was then resuspended in 0.1 ml of ice-cold TEN buffer, and the amount of viral p24 antigen was determined using an enzyme-linked immunosorbent assay (ELISA) as described above. Reactions were performed in 30l, and reaction mixtures contained 10 ng of p24 in 0.01% Triton X-100, 50 mM NaCl, 50 mM Tris-HCl (pH 8.0), 10 mM dithiothreitol, 5 mM MgCl2, and 100M (each) dATP, dGTP, dCTP, and dTTP. As negative
controls for synthesis of the reverse transcription products during the endoge-nous reaction, parallel reactions were performed without the addition of dTTP. After incubation of the reaction mixtures at 37°C for 2 h, 270l of stop buffer (50
g of proteinase K/ml, 20g of yeast RNA/ml, 1.5 mM EDTA [pH 8.0]) was added and the incubation continued at 60°C for 1 h. Proteinase K was inactivated by incubation of reaction mixtures at 95°C for 15 min. Hot PCR was performed on 5-l aliquots of the reaction mixtures as described above.
RESULTS
We have introduced a series of mutations into the R se-quences of the HIV-1 viral construct HIV-gpt. This consists of a full-length provirus (strain HXB2) into which a selectable marker gene (gpt) has been inserted in the place ofenv se-quences. We have pseudotyped our HIV-1 core particles with the AMLV envelope protein (35, 40). These virions are capa-ble of undergoing one round of replication after infection of susceptible cells. The extent of DNA synthesis in infected cells
on November 9, 2019 by guest
http://jvi.asm.org/
was determined using a semiquantitative PCR assay that am-plifies early, intermediate, or late products of reverse transcrip-tion (52). Infectivities of the various mutants were determined by their ability to stably transduce the marker gene-producing colonies during drug selection. The ability of selected mutants to undergo intravirion reverse transcription was determined using an endogenous reverse transcriptase assay in which nu-cleotides and small amounts of detergent are incubated with sucrose-purified virion particles (38). DNA products were then amplified from disrupted virions using semiquantitative PCR.
Certain 5ⴕ or 3ⴕ R mutations reduce reverse transcription
efficiency in infected cells.It has been previously reported that
certain mutations in the 5⬘ TAR element result in apparent defects in reverse transcription initiation that were not attrib-utable to defects in RNA packaging (mS-1, mS-2, and mS-3) (9). These mutations could, however, introduce mismatches between the mutant⫺sssDNA and wild-type 3⬘R during the first-strand transfer of reverse transcription. To determine if the putative mismatches were causing these DNA synthesis defects, we introduced these three mutations (Fig. 1B) into the 3⬘R (3⬘ TAR mS-1, mS-2, and mS-3) or created double mu-tations in both the 5⬘and the 3⬘R sequences of HIV-gpt (5⬘-3⬘ TAR mS-1, mS-2, and mS-3). These viral constructs were co-transfected into 293T cells along with the AMLV expression vectors as previously described (9–11). Supernatants were col-lected 48 h posttransfection and assayed for the viral capsid protein p24, as well as for exogenous reverse transcriptase activity. All of these mutants produced levels of p24, with associated reverse transcriptase activity, which were similar to that produced by wild-type HIV-gpt (data not shown). Vi-ral supernatants containing equivalent amounts of p24 were treated with DNase I to remove any potentially contaminating plasmid DNA. These supernatants were then used to infect COS-7 cell cultures in either the presence or absence of AZT (10 M). The AZT controls served to show that the DNA products we were observing were synthesized post-cell entry and were not being made inside virion particles prior to infec-tion, which can occur in HIV-1. After 90 min at 37°C, cell monolayers were extensively washed and refed with media with or without AZT and then harvested about 20 h postinfection. Appropriate dilutions of total-cell lysates which contained ap-proximately equal amounts of the cellular gene CCR5 were prepared. These lysates were then used in hot PCRs to amplify the various products of reverse transcription (Fig. 1C). As previously reported, a virus containing the mS-1 mutation in only the 5⬘ copy of TAR accumulated markedly reduced amounts of all the products of reverse transcription in infected cells (Fig. 2A to C) (9). A virus harboring this same mutation in only the 3⬘ copy of TAR (3⬘ TAR mS-1) had a similar phenotype, accumulating reduced amounts of all reverse tran-scription products in cells (Fig. 2A to C). In sharp contrast, a double mutant containing the mS-1 mutation in both the 5⬘ and 3⬘R elements (5⬘-3⬘TAR mS-1) produced wild-type levels of DNA in infected cells (Fig. 2A to C). Phosphorimaging the gels indicated that the single-mutant-infected (5⬘TAR mS-1 or 3⬘TAR mS-1) cellular lysates contained at least 10-fold-less DNA products than either the wild-type or the 5⬘-3⬘TAR mS-1 lysates. Virions harboring either the mS-2 or mS-3 mutation in only the 5⬘ R also accumulated reduced amounts of reverse tran-scription products, compared to the wild type, at 20 h postin-fection, as previously described (Fig. 3A and B) (9). Virions containing these identical mutations in only the 3⬘R, 3⬘TAR mS-2 and 3⬘ TAR mS-3, accumulated reduced amounts of reverse transcription products, at about the same level as the 5⬘ TAR mutants (Fig. 3A and B). In contrast, lysates from cells infected with the double mutants 5⬘-3⬘ TAR mS-2 and mS-3
each harbored essentially wild-type levels of reverse transcrip-tion products (Fig. 3A and B). Phosphorimager analysis indi-cated that the single mutants, in either the 5⬘ or the 3⬘ R, produced at least 10-fold-less reverse transcription products than either the wild-type or double-mutant virions.
The infectivities of these mutants were then determined based on their abilities to stably transduce thegpt⫹gene into human
HOS cells. Infected-cell cultures were grown for approximately 18 days under selection before visible colonies were quantitated. As previously reported, the 5⬘ TAR mS-1 and mS-2 virions were about 100-fold less infectious than wild-type HIV-gpt, while the 5⬘ TAR mS-3 mutant had about a 15-fold reduction in infectivity (Fig. 4) (9). Similar reductions in infectivities were observed for the 3⬘ TAR mS-1, mS-2, and mS-3 TAR mutants (Fig. 4). In contrast, the infectivities of all three of the 5⬘-3⬘ TAR double mutants were greater than those of the single mutants and were only slightly less than that of wild-type virions (Fig. 4). Therefore, the infectivities correlated well with the results obtained using the semiquantitative PCR analysis, although the semiquantitative PCR appeared to underestimate the extent of the infectivity de-fect. These results are consistent with the idea that certain mis-matches between the⫺sssDNA and the 3⬘R interfere with re-verse transcription, most likely during the first-strand transfer, since these defects are not observed with virions containing iden-tically mutated repeats (Fig. 5A and B).
FIG. 2. Semiquantitative PCR analysis of the efficiency of reverse transcrip-tion by the 5⬘, the 3⬘, and the 5⬘-3⬘mS-1 mutant virions. Equal amounts of DNase I-treated viral supernatants (containing equivalent amounts of p24) were used to infect cell monolayers as described in Materials and Methods. One-half of the cells were treated with 10M reverse transcriptase inhibitor AZT. Total-cell lysates, harvested 20 h postinfection, were assayed for the presence of strong-stop (A), minus-strand jump (B), or full-length (C) viral DNAs. PCR standards are shown for reaction mixtures that contained 10, 50, 250, and 2,500 copies of viral DNA in an HIV-gpt vector. To verify that approximately equal amounts of host cell-derived nucleic acids were present in the samples, PCR was performed using a primer pair that amplifies the cellular gene CCR5 (D). PCR standards for CCR5 were generated from reaction mixtures containing 10-fold serial dilutions of cell lysate, from undiluted (0) to a 104dilution. The 10-fold dilution of lysate
was used to amplify CCR5 in the various samples. Mock, control plates of cells were incubated with DNase I-treated supernatants from mock-transfected 293T cells and then processed in parallel with the other samples.
on November 9, 2019 by guest
http://jvi.asm.org/
Some virions containing mismatched 5ⴕand 3ⴕR sequences
can efficiently undergo reverse transcription. Previous work
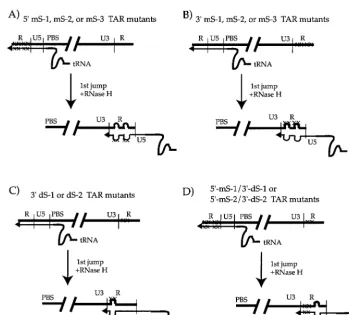
had indicated that mismatches in the 3⬘ part of the R se-quences, in the poly(A) hairpin region, did not significantly reduce viral infectivity (9). Based on this previous work and the above observations we wanted to test the hypothesis that it was the proximity of the mismatches to the U3-R junction which was causing the observed defects in reverse transcription (Fig. 5A and B). We created two double-mutant HIV-1 virions har-boring the mS-1 or mS-2 mutations in the 5⬘ copy of TAR while harboring mutations that would disrupt the base pairing in the 3⬘TAR stem, dS-1 and dS-2 (Fig. 1B). These new TAR double mutants were named 5⬘mS-1/3⬘ dS-1 and 5⬘ mS-2/3⬘ dS-2 (Fig. 5D). These combinations of mutations would have the effect of moving potential mismatches farther away from the U3-R junction as diagrammed in Fig. 5D. The 5⬘mS-1/3⬘ dS-1 construct would produce a 5-bp mismatch between the ⫺sssDNA and 3⬘ R sequence that is 51 bp 3⬘ of the U3-R junction. The 5⬘ mS-2/3⬘ dS-2 viruses would produce a 9-bp mismatch that is 47 bp from this junction. We also introduced these two disruptive mutations into only the 3⬘TAR element of HIV-gpt, creating constructs named 3⬘TAR dS-1 and dS-2. The 3⬘ TAR dS-1 mutant would produce a 5-bp mismatch, while the 3⬘TAR dS-2 mutant would form a 9-bp mismatch, that would be within 4 bp of the U3-R junction for each mutant (Fig. 5C).
These viral constructs were cotransfected into 293T cells along with the AMLVenvexpression vectors to produce in-fectious stocks containing pseudotyped virions. The concentra-tion of the viral capsid antigen p24 was determined for each stock, as was the pelletable reverse transcriptase activity asso-ciated with these virions. All of these mutants produced levels of p24, with associated reverse transcriptase activities, which were similar to those of wild-type HIV-gpt (data not shown). Because it was previously shown that the two disruptive mu-tations near the bottom of the 5⬘TAR stem resulted in virions that were defective for proper genomic RNA encapsidation,
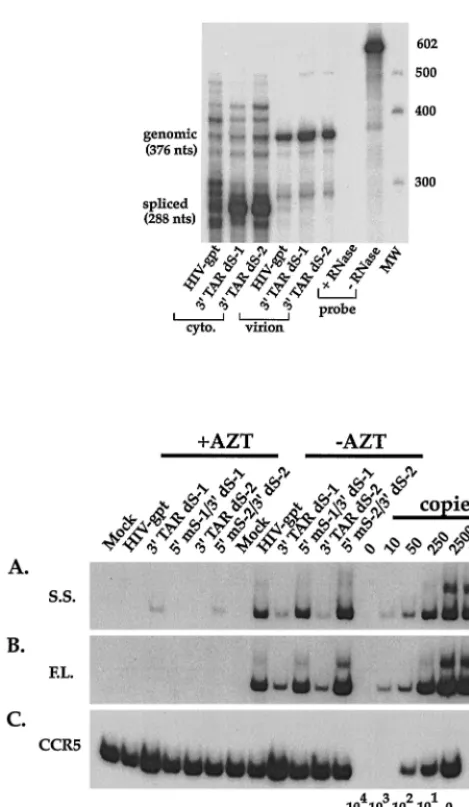
we needed to determine their effects on RNA packaging when present in only the 3⬘TAR element (9). Wild-type HIV-gpt, 3⬘ TAR dS-1, and 3⬘TAR dS-2 virions were partially purified by being pelleted through 20% sucrose cushions. RNA extracted from known quantities of virions was subjected to the previ-ously described quantitative RNase protection assay (9–11). This analysis, which detects both genomic and subgenomic HIV-1 RNAs, revealed that 3⬘ TAR dS-1 and 3⬘TAR dS-2 virions packaged wild-type amounts of genomic RNA and properly excluded spliced RNAs, similarly to wild-type HIV-gpt (Fig. 6, upper). Therefore disrupting the base pairing near the bottom of the 3⬘TAR hairpin does not affect HIV-1 RNA encapsidation, in contrast to what was found for the 5⬘TAR element (9). Based on these results, we would not expect 5⬘ mS-1/3⬘dS-1 or 5⬘mS-2/3⬘dS-2 to have defects in RNA pack-aging either.
We then determined the abilities of these mutants to un-dergo efficient reverse transcription in infected-cell cultures using the semiquantitative PCR assay. DNase I-treated stocks were used to infect COS cell cultures as described above. After 90 min at 37°C, cell monolayers were extensively washed and refed with media with or without AZT and then harvested about 20 h postinfection. Appropriate dilutions of total-cell lysates, which contained approximately equal amounts of the cellular gene CCR5, were prepared (Fig. 6C). These lysates were then used in hot PCRs to amplify the various products of reverse transcription. Lysates from both the 3⬘TAR dS-1- and 3⬘ TAR dS-2-infected cells contained significantly less DNA products than lysates from wild-type HIV-gpt-infected cells (Fig. 6A and B). Phosphorimaging the gels indicated that the 3⬘TAR dS-1 and dS-2 lysates contained at least 5- to 10-fold-less DNA products than the wild-type lysates. Double mu-tants 5⬘mS-1/3⬘dS-1 and 5⬘mS-2/3⬘dS-2 produced wild-type amounts of DNA products in infected cells at about 20 h postinfection, indicating that there was no obvious defect in reverse transcription in either mutant (Fig. 6A and B). The infectivities of the 3⬘ TAR dS-1 and 5⬘ TAR mS-1/3⬘ dS-1
FIG. 3. Semiquantitative PCR analysis of the efficiency of reverse transcrip-tion by the 5⬘, the 3⬘, and the 5⬘-3⬘mS-2 and mS-3 mutant virions. Infections were performed as described for Fig. 2. Total-cell lysates, harvested 20 h postinfection, were assayed for the presence of strong-stop (A) or full-length (B) viral DNAs. To verify that approximately equal amounts of host-cell derived nucleic acids were present in the samples, PCR was performed using a primer pair that am-plifies the cellular gene CCR5 (C), as for Fig. 2.
FIG. 4. Infectivities of the mS-1, mS-2, and mS-3 series mutants compared to the wild-type HIV-gpt. Infectivities of the constructs are expressed as thegpt⫹ CFU per microgram of the viral capsid protein p24 on HOS cells. The standard deviations from duplicate infection assays are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
virions were determined using the colony formation assay on HOS cell cultures. The 3⬘ TAR dS-1 virions had about a 10-fold reduction in infectivity compared to the parental HIV-gpt virions (Fig. 7). In contrast, the double mutant 5⬘mS-1/3⬘dS-1 had an essentially wild-type infectivity (Fig. 7). Therefore, these data are consistent with the idea that it is the proximity of the mismatches to the U3-R junction, and not the destabi-lization of the overall base pairing between the⫺sssDNA and 3⬘ acceptor, that causes the defects in reverse transcription. This is based on the fact that the 3⬘ TAR dS-1 and dS-2 mutants have the same number of base pair mismatches as the double 5⬘mS-1/3⬘dS-1 and 5⬘mS-2/3⬘dS-2 mutants; however, only the double R mutants are able to efficiently undergo reverse transcription (Fig. 5C and D). We can also conclude that certain mutations farther than 47 bp from the U3-R junc-tion do not cause defects in reverse transcripjunc-tion efficiency or in stable provirus formation.
Mutations farther than 10 nucleotides downstream from the U3-R junction do not cause defective reverse transcription in infected cells and define a region of R that is sensitive to
mismatches.In order to define the region of R in which
mis-matches cause defects in reverse transcription, we introduced a series of additional mutations into the 5⬘, the 3⬘, or both R elements. We called these new mutations, which introduce altered stem sequences into the 5⬘and 3⬘TAR elements, mS-4, mS-5, and mS-6 (Fig. 1B). We introduced this type of muta-tion, which maintains 5⬘TAR stem base pairing, so as not to
[image:6.612.127.477.67.382.2]cause defects in genomic RNA encapsidation (9, 29). Those constructs in which the mutations were introduced into only the 5⬘ R were called 5⬘ TAR mS-4, mS-5, and mS-6. Those constructs harboring the mutations in only the 3⬘ R were named 3⬘TAR mS-4, mS-5, and mS-6, while the double mu-tants containing the altered sequences in both repeats were called 5⬘-3⬘TAR mS-4, mS-5, and mS-6. These viral constructs were transfected into 293T cells along with the AMLV enve-lope expression vector, as described above. After about 48 h, supernatants were harvested and filtered through 0.45- m-pore-size filters and assayed for p24 concentrations by enzyme-linked immunosorbent assay as described above. Infectious supernatants containing equal amounts of p24 were treated with DNase I and then used to infect COS cell cultures, in either the presence or absence of AZT (10M), as before. Total-cell lysates were prepared about 20 h postinfection, and appropriate dilutions were assayed for the presence of approx-imately equal amounts of host cell CCR5 by hot semiquanti-tative PCR (Fig. 8B [lower two panels] and C). Lysates were then used in the semiquantitative PCR assay with primers specific for either strong-stop or nearly full-length HIV-1 DNA (Fig. 8). We initially characterized the 5⬘TAR mS-4, mS-5, and mS-6 mutants with primers specific for strong-stop and full-length DNAs (Fig. 8A and B, upper panel). None of these single 5⬘ R mutants appeared to have reduced amounts of reverse transcription products in host cell lysates compared to wild-type HIV-gpt (Fig. 8, upper panel). We next tested all FIG. 5. Diagram of the first-strand transfer of reverse transcription with some of the R mutants used in this study. Potential mismatches between the⫺sssDNA and 3⬘acceptor R sequences are indicated as bulges. The 5⬘TAR mS-1, mS-2, and mS-3 virions would have two mismatched regions (A), as would the 3⬘TAR mS-1, mS-2, and mS-3 mutants (B). The 3⬘TAR dS-1 and dS-2 virions would have one mismatched region within five nucleotides of the U3-R junction (C). The double mutants 5⬘mS-1/3⬘dS-1 and 5⬘mS-2/3⬘dS-2 would also contain only one mismatched region that would be 51 and 47 bp from the U3-R junction, respectively (D). Thick lines, RNA; thin lines, newly synthesized DNA. The relative positions of the U3, R, U5, and primer binding site (PBS) elements are indicated. The primer tRNAlysis shown
as a curved line (tRNA), and point mutations are denoted with an X. The diagram is not drawn to scale.
on November 9, 2019 by guest
http://jvi.asm.org/
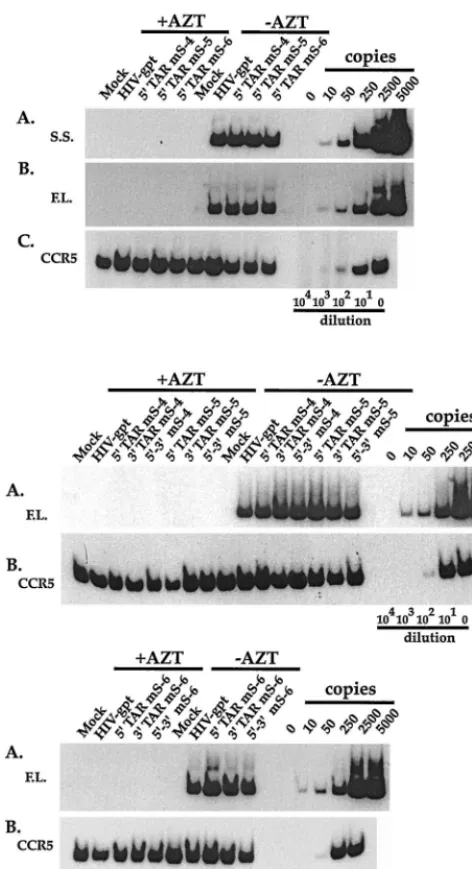
nine of the 5⬘, 3⬘, and double R mutants (Fig. 8, lower two panels). None of the six single (5⬘TAR mS-4 to mS-6 and 3⬘ TAR mS-4 to mS-6) or 3 double (5⬘-3⬘TAR mS-4 to mS-6) R mutants appeared to have consistently reduced amounts of full-length DNA in infected-cell lysates compared to wild-type HIV-gpt (Fig. 8A, lower two panels). To confirm these obser-vations and to determine if this proviral DNA could be stably maintained in infected cells, the infectivities of these mutants were determined using the colony formation assay. Equal amounts of p24 were used to infect HOS cell cultures. After 24 h, infected cells were placed into drug selection media and
grown for approximately 18 days before being fixed, stained, and counted. All nine mutants had approximately wild-type infectivities, confirming that these mutants were not defective for reverse transcription (Fig. 9). Therefore, certain mutations greater than 10 bp downstream from the U3-R junction do not interfere with reverse transcription or viral infectivity.
The inability of mutants to efficiently undergo reverse tran-scription in infected cells is not an intrinsic property of the
virion particles.Our results suggest that mismatches between
the ⫺sssDNA and 3⬘ R sequences near the U3-R boundary cause defects in reverse transcription during infection. While these mutations likely cause defects before or at the time of the first-strand transfer, it is possible that they somehow interfere with the initiation of reverse transcription in infected cells. We have not detected wild-type levels of strong-stop DNA in ly-sates from cells infected with the 5⬘ or the 3⬘ mS-1 to mS-3 single R mutants even at earlier times postinfection (data not shown). Therefore, if the⫺sssDNAs are being synthesized at normal levels by these single mutants, they are being rapidly degraded. This putative degradation of mismatched⫺sssDNA to the 3⬘repeat RNA might be occurring through the actions of cellular nucleases. If this were true, these defects in reverse transcription would not be expected to occur during the en-dogenous reverse transcriptase reaction, since no cellular nucleases would be present.
The ability of these mutants to undergo reverse transcription was determined using a previously described semiquantitative endogenous reverse transcriptase assay that utilizes virions which have been disrupted with small amounts of detergent (38). Viral stocks containing wild-type HIV-gpt or the 5⬘, the 3⬘, or the 5⬘-3⬘mS-1, mS-2, and mS-3 virions were treated with DNase I to remove extravirion DNA. Viral particles in these supernatants were then purified by centrifugation through 20% sucrose. After p24 capsid concentrations were determined, equal amounts of virions were used in the endogenous reaction without the addition of exogenous primers or templates. As a control for the synthesis of viral DNA during the endogenous reaction, identical incubations were performed without the addition of deoxynucleotide dTTP. DNA products in the re-actions were then quantitated by using the above-described semiquantitative PCR analysis, with a primer pair that could amplify strong-stop DNAs (Fig. 10, S.S.). In contrast to what was found for infections, all nine of these mutants produced approximately wild-type levels of strong-stop DNA in the en-dogenous reaction (Fig. 10, S.S.). Therefore, the ability of these R mutants to initiate reverse transcription is not
[image:7.612.55.290.65.469.2]inher-FIG. 6. (Upper) Quantitative RNase protection assay. Cytoplasmic (cyto.) or virion-derived RNAs were annealed to an excess of radiolabeled riboprobe and treated with single-strand-specific RNases; protected fragments were then sep-arated on denaturing polyacrylamide gels. The positions and sizes (in nucleo-tides) of the genomic and spliced fragments are indicated to the left, while the positions of molecular weight markers (nucleotides) are shown on the right. Phosphorimaging the genomic fragments from virions revealed that 3⬘TAR dS-1 contained 160%, and 3⬘TAR dS-2 contained 115%, of the levels of genomic RNA in wild-type HIV-gpt virions. (Lower) Semiquantitative PCR analysis of the efficiency of reverse transcription by the 3⬘TAR dS-1 and dS-2 single-mutant virions and the 5⬘ mS-1/3⬘dS-1 and 5⬘mS-2/3⬘dS-2 double-mutant virions. Infections were performed as for Fig. 2. Total-cell lysates, harvested 20 h postin-fection, were assayed for the presence of strong-stop (A) or full-length (B) viral DNAs. To verify that approximately equal amounts of host cell-derived nucleic acids were present in the samples, PCR was performed using a primer pair that amplifies the cellular gene CCR5 (C), as for Fig. 2.
FIG. 7. Infectivities of the 3⬘TAR dS-1 and 5⬘TAR mS-1/3⬘dS-1 mutants compared to that of the wild-type HIV-gpt. Infectivities of the constructs are expressed as thegpt⫹CFU per microgram of the viral capsid protein p24 on HOS cells. The standard deviations from duplicate infection assays are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.314.546.557.693.2]ently reduced in disrupted virions. This also suggests that the amount of primer tRNA incorporated into these virions is not significantly altered and is not the cause of their reduced ability to synthesize DNA in infected cells. The ability of the mS-3 series virions to synthesize reverse transcription products after the second-strand transfer was also determined by using a primer pair that amplifies nearly full-length DNAs. This anal-ysis revealed that mS-3 mutant virions were as efficient as wild-type HIV-gpt in synthesizing late DNA products (Fig. 10, F.L.). Therefore, these 5⬘ or 3⬘ R mutant virions are not in-herently defective for DNA synthesis, but the phenotype ap-pears during infection of host cells. These results are consistent
with the idea that the strong-stop DNAs are being degraded in host cells as a result of certain mismatches between the ⫺sssDNA and 3⬘R sequences that are located near the U3-R boundary. Alternatively, the virion-disrupted endogenous
[image:8.612.54.290.71.505.2]re-FIG. 8. (Upper panel) Semiquantitative PCR of the efficiency of reverse transcription by the 5⬘TAR mS-4, mS-5, and mS-6 mutant virions. (Lower two panels) Semiquantitative PCR analysis of the efficiency of reverse transcription by the 5⬘, the 3⬘, and the 5⬘-3⬘mS-4, mS-5, and mS-6 mutant virions. Infections were performed as for Fig. 2. Total-cell lysates, harvested 20 h postinfection, were assayed for the presence of strong-stop (upper panel, A) and full-length (upper panel, B; lower two panels, A) viral DNAs. To verify that approximately equal amounts of host-cell derived nucleic acids were present in the samples, PCR was performed using a primer pair that amplifies the cellular gene CCR5 (upper panel, C; lower two panels, B), as for Fig. 2.
FIG. 9. Infectivities of the mS-4, mS-5, and mS-6 series mutants compared to that of the wild-type HIV-gpt. Infectivities of the constructs are expressed as the
gpt⫹CFU per microgram of the viral capsid protein p24 on HOS cells. The standard deviations from duplicate infection assays are indicated.
FIG. 10. Semiquantitative PCR analysis of endogenous reverse transcriptase reactions using purified 5⬘, 3⬘, or 5⬘-3⬘mS-1, mS-2, and mS-3 virions. Viral stocks (12 ml) were treated with DNase I before pseudotyped virions were pelleted through 20% sucrose. Pellets were resuspended in 100l of TEN buffer (Ma-terials and Methods). Aliquots containing 10 ng of p24 were incubated in en-dogenous reaction mixtures containing 100 M concentrations of each de-oxynucleotide with or without dTTP. As an additional control, reactions were performed on “pelleted” mock-transfected supernatants that were treated ex-actly as the virion-containing samples (Mock). After the endogenous reaction, virions were digested with proteinase K. To detect newly synthesized viral DNAs, aliquots were amplified using the hot semiquantitative PCR assay, as described in Materials and Methods. Samples were assayed for the presence of strong-stop DNA products (S.S.). The mS-3 series mutants were also assayed for nearly full-length DNAs (F.L.). PCR standards are shown for reaction mixtures that contained 10, 50, 250, 2,500, and 5,000 copies of viral DNA in an HIV-gpt vector.
on November 9, 2019 by guest
http://jvi.asm.org/
verse transcription reaction is not reflecting what is happening in infected cells.
DISCUSSION
We have utilized a single-round replication system to study the effects of mutations in the HIV-1 terminal repeats on reverse transcription efficiency. We provide genetic evidence indicating that mutations which introduce mismatches of 3 bp or more within 10 nucleotides downstream of the HIV-1 U3-R border, between the⫺sssDNA and the 3⬘ repeat RNA se-quences, result in viruses that are unable to efficiently undergo reverse transcription in infected cells. We detected greatly reduced levels of even the initial products of reverse transcrip-tion, the⫺sssDNAs, in lysates from cells infected with these mutants. However, these same mutations do not cause defects in initial DNA synthesis in disrupted virions during an endog-enous reverse transcription reaction. These data indicate that reverse transcription is not intrinsically defective in these mu-tants, at least in the presence of detergent. They are consistent with the idea that⫺sssDNAs which have misalignments near the polymerization site at the 3⬘R, near the U3-R junction, are rapidly degraded in infected cells. These data further indicate that, during an infection, most HIV-1 virions synthesize full-length or near-full-full-length⫺sssDNAs that have copied the en-tire 5⬘R and that are then transferred to the 3⬘ R acceptor leading to full-length minus-strand cDNA, in agreement with previous reports (33). Otherwise, we would not have observed defective reverse transcription with most of the mutants used in this study which introduce near-terminal mismatches be-tween full-length⫺sssDNA and the 3⬘ R. Premature “jump-ing” would not lead to mismatches with most of the mutations used here. These results also have implications for the posi-tions of important contacts between the reverse transcriptase enzyme and primer template strands during the first-strand transfer reaction.
In vitro studies, with synthetic donor and acceptor molecules and purified reverse transcriptase, have been used to examine aspects of strand switching in many retroviral systems (1–3, 6, 16, 22–25, 27, 42, 51). For HIV-1, it has been shown that the first-strand transfer is greatly facilitated by the nucleocapsid (NC) protein, which is known to have nucleic acid chaperone-like activities (7, 16, 21, 27, 30, 36, 41, 51). A study in which mismatches were introduced between the donor DNA and acceptor RNA molecules revealed that misalignments at the 3⬘ polymerization site of the donor were especially detrimental to continued reverse transcription. HIV-1 reverse transcriptase was able to extend primers containing 5-bp terminal mis-matches at about 3 to 5% of the efficiency of wild-type controls, depending on the concentration of NC protein (36). Others have also observed that reverse transcriptase is able to extend mismatched primer termini in vitro (36, 43, 44, 53).
Studies have examined the effects of mutations in R se-quences on replication and provirus formation using sev-eral retroviral systems. A study using replication-competent MMLV showed that the 5⬘ repeat element was not always completely copied before it was translocated to the 3⬘acceptor (37). This led to the eventual loss of insertion and deletion mutations introduced into either the 5⬘or the 3⬘R and caused the eventual outgrowth of wild-type virus. Virions harboring these same mutations in both R elements stably maintained them (37). A spleen necrosis virus (SNV) single-round repli-cation system was used to determine the frequency of prema-ture first-strand transfers (45). By placing a genetic marker into the middle of the 5⬘or 3⬘SNV R, it was determined that about 10% of progeny virions resulted from premature strand
trans-fers which occurred before the entire 5⬘ R was copied (45). Utilizing point mutations near the U3-R border, another group determined the sites of the first-strand transfers in an MMLV-based system and showed that premature transfers occurred in 1 to 2% of DNA synthesized during reverse transcription (34). Studies with HIV-1 have also revealed that the first-strand transfer occurs prematurely in a small number of cases (33). Our data are in general agreement with these studies, because the infectivities of our mutants, which introduced near-termi-nal mismatches between the⫺sssDNA and 3⬘R, were reduced by between 90 and 99% compared to that of the wild type. This suggests that in 1 to 10% of viruses, a premature first-strand transfer occurred before the mutations were copied, thus in-troducing no mismatches. Alternatively, our results may indi-cate that in 1 to 10% of viruses, reverse transcription was able to bypass the nearly terminal mismatches in some manner. A study using an infectious clone of HIV-1 in which the 3⬘R was progressively truncated from its 3⬘ end showed that much shorter acceptors, as short as 30 nucleotides, could work effi-ciently during the first-strand transfer (5). These mutations did not generally introduce mismatches near the U3-R border. More recently a series of viruses with deletion and substitution mutations spanning the 3⬘U3-R region were used to examine the first-strand transfer in MMLV by means of a single-round replication system (49). Small and large deletions in the 3⬘R sequences slightly downstream of the U3-R junction reduced viral titers between one- and fivefold. A virus harboring a mutation altering the first five bases of the 3⬘R had a fivefold titer reduction compared to the wild type. Fairly large deletion mutations that spanned the U3-R junction, in contrast, re-duced titers between 25- and 200-fold. Based on these and other data, the authors concluded that complementarity-inde-pendent mechanisms at the U3-R junction were sufficient to direct the first-strand transfer in MMLV (49). Taking these results together with our results, it seems that the first-strand transfer in HIV-1 has significant differences with that in MMLV. Reverse transcription in HIV-1 appears to be more sensitive to small mutations near the U3-R border, significantly reducing viral infectivities. A 3-bp mutation reduced titers by 10-fold, while a 4-bp mutation reduced viral infectivity by 100-fold in our HIV-1 system. Therefore, small mismatches be-tween the⫺sssDNA and the 3⬘acceptor are quite deleterious to efficient reverse transcription in HIV-1. Although our study did not exhaustively address the contribution(s) of the TAR hairpin structures to the first-strand transfer of reverse tran-scription, we can make several conclusions. Two mutants used in this study introduced disruptions into the lower stem of the 3⬘TAR hairpin, 5⬘mS-1/3⬘dS-1 and 5⬘mS-2/3⬘dS-2. Both of these double mutants appeared to undergo efficient reverse transcription (Fig. 6), and the 5⬘ TAR mS-1/3⬘ dS-1 mutant had wild-type infectivity (Fig. 7). We conclude that these two disruptions, of four and eight stem base pairs, did not signifi-cantly reduce reverse transcription efficiency. Therefore, our results agree with a recent study that found no evidence that TAR structure had a significant effect on the mechanism of reverse transcription (18).
These data agree well with previous studies that have exam-ined the effects of terminal or near-terminal misalignments between the primer tRNAlys and the primer binding site of HIV-1 (17, 46, 50). Previous studies have shown that the first six nucleotides of the primer binding site are sufficient for primer tRNA binding and initiation of reverse transcription in HIV-1 mutants (50). When two- to four-nucleotide insertion and deletion mutations were introduced into the HIV-1 primer binding site, those that were closest to the 3⬘tRNA polymer-ization site had the most severe defects in viral replication (17).
on November 9, 2019 by guest
http://jvi.asm.org/
In vitro experiments revealed that tRNA priming was reduced because of these misalignments (17). Mutations in the primer binding site also affect the second-strand transfer of reverse transcription which utilizes the 18-nucleotide primer binding site sequence. This can cause even further reductions in viral replication in some primer binding site mutant virions. Our results suggest that the first-strand transfer in HIV-1 is similar in certain respects to the initiation of reverse transcription and to the second-strand transfer; mutations that lead to near-terminal mismatches between the primer and template strands cause marked defects in reverse transcription efficiency and viral infectivity. Our results are consistent with the idea that before or during the first-strand switch in HIV-1, these mis-matches lead to the rapid degradation of the⫺sssDNAs in infected cells by host-encoded nucleases. However, we realize that we have presented no direct evidence that the⫺sssDNAs are being rapidly turned over in infected cells. It is possible, although unlikely, that the initiation of reverse transcription is reduced in these mutants in cells and that this is not reflected in the disrupted-virion endogenous reverse transcription assay. Nonetheless, our data clearly indicate that sequences within the first 10 nucleotides of either repeat are crucial for efficient reverse transcription. The precise mechanism by which these mutations reduce reverse transcription awaits the results of further studies. These data may be useful is designing novel strategies for interfering with reverse transcription, a critical step in the viral life cycle.
ACKNOWLEDGMENTS
We thank T. G. Parslow for the use of certain reagents such as plasmid DNA, and we thank I. S. Y. Chen and E. Hunter for helpful discussions.
This research was supported, in part, by an award to the University of Texas Health Science Center at San Antonio from the Research Resources Program for Medical Schools from the Howard Hughes Medical Institute. Additional support was provided by a grant from the UTHSCSA CREF fund for new faculty.
REFERENCES
1.Allain, B., M. Lapadat-Tapolsky, C. Berlioz, and J. L. Darlix.1994. Trans-activation of the minus-strand DNA transfer by nucleocapsid protein during reverse transcription of the retroviral genome. EMBO J.13:973–981. 2.Allain, B., J. B. Rascle, H. de Rocquigny, B. Roques, and J. L. Darlix.1998.
CIS elements and acting factors required for minus strand DNA trans-fer during reverse transcription of the genomic RNA of murine leukemia virus. J. Mol. Biol.277:225–235.
3.Anderson, J. A., R. J. Teufel II, P. D. Yin, and W. S. Hu.1998. Correlated template-switching events during minus-strand DNA synthesis: a mechanism for high negative interference during retroviral recombination. J. Virol.72:
1186–1194.
4.Berkhout, B.1996. Structure and function of the human immunodeficiency virus leader RNA. Prog. Nucleic Acid Res. Mol. Biol.54:1–34.
5.Berkhout, B., J. van Wamel, and B. Klaver.1995. Requirements for DNA strand transfer during reverse transcription in mutant HIV-1 virions. J. Mol. Biol.252:59–69.
6.Buiser, R. G., R. A. Bambara, and P. J. Fay.1993. Pausing by retroviral DNA polymerases promotes strand transfer from internal regions of RNA donor templates to homopolymeric acceptor templates. Biochim. Biophys. Acta.
1216:20–30.
7.Cameron, C. E., M. Ghosh, S. F. Le Grice, and S. J. Benkovic.1997. Muta-tions in HIV reverse transcriptase which alter RNase H activity and decrease strand transfer efficiency are suppressed by HIV nucleocapsid protein. Proc. Natl. Acad. Sci. USA94:6700–6705.
8.Clever, J., C. Sassetti, and T. G. Parslow.1995. RNA secondary structure and binding sites forgaggene products in the 5⬘packaging signal of human immunodeficiency virus type 1. J. Virol.69:2101–2109.
9.Clever, J. L., D. A. Eckstein, and T. G. Parslow.1999. Genetic dissociation of the encapsidation and reverse transcription functions in the 5⬘R region of human immunodeficiency virus type 1. J. Virol.73:101–109.
10. Clever, J. L., and T. G. Parslow.1997. Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. J. Virol.71:3407–3414.
11. Clever, J. L., R. A. Taplitz, M. A. Lochrie, B. Polisky, and T. G. Parslow.
2000. A heterologous, high-affinity RNA ligand for human immunodefi-ciency virus Gag protein has RNA packaging activity. J. Virol.74:541–546. 12. Coffin, J. M.1979. Structure, replication, and recombination of retrovirus
genomes: some unifying hypotheses. J. Gen. Virol.42:1–26.
13. Coffin, J. M., S. H. Hughes, and H. E. Varmus.1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
14. Collin, M., G. Herbein, L. Montaner, and S. Gordon.1993. PCR analysis of HIV1 infection of macrophages: virus entry is CD4-dependent. Res. Virol.
144:13–19.
15. Cullen, B. R.1998. HIV-1 auxiliary proteins: making connections in a dying cell. Cell93:685–692.
16. Darlix, J. L., A. Vincent, C. Gabus, H. de Rocquigny, and B. Roques.1993. Trans-activation of the 5⬘to 3⬘viral DNA strand transfer by nucleocapsid protein during reverse transcription of HIV1 RNA. C. R. Acad. Sci.316:763– 771.
17. Das, A. T., and B. Berkhout.1995. Efficient extension of a misaligned tRNA-primer during replication of the HIV-1 retrovirus. Nucleic Acids Res.23:
1319–1326.
18. Das, A. T., B. Klaver, and B. Berkhout.1998. The 5⬘and 3⬘TAR elements of human immunodeficiency virus exert effects at several points in the virus life cycle. J. Virol.72:9217–9223.
19. Das, A. T., B. Klaver, and B. Berkhout.1999. A hairpin structure in the R region of the human immunodeficiency virus type 1 RNA genome is instru-mental in polyadenylation site selection. J. Virol.73:81–91.
20. Das, A. T., B. Klaver, B. I. Klasens, J. L. van Wamel, and B. Berkhout.1997. A conserved hairpin motif in the R-U5 region of the human immunodefi-ciency virus type 1 RNA genome is essential for replication. J. Virol.71:
2346–2356.
21. DeStefano, J. J.1996. Interaction of human immunodeficiency virus nucleo-capsid protein with a structure mimicking a replication intermediate. Effects on stability, reverse transcriptase binding, and strand transfer. J. Biol. Chem.
271:16350–16356.
22. DeStefano, J. J., R. G. Buiser, L. M. Mallaber, T. W. Myers, R. A. Bambara, and P. J. Fay.1991. Polymerization and RNase H activities of the reverse transcriptases from avian myeloblastosis, human immunodeficiency, and Moloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem.
266:7423–7431.
23. DeStefano, J. J., L. M. Mallaber, L. Rodriguez-Rodriguez, P. J. Fay, and R. A. Bambara.1992. Requirements for strand transfer between internal regions of heteropolymer templates by human immunodeficiency virus re-verse transcriptase. J. Virol.66:6370–6378.
24. Diaz, L., and J. J. DeStefano.1996. Strand transfer is enhanced by mis-matched nucleotides at the 3⬘primer terminus: a possible link between HIV reverse transcriptase fidelity and recombination. Nucleic Acids Res.24:
3086–3092.
25. Garces, J., and R. Wittek.1991. Reverse-transcriptase-associated RNaseH activity mediates template switching during reverse transcription in vitro. Proc. R. Soc. London Ser. B243:235–239.
26. Gilboa, E., S. W. Mitra, S. Goff, and D. Baltimore.1979. A detailed model of reverse transcription and tests of crucial aspects. Cell18:93–100. 27. Guo, J., L. E. Henderson, J. Bess, B. Kane, and J. G. Levin.1997. Human
immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-depen-dent self-priming from minus-strand strong-stop DNA. J. Virol.71:5178– 5188.
28. Harrich, D., C. Ulich, and R. B. Gaynor.1996. A critical role for the TAR element in promoting efficient human immunodeficiency virus type 1 reverse transcription. J. Virol.70:4017–4027.
29. Helga-Maria, C., M. L. Hammarskjold, and D. Rekosh.1999. An intact TAR element and cytoplasmic localization are necessary for efficient packaging of human immunodeficiency virus type 1 genomic RNA. J. Virol.73:4127–4135. 30. Kim, J. K., C. Palaniappan, W. Wu, P. J. Fay, and R. A. Bambara.1997. Evidence for a unique mechanism of strand transfer from the transactivation response region of HIV-1. J. Biol. Chem.272:16769–16777.
31. Klasens, B. I., A. T. Das, and B. Berkhout.1998. Inhibition of polyadenyl-ation by stable RNA secondary structure. Nucleic Acids Res.26:1870–1876. 32. Klasens, B. I., M. Thiesen, A. Virtanen, and B. Berkhout.1999. The ability of the HIV-1 AAUAAA signal to bind polyadenylation factors is controlled by local RNA structure. Nucleic Acids Res.27:446–454.
33. Klaver, B., and B. Berkhout.1994. Premature strand transfer by the HIV-1 reverse transcriptase during strong-stop DNA synthesis. Nucleic Acids Res.
22:137–144.
34. Kulpa, D., R. Topping, and A. Telesnitsky.1997. Determination of the site of first strand transfer during Moloney murine leukemia virus reverse tran-scription and identification of strand transfer-associated reverse transcrip-tase errors. EMBO J.16:856–865.
35. Landau, N. R., K. A. Page, and D. R. Littman.1991. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency virus host range. J. Virol.65:162–169.
36. Lapadat-Tapolsky, M., C. Gabus, M. Rau, and J. L. Darlix.1997. Possible roles of HIV-1 nucleocapsid protein in the specificity of proviral DNA synthesis and in its variability. J. Mol. Biol.268:250–260.
on November 9, 2019 by guest
http://jvi.asm.org/
37.Lobel, L. I., and S. P. Goff.1985. Reverse transcription of retroviral ge-nomes: mutations in the terminal repeat sequences. J. Virol.53:447–455. 38. Masuda, T., V. Planelles, P. Krogstad, and I. S. Y. Chen.1995. Genetic
analysis of human immunodeficiency virus type 1 integrase and the U3att
site: unusual phenotype of mutants in the zinc finger-like domain. J. Virol.
69:6687–6696.
39. McBride, M. S., M. D. Schwartz, and A. T. Panganiban.1997. Efficient encapsidation of human immunodeficiency virus type 1 vectors and further characterization ofciselements required for encapsidation. J. Virol.71:
4544–4554.
40. Page, K. A., N. R. Landau, and D. R. Littman.1990. Construction and use of a human immunodeficiency virus vector for analysis of virus infectivity. J. Virol.64:5270–5276.
41. Peliska, J. A., S. Balasubramanian, D. P. Giedroc, and S. J. Benkovic.1994. Recombinant HIV-1 nucleocapsid protein accelerates HIV-1 reverse tran-scriptase catalyzed DNA strand transfer reactions and modulates RNase H activity. Biochemistry33:13817–13823.
42. Peliska, J. A., and S. J. Benkovic.1992. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science258:1112–1118. 43. Perrino, F. W., B. D. Preston, L. L. Sandell, and L. A. Loeb.1989. Extension of mismatched 3⬘termini of DNA is a major determinant of the infidelity of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA86:8343–8347.
44. Preston, B. D., and J. P. Dougherty.1996. Mechanisms of retroviral muta-tion. Trends Microbiol.4:16–21.
45. Ramsey, C. A., and A. T. Panganiban.1993. Replication of the retroviral
terminal repeat sequence during in vivo reverse transcription. J. Virol.67:
4114–4121.
46. Rhim, H., J. Park, and C. D. Morrow.1991. Deletions in the tRNA(Lys) primer-binding site of human immunodeficiency virus type 1 identify essen-tial regions for reverse transcription. J. Virol.65:4555–4564.
47. Swanstrom, R., H. E. Varmus, and J. M. Bishop.1981. The terminal redun-dancy of the retrovirus genome facilitates chain elongation by reverse tran-scriptase. J. Biol. Chem.256:1115–1121.
48. Telesnitsky, A., and S. P. Goff.1993. Strong-stop strand transfer during reverse transcription, p. 49–83.InA. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. 49. Topping, R., M. A. Demoitie, N. H. Shin, and A. Telesnitsky.1998. Cis-acting elements required for strong stop acceptor template selection during Molo-ney murine leukemia virus reverse transcription. J. Mol. Biol.281:1–15. 50. Wakefield, J. K., H. Rhim, and C. D. Morrow.1994. Minimal sequence
requirements of a functional human immunodeficiency virus type 1 primer binding site. J. Virol.68:1605–1614.
51. You, J. C., and C. S. McHenry.1994. Human immunodeficiency virus nu-cleocapsid protein accelerates strand transfer of the terminally redundant sequences involved in reverse transcription. J. Biol. Chem.269:31491–31495. 52. Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Chen.
1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell61:213–222.
53. Zinnen, S., J. C. Hsieh, and P. Modrich.1994. Misincorporation and mis-paired primer extension by human immunodeficiency virus reverse transcrip-tase. J. Biol. Chem.269:24195–24202.