NP1 Protein of the
Bocaparvovirus
Minute Virus of Canines Controls
Access to the Viral Capsid Genes via Its Role in RNA Processing
Olufemi O. Fasina, Yanming Dong, David J. Pintel
Department of Molecular Microbiology and Immunology, University of Missouri—Columbia, School of Medicine, Bond Life Sciences Center, Columbia, Missouri, USA
ABSTRACT
Minute virus of canines (MVC) is an autonomous parvovirus in the genusBocaparvovirus. It has a single promoter that gener-ates a single pre-mRNA processed via alternative splicing and alternative polyadenylation to produce at least 8 mRNA tran-scripts. MVC contains two polyadenylation sites, one at the right-hand end of the genome, (pA)d, and another complex site, (pA)p, within the capsid-coding region. During viral infection, the mRNAs must extend through (pA)p and undergo additional splicing of the immediately upstream 3D⁄3A intron to access the capsid gene. MVC NP1 is a 22-kDa nuclear phosphoprotein unique to the genusBocaparvovirusof theParvovirinaewhich we have shown governs suppression of (pA)p independently of viral genome replication. We show here that in addition to suppression of (pA)p, NP1 is also required for the excision of the MVC 3D⁄3A intron, independently of its effect on alternative polyadenylation. Mutations of the arginine⁄serine (SR) di-repeats within the intrinsically disordered amino terminus of NP1 are required for splicing of the capsid transcript but not suppression of polyadenylation at (pA)p. 3=-end processing of MVC mRNAs at (pA)p is critical for viral genome replication and the optimal expression of NP1 and NS1. Thus, a finely tuned balance between (pA)p suppression and usage is necessary for efficient virus replication. NP1 is the first parvovirus protein implicated in RNA processing. Its characterization reveals another way that par-voviruses govern access to their capsid protein genes, namely, at the RNA level, by regulating the essential splicing of an intron and the suppression of an internal polyadenylation site.
IMPORTANCE
TheParvovirinaeare small nonenveloped icosahedral viruses that are important pathogens in many animal species, including humans. Although parvoviruses have only subtle early-to-late expression shifts, they all regulate access to their capsid genes. Minute virus of canines (MVC) is an autonomous parvovirus in the genusBocaparvovirus. It has a single promoter generating a single pre-mRNA which is processed via alternative splicing and alternative polyadenylation to generate at least 8 mRNA tran-scripts. MVC contains two polyadenylation sites, one at the right-hand end of the genome, (pA)d, and another, (pA)p, within the capsid-coding region. It had not been clear how the potent internal polyadenylation motif is suppressed to allow processing, ex-port, and accumulation of the spliced capsid protein-encoding mRNAs. We show here that MVC NP1, the first parvovirus pro-tein to be implicated in RNA processing, governs access to the MVC capsid gene by facilitating splicing and suppressing internal polyadenylation of MVC pre-mRNAs.
M
inute virus of canines (MVC) is an autonomous parvovirus in the genusBocaparvovirus. Infection in young dogs can result in neonatal mortality (1) and respiratory and gastrointesti-nal distress (2) and can cause infertility and stillbirths in adult pregnant female dogs (3). The parvovirus genusBocaparvovirushas 12 currently known species. MVC, bovine parvovirus (BPV), and the recently identified human bocavirus 1 (HBoV1), which has been suggested to be a human pathogen, are the most com-monly studied (4,5). MVC, which can be propagated readily in tissue culture and for which there is an infectious clone, has proven to be a useful prototype for characterization of members of this genus. Parvoviruses have very small single-stranded DNA ge-nomes (⬃5 kb) and use extensive RNA processing strategies to maximize their coding capacity (6).The MVC genome generates a single pre-mRNA that is alternatively spliced and alternatively polyadenylated to generate at least 8 mRNAs (seeFig. 1) (7,8). MVC encodes two sets of nonstructural proteins essential for rep-lication, a larger set of nonstructural proteins that share overlap-ping coding open reading frames (ORFs) and are analogous to the MVM NS and AAV Rep proteins, and another 186 amino acid nonstructural protein, NP1, unique to theBocaparvovirusgenus (9–12). MVC has two polyadenylation sites, a proximal site,
(pA)p, near the center of the genome within the VP1 capsid cod-ing region, and a distal site, (pA)d, at the right-hand end (8). While mRNAs using the internal polyadenylation site could po-tentially encode the viral nonstructural proteins, approximately 60 to 70% of mRNAs read through (pA)p and undergo additional splicing of the immediately upstream 3D⁄3A intron to access the capsid gene and terminate using (pA)d (7).
Although parvoviruses have only subtle early-to-late expres-sion shifts, they all regulate access to their capsid genes, and dif-ferent parvoviruses do so in difdif-ferent ways. The Dependoparvovi-rusadeno-associated virus type 2 (AAV2) and Protoparvovirus
Received12 October 2015Accepted20 November 2015
Accepted manuscript posted online4 December 2015
CitationFasina OO, Dong Y, Pintel DJ. 2016. NP1 protein of theBocaparvovirus
minute virus of canines controls access to the viral capsid genes via its role in RNA processing. J Virol 90:1718 –1728.doi:10.1128/JVI.02618-15.
Editor:G. McFadden
Address correspondence to David J. Pintel, [email protected]. Copyright © 2016, American Society for Microbiology. All Rights Reserved.
on November 7, 2019 by guest
http://jvi.asm.org/
minute virus of mice (MVM) use transactivation of a capsid gene promoter to generate capsid protein-encoding mRNAs (13–15). For other parvoviruses that have an internal polyadenylation site, such as adeno-associated virus type 5 (AAV5),Erythroparvovirus
B19, and theDependoparvovirusgoose parvovirus (GPV), this site typically lies within an intron whose excision allows extension of the spliced RNA into the capsid gene (6,16–18). For the bocapar-voviruses, however, these potent internal polyadenylation motifs are retained in the capsid protein-encoding cytoplasmic mRNA; thus, they must be suppressed to allow processing, export, and accumulation of the spliced capsid-encoding mRNAs in the cyto-plas, and hence production of the capsid proteins.
We recently showed that MVC NP1, a 22-kDa nuclear phos-phoprotein unique to the genusBocaparvovirusof the Parvoviri-nae, is essential for the accumulation of capsid mRNAs and capsid protein (7). This effect was independent of the viral replicator protein NS1 and viral genome replication itself. Here, we demon-strate that, in addition to suppression of (pA)p, NP1 is also spe-cifically required for the excision of the MVC 3D⁄3A intron, which lies immediately upstream of (pA)p and controls access to the VP1 open reading frame. NP1 appears to govern definition of the up-stream intron donor site, as NP1 still regulated excision of the upstream intron using a new proximal splice donor sequence in-troduced downstream of the authentic 3D signal. Mutation of viral RNA processing signals demonstrated that NP1 could affect alternative polyadenylation and splicing independently, and con-sistent with this, mutations in the SR domain of NP1 dissociated its role in splicing from its role in polyadenylation. We also show that MVC internal polyadenylation is required for viral genome replication and supports expression of viral nonstructural pro-teins NP1 and, to a lesser extent, NS1. Although NP1 is necessary to suppress internal polyadenylation, some accumulation of mRNAs using (pA)p was required to provide a continuing source of NS1 and NP1. Thus, a finely tuned balance between (pA)p read-through and usage appears to be necessary for efficient virus replication.
NP1 is the first parvovirus protein implicated in RNA process-ing, and its characterization reveals yet another way in which par-voviruses govern essential access to its capsid protein gene, namely, by the action of NP1 to both facilitate splicing and allow read-through of the internal (pA)p.
MATERIALS AND METHODS
Cells, viruses, infections, and transfections.All experiments were car-ried out as specified in the text in either Walter Reed canine cell/3873D (WRD) or 293T cells, which were propagated as previously described in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal calf serum. The minute virus of canines (MVC) used in this study was the original strain (GA3), obtained from Colin Parrish at Cornell University. The infectious molecular clone, pIMVC (GenBank accession number
FJ214110.1) used for transfection and generation of the mutants was de-scribed previously (8).The HBoV1 nonreplicating construct pHBoVm630 was kindly provided by Jianming Qiu, University of Kansas Medical Cen-ter. Transfections were performed with WRD or 293T cells using either Lipofectamine Plus (Life Technologies, CA) or LipoD293 transfection reagent (SignaGen Laboratories, MD).
RNase protection assays.Total RNA was isolated using the TRIzol reagent (Invitrogen), and RNase protection was performed as previously described (19). A number of different probes were utilized in this study: a proximal polyadenylation probe which spanned MVC nucleotides (nt) 3107 to 3333; appropriate homologous probes when the MVC proximal
polyadenylation region was replaced with heterologous sequences; the 2A3D probe (nt 2344 to 2550), used to analyze the splicing across the MVC second and third introns; the 3D=probe (nt 2660 to 2960), used to analyze splicing across the 3D=splice donor (nt 2790); and the HBoV 3A probe (nt 2945 to 3145), which protected HBoV RNA transcripts across the third intron acceptor (nt 2357 to 2955) of HBoV (GenBank accession numberDQ000496).
Antibodies.Polyclonal antibodies directed against MVC NS1, MVC NP1, and tubulin were used for immunoblotting as previously described (7). Anti-capsid antibodies produced in rabbits were kindly provided by Colin Parrish (Cornell University).
Immunoblot analyses.Cells grown and transfected in 60-mm dishes with various constructs as described below and in the figure legends were harvested 48 h posttransfection and lysed in either Laemmli buffer with 2% SDS or radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) which contained protease and phosphatase inhibitors. Bradford assay of RIPA cell lysates was carried out to deter-mine total protein concentrations in each lysate. SDS-PAGE and Western blotting were performed as previously described (7).
Southern blotting.WRD cells were transfected with pIMVC and the pIMVC-derived constructs as described below and figure legends. DNA was extracted 48 h posttransfection, and DNA replication was assayed by Southern blotting as described previously (20) using the randomly primed radiolabeled NotI-digested genomic fragment of pIMVC as the probe. Loading of samples was standardized using a Nanodrop spectro-photometer. Hybridization to mitochondrial DNA using a homologous probe was used as a loading control.
Plasmid constructs.The generation of the multiple plasmid con-structs used in this study was as follows.
pIMVC (WT), the wild-type (WT) infectious clone, was generated as previously described (8).
pIMVC 3Am, including the splice acceptor site of the third intron of MVC (nt 3037), was generated as an in-frame G-to-A substitution.
pIMVC NP1 2738TAAm has the NP1 open reading frame terminated via a TAA stop codon substitution for the codon from nt 2738 to 2740.
pIMVC NP1 5XPro mutant (5X), a functionally defective mutant of NP1, was generated by introducing five consecutive proline substitutions into the NP1 ORF at nt 2780.
The pIMVC NP1 cap fusion mutant was generated by a single nucle-otide deletion at nt 3089, resulting in fusion of NP1 and the capsid ORFs. The site of the deletion lies downstream of the 3D⁄3A intron.
pIMVC 3D=contains a substitution of the third nucleotide between nt 2780 and 2794, CACAGCACTAGATTA to CATAGTACAAGGTTG, which generated a new 5=splice site (in bold) within the NP1 ORF.
pIMVC 3D=TAA is an NP1 termination mutant which was generated in the pIMVC 3D=background via a nucleotide insertion at nt 2730 that caused premature termination in NP1 translation.
pIMVC 3DmAm (WT, 5X) was generated by debilitating the splice donor at nt 2491 via an in-frame G-to-A substitution in the NS1 ORF coupled with the third-nucleotide substitution between nt 3026 and 3040, GTTCTCACACAGGAT to GTGCTGACACAAGAC, around the third intron acceptor (in bold). The mutant was generated in both the pIMVC WT and pIMVC NP1 5XPro backgrounds.
pIMVC SK (WT, 5X), created both in the wild-type and NP1 5XPro mutant infectious-molecular-clone backgrounds, was generated by site-directed mutagenesis which introduced a SacI site and a KasI site to re-place nt 3105 to 3111 and 3348 to 3353, respectively, in order to generate the mutants described below, where the MVC proximal polyadenylation region from nt 3111 to 3353 was replaced with various gene block-synthe-sized oligonucleotides (Integrated DNA Technologies, IA).
For pIMVC BGH (WT, 5X), the bovine growth hormone polyadenyl-ation sequence (nt 1021 to 1263) was amplified from pcDNA3.1(⫹) (Life Technologies, CA) and cloned into pIMVC SK in place of MVC proximal polyadenylation elements.
on November 7, 2019 by guest
http://jvi.asm.org/
For pIMVC NP1 SRm1-5, the NP1 RS dipeptides R9, S10, R13, S14, R15, S16, R17, S18, S22, R23, S48, R49, S67, and R68 were mutated to alanine residues (seeFig. 4A).
pIMVC NP1 SRsm is a silent NP1 SR mutant generated by third-nucleotide substitution of the SR dipeptides described above (nt 2550 to 2740) (seeFig. 4A).
pIMVC pApmVP1csm (WT, 5X). The AAUAAA and GU-rich core elements within the MVC proximal polyadenylation region (nt 3111 to 3353) were disrupted by third-nucleotide substitution in these constructs. VP1 amino acid composition was conserved, in both a wild-type NP1 and 5X mutant backgrounds.
MVC RepCap RC. This is a nonreplicating construct of MVC genome that lacks the left-end hairpin, cloned into the pSK-Bluescript vector (Agi-lent, CA).
pIMVC 1D=m, an MVC nonreplicating construct, was generated by silent third-nucleotide substitutions (nt 1266 to 1280; GCCTAAGGTAC CTGA to GCCTAAAGTCCCTGA) in the NS1 ORF that inactivated the 5=
splice site required to generate spliced NS1 transcripts.
HBoV1 NP1 5XPro, a structurally defective mutant of HBoV 1NP1, was generated by introducing five consecutive proline substitutions into the NP1 ORF at nt 2649 (HBoV1 GenBank accession number
DQ000496).
HBoV1 NP1 TAAm is a translation termination mutant of NP1 cloned by a G-A nucleotide substitution at nt 2493 (HBoV1 GenBank accession numberDQ000496), resulting in an amber mutation.
HBoV1 NP1 CapF is a mutant generated by a single nucleotide dele-tion at nt 3052 (HBoV1 GenBank accession numberDQ000496), result-ing in fusion of NP1 and the capsid ORFs. The site of the deletion lies downstream of the HBoV1 3D⁄3A intron acceptor.
RESULTS
MVC and HBoV1 NP1 proteins are required for splicing the viral 3D⁄3A intron, which allows access to the VP1 AUG.We previously demonstrated that the MVC NP1 protein plays a role in alternative polyadenylation of MVC pre-mRNA (7). During wild-type infection of canine WRD cells and following transfection of either WRD or 293 cells, approximately 60 to 70% of MVC mRNAs extend through the internal polyadenylation site (pA)p into the capsid gene and terminate at (pA)d at the 3=end of the genome (7). However, in the absence of a fully functional NP1, more than 80% of MVC mRNAs accumulate as species internally polyadenylated at (pA)p (7). This implicated the NP1 protein in suppressing cleavage and or polyadenylation of MVC pre-mRNAs at the internal (pA)p site (7).
It is becoming increasingly clear that there can be important connections between splicing and polyadenylation during pro-cessing of pre-mRNAs (21–23). In addition to read-through of (pA)p, access to the VP1 AUG and hence the capsid gene open reading frame requires splicing of the proximal 3D⁄3A intron (Fig. 1A). Therefore, we sought to determine whether NP1 might also play a role in the splicing of MVC pre-mRNAs. We developed an RNase protection assay using the 2A3D probe, which spans the 3=
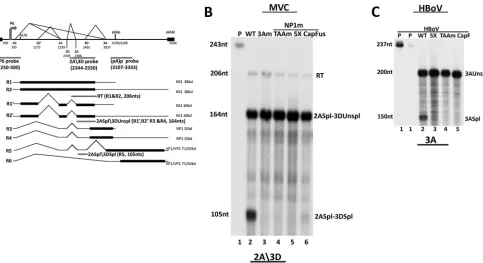
splice site of the 2D⁄2A intron and the 5=splice site of the 3D⁄3A intron (diagrammed inFig. 1A). This probe allowed us to analyze splicing of the MVC 2D⁄2A and 3D⁄3A introns lying upstream of the proximal polyadenylation site. As can be seen inFig. 1B, RNAs generated following transfection of three different NP1 mutant full-length MVC plasmid clones, TAAm, 5X, and CapFus (the last of these mutations lies downstream of the 3D⁄3A intron; all mu-tants are described in Materials and Methods), previously charac-terized as deficient in (pA)p read-through (7), were also dramat-ically deficient in splicing of the 3D⁄3A intron compared to the
wild type (Fig. 1B, compare lanes 4 to 6 to lane 2). A mutant in which the acceptor of the 3D⁄3A intron was debilitated served as the control (3Am) (Fig. 1B, lane 3). The 2A3D probe also dem-onstrated that splicing of the upstream 2D⁄2A intron was not de-creased in mutant-derived RNA (splicing was in fact slightly in-creased relative to the read-through product [RT] for reasons that are not yet clear) (Fig. 1B, compare lanes 4 to 6 to lanes 2 and 3). In addition, assays using the P6 probe, which spans the pre-mRNA start site and first intron donor D1 (also diagrammed inFig. 1A), demonstrated that mutation of NP1 had no detectable effect on splicing at D1 (data not shown). These results suggested that MVC NP1 is required for efficient splicing specifically of the MVC in-tron immediately upstream of the proximal polyadenylation sites. We have also found that NP1 of the human bocavirus HBoV1 was also required for efficient splicing of the analogous 3D⁄3A intron upstream of its internal polyadenylation site (also observed by J. Qiu [personal communication]). For this purpose, we devel-oped an RNase protection assay for HBoV1 RNA using an HBoV1-specific probe that spans the 3=splice site of the 3D⁄3A intron of HBoV1. As can be seen inFig. 1C, following transfection of nonreplicating clones of HBoV1 in 293T cells, splicing of the HBoV1 3D⁄3A intron was lost in RNA generated by three similar HBoV1 NP1 mutants (5X, TAAm, and CapF) (Fig. 1C, compare lanes 3 to 5 to lane 2) (mutant construction is described in Mate-rials and Methods). This suggested that the requirement of NP1 for splicing of the (pA)p-proximal intron is likely conserved among theBocaparvoviruses.
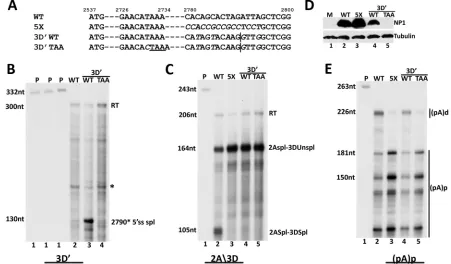
As mentioned above, we found that MVC NP1 enhanced splic-ing of only the intron directly upstream of (pA)p: neither the 2D⁄2A intron nor introns using 1D and 1D=were affected by NP1. To examine this distinction and to determine the specificity of this regulation further, we inserted an additional U1snRNP binding site and splicing donor motif downstream of the authentic 3D donor, making it the most proximal donor to both the 3A acceptor and to (pA)p (3D=) (Fig. 2A). The 3D=donor was found to be used exclusively in this construct: an RNase protection assay using a probe specific for the new donor (3D=probe) demonstrated its use in the presence of wild-type NP1 (Fig. 2B, lane 3), and an RNase protection assay using the previously mentioned 2A3D probe, which spans the original 3D donor, demonstrated that the original donor was no longer used when 3D=had been introduced (Fig. 2C, compare lane 4 to lane 2). Splicing of the 3D=donor was also NP1 dependent; it was not used in the presence of a termination mu-tant of NP1 (Fig. 2B, lane 4). Interestingly, in the construct con-taining both donors, use of 3D was not regained when NP1 was inactivated (Fig. 2C, lane 5). The reason for this is not yet clear.
Figure 2Dshows similar NP1 protein expression in these experi-ments. Thus, it seems that NP1 likely helped define the intron immediately upstream of (pA)p based on a proximal donor-ac-ceptor pair and independently of the nature of the specific donor. Introduction of the additional donor sequence had no detectable effect on NP1-dependent polyadenylation at (pA)p (Fig. 2E, com-pare lanes 4 and 5 to lanes 2 and 3).
MVC NP1 can affect polyadenylation at (pA)p indepen-dently of splicing of the MVC 3D⁄3A intron.Interaction between polyadenylation sites and proximal upstream introns has been shown to be important in definition of 3=-terminal exons (24–27). To first determine whether NP1’s role in regulating alternative polyadenylation at (pA)p was influenced by processing of the up-stream intron, we generated a mutant in which both donor and
on November 7, 2019 by guest
http://jvi.asm.org/
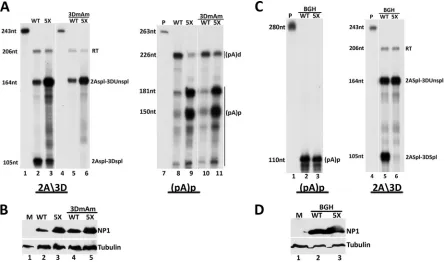
acceptor sites of that intron were abolished to best prevent engage-ment with the spliceosome (3DmAm; described in Materials and Methods). The mutation was placed in both wild-type and mutant NP1 backgrounds. RNase protection assays using the 2A3D probe diagrammed inFig. 1confirmed that the 3DmAm mutation prevented splicing of the 3D⁄3A intron in both wild-type and mu-tant NP1 backgrounds (Fig. 3A, left, compare lanes 5 and 6 to lanes 2 and 3). RNase protection assays of the same RNA samples using the (pA)p probe, also diagrammed inFig. 1, showed that debilitating the 3D⁄3A intron had no significant effect on alterna-tive polyadenylation. In the presence of wild-type NP1, the ma-jority of RNA generated by the 3DmAm mutant extended to the right-hand end, similar to constructs with an unaltered intron (Fig. 3A, right, compare lane 10 to lane 8). Similarly, in the mutant NP1 background, RNA generated by the 3Dm3Am mutant showed a relative increase in accumulation of internally polyade-nylated RNA similar to that generated by constructs with an un-altered intron (Fig. 3A, right, compare lane 11 to lane 9). These results indicated that NP1 can modulate alternative polyadenyla-tion of MVC pre-mRNAs independently of a funcpolyadenyla-tional upstream intron. The levels of NP1 expression in this experiment are shown inFig. 3B.
The direct determination of whether a functional (pA)p motif is required incisfor upstream splicing is confounded because, as described more fully below, NP1, which is required for 3D⁄3A splicing is produced primarily from RNAs polyadenylated at (pA)p. However, the experiments described below demonstrate that NP1 can modulate 3D⁄3A splicing in the presence of a heter-ologous polyadenylation signal. For these experiments, we re-placed the MVC (pA)p region with the polyadenylation site from the bovine growth hormone (bGH) gene, often used in transient expression vectors. This replacement resulted in essentially full polyadenylation at this internal site, under both NP1-replete and NP1 mutant conditions (Fig. 3C, left), indicating that NP1 had little effect on this heterologous polyadenylation signal. That poly-adenylation at the bGH polypoly-adenylation signal was not affected by NP1 may have been due either to the intrinsic strength of its core elements or, alternatively, that additional elements within the (pA)p motif were required for NP1 responsiveness. This is cur-rently being investigated. RNase protections of the same samples using the 2A3D probe indicated that splicing of the 3D⁄3A intron in the bGH replacement construct showed the same dependence on NP1 as when MVC (pA)p was present (Fig. 3C, right, compare lane 6 and lane 5). The expression of NP1 in this experiment is FIG 1MVC and HBoV NP1s are required for splicing of the intron that lies directly upstream of their proximal polyadenylation sites. (A) Transcription profile of minute virus of canine (MVC) showing the P6 promoter, splice donors (D) and acceptors (A), and the proximal [(pA)p] and distal [(pA)d] polyadenylation sites. The annotated nucleotides delineate the boundaries of the transcription landmarks indicated within the MVC genome (GenBank accession number
FJ214110.1). The position of the RNase protection probes, P6 (nt 250 to 500), 2A3D (nt 2344 to 2550) and (pA)p (3107 to 3333) are indicated. The expected sizes of MVC transcripts protected by the 2A3D probe are shown. The read-through product (RT) (206 nt) is protected by R1 and R2; the 2A spliced3D unspliced (2ASpl3DUnspl) product (164 nt) is protected by species of R1=, R2=, R3, and R4 spliced at the second intron but not at the third; and the 2A spliced3D spliced (2ASpl3Dspl) product (105 nt) is protected by R5 species in which both the second and third intron are spliced. (B) RNase protection assays of total RNA extracted from 293T cells transfected with pIMVC WT (lane 2), pIMVC 3Am (lane 3), and pIMVC NP1mutants [TAAm (lane 4), 5X (lane 5), and CapFus (lane 6)], analyzed by RNase protection assay (RPA) with the 2A3D probe. The sizes of the probe (243 nt) and protected fragments (206 nt, 164 nt, and 105 nt, respectively) are shown on the left. Bands reflecting RNA species polyadenylated at (pA)d (RT), RNAs spliced at the second intron acceptor but not at the third intron donor (2Aspl3DUnspl), and RNAs spliced at the second intron acceptor and also the third intron donor (2Aspl3Dspl), as described more fully in the text, are indicated on the right. (C) RNase protection assays of total RNA extracted from 293T cells transfected with HBoV plasmid constructs pHBoVm630 WT (lane 2) and HBoV NP1 mutants [5X (lane 3), TAAm (lane 4), CapF (lane 5)], using a 200-nt probe across the 3=splice site of the HBoV third intron (nt 2357 to 2955) (HBoV GenBank accession numberDQ000496). The sizes of the probe (237 nt) and protected transcripts (200 nt and 150 nt) are shown on the left, while the corresponding RNA species (3AUspl and 3Aspl) are indicated on the right.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.55.538.64.327.2]shown inFig. 3D. These results indicated that NP1 was still nec-essary for 3D⁄3A splicing in the presence of a heterologous poly-adenylation signal downstream, which itself was not affected by NP1. Taken together, our results suggest that NP1 can dramati-cally affect alternative polyadenylation at (pA)p and splicing of its proximal upstream intron independently of each other.
The S⁄R domain of NP1 is required for its effect on splicing but not polyadenylation.In silicostructural prediction of NP1 (GenBank accession numberFJ214110.1) with the PSIPRED (ver-sion 3.3) (28) and DISOPRED3 (28) programs revealed that the N terminus (amino acids 1 to 60) of the protein is likely to be unstructured, with a high disorder confidence score (data not shown). This region of NP1 has a series of arginine⁄serine di-re-peats (often also found as SR di-redi-re-peats) (Fig. 4A), which are a signature of the SR protein and SR-related group of proteins, many of which are involved in cellular and viral RNA processing (29–33).
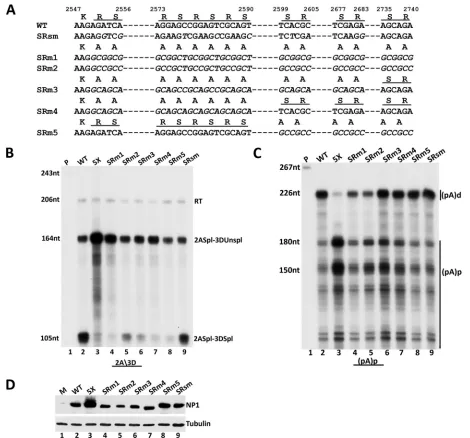
Mutation of the seven RS dipeptides in the amino terminus of NP1 to alanine, generated using two separate changes of primary nucleotide sequence (SRm1 and SRm2) (diagrammed inFig. 4A), resulted in NP1 proteins which were substantially deficient in en-hancing splicing of the 3D⁄3A intron (Fig. 4B, compare lanes 4
[SRm1] and 5 [SRm2] to lane 2 [wild type] and lane 3 [prototype 5X-pro NP1 mutant]). Retention in NP1 of the single 3=-most SR dipeptide, the last three dipeptides, or the first three dipeptides (SRm3, SRm4, and SRm5, respectively) (diagrammed inFig. 4A) each retained the mutant phenotype for splicing of the 3D⁄3A intron; the presence of the remaining SR residues in these mutants was not sufficient to sustain their ability to enhance splicing (Fig. 4B, compare lanes 6, 7, and 8 to lane 2 [wild type] and lane 3 [prototype 5X-pro NP1 mutant]). (The SRm2 mutant exhibited somewhat greater splicing of the 3D⁄3A intron for reasons that are currently unclear. The particular sequence introduced in this mu-tant may have conferred a splicing enhancer activity, known to be present in some G⁄C-rich sequences [34–36]. Some of these se-quences are shared in the SRm3 mutant, which also shows slightly increased splicing.) Importantly, all of these SR mutants remained functional for suppression of (pA)p (Fig. 4C, compare lanes 6, 7, and 8 to lanes 2 and 3), although the seven-amino-acid substitu-tion mutants (SRm1 and SRm2) were slightly less effective in this regard (Fig. 4C, compare lanes 4 and 5 to lanes 2 and 6 to 8). All mutant proteins were expressed at similar levels (Fig. 4D). The sequences of all mutant genes were confirmed, and so the differ-ences in the migration of the various NP1s seen inFig. 4Dmay FIG 2MVC NP1 is required for splicing of the intron that lies directly upstream from the proximal polyadenylation sites. (A) Alignment of pIMVC WT, pIMVC 5X, pIMVC 3D=WT, and pIMVC 3D=TAA sequences. Proline codon substitutions in pIMVC 5X and silent mutations in pIMVC 3D=WT and pIMVC 3D=TAA between nt 2780 to 2800 are shown in italics. The 5=splice site at nt 2790 in pIMVC 3D=WT and pIMVC 3D=TAA is depicted with a vertical line, while the premature termination codon in pIMVC 3D=TAA is shown with a horizontal line. (B) RNase protection assays of total RNA extracted from 293T cells transfected with pIMVC WT (lane 2), pIMVC 3D=WT (lane 3), and pIMVC 3D=TAA (lane 4) analyzed by RNase protection assay (RPA) with the 3D=(nt 2660 to 2960) probe. The sizes of the probe (332 nt) and protected fragments (300 nt and 130 nt) are shown on the left. The read-through (RT) band and the spliced RNA product (2790*5=ss spl) are indicated on the right, while the asterisk denotes a nonspecific unidentified fragment. (C) RNase protection assays of total RNA extracted from 293T cells transfected with pIMVC WT (lane 2), pIMVC 5X (lane 3), and pIMVC 3D=WT (lane 4) and pIMVC 3D=TAA (lane 5) analyzed by RNase protection assay (RPA) with the 2A3D probe. The sizes of the probe (243 nt) and protected fragments (206 nt, 164 nt, and 105 nt) are shown on the left. Bands reflecting RNA species as described in the text andFig. 1are indicated on the right. (D) 293T cells transfected with constructs described for panels A and C were harvested 48 h after transfection and analyzed by immunoblotting with NP1 and tubulin antibodies. (E) RNase protection assays of total RNA extracted from 293T cells transfected with pIMVC WT (lane 2), pIMVC 5X (lane 3), pIMVC 3D=WT (lane 4), and pIMVC 3D=TAA (lane 5) analyzed by RNase protection assay (RPA) with the (pA)p probe (nt 3107 to 3333). The sizes of the probe (263 nt) and the protected fragments (226 nt, 181 nt, and 150 nt) are indicated on the left. The RNA species polyadenylated at the distal [(pA)d] and proximal [(pA)p] polyadenylation sites are shown on the right.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.65.516.68.332.2]have been due to posttranslational modifications, typically seen for SR residues (37,38). As an additional control, a mutant bear-ing third-nucleotide changes that altered the primary nucleotide sequence, but not the amino acid sequence, in this region, using a much reduced G⁄C-rich content, performed like the wild type (SRsm) (diagrammed inFig. 4A, B, andC, lanes 9). The SR mutations overlap the MVC NS1 coding region and change the amino acids of NS1 in this region. However, we previously confirmed (7), and show again below (Fig. 5), that MVC NS1 does not play a role in either alternative splicing or polyadenyl-ation of MVC pre-mRNA.
These results demonstrated that the SR domain of NP1 is nec-essary for enhancement of splicing of the 3D⁄3A intron and con-firmed that the effects of NP1 on splicing and polyadenylation were separable. This is consistent with the genetic experiments described above showing that splicing of the 3A⁄3D intron and polyadenylation at (pA)p could be substantially independent (Fig. 2). As expected, the splicing deficient NP1 SR mutants were
un-able to generate detectun-able levels of capsid proteins, which is de-pendent upon proper 3D⁄3A splicing (data not shown).
Additionally, the NP1 SR mutants showed no detectable defi-ciency in splicing of the upstream 2D⁄2A MVC introns (Fig. 4B; compare levels of the 2ASpl-3DUnSpl band to the read-through band [RT]) and generated wild-type levels of the 66-kDa NS1 protein, which is generated from RNAs multiply spliced upstream (data not shown). Additional characterization of the role of NP1 in these processes is in progress.
The MVC proximal polyadenylation element is essential for viral genome replication.To determine the importance of alter-native RNA processing at (pA)p in the MVC life cycle, we gener-ated third-position mutations in the infectious clone of MVC that debilitated the core (pA)pcis-acting signals required for internal polyadenylation while conserving the capsid protein coding se-quence (pIMVC pApmVP1csm) (Fig. 5A). As can be seen inFig. 5B, RNase protection assays using a homologous (pA)p probe showed that, as expected, the great majority of the transcripts FIG 3MVC NP1 can affect splicing of the MVC 3D⁄3A intron and alternative polyadenylation at (pA)p independently. (A) (Left) RNase protection assay of total RNA extracted from 293T cells transfected with pIMVC WT (lane 2), pIMVC NP1 5XP (lane 3), and pIMVC 3DmAm bearing either a wild-type WT (lane 5) or 5XP mutant (lane 6) NP1 gene, using homologous 2A3D probes (lanes 1 and 4, respectively). The sizes of the probe (243 nt) and protected RNA fragments (206 nt, 164 nt, and 105 nt) are shown on the left. Bands reflecting RNA species polyadenylated at (pA)d (RT), RNAs spliced at the second intron acceptor but not at the third intron donor (2Aspl3DUnspl), and RNAs spliced at the second intron acceptor and also the third intron donor (2Aspl3Dspl), as described in detail in the text, are indicated on the right. (Right) RNase protections of the same RNA samples described above using the proximal polyadenylation probe pAp (nt 3107 to 3333). The (pA)p probe alone is shown in lane 7. The sizes of the probe (263 nt) and protected fragments (226 nt, 181 nt, and 150 nt) are shown on the left, while the corresponding RNAs polyadenylated proximally at (pA)p and distally at (pA)d are shown on the right. Lane headings are the same as in in the left part of panel A. (B) Protein samples taken from the transfections described for panel A were loaded as indicated, run on 12% polyacrylamide gels, and subjected to immunoblotting using antibodies directed against NP1 and tubulin. (C) (Left) RNase protection assays of total RNA isolated from 293T cells transfected with pIMVC BGH constructs bearing either the wild-type (WT, lane 2), or mutant (5X, lane 3) NP1 gene, using a homologous proximal polyadenylation probe. The probe alone is shown in lane 1. The sizes of the probe (280 nt) and the protected fragment (110 nt) are shown on the left. The corresponding proximal polyadenylated transcript is indicated on the right [(pA)p]. (Right) RNase protection assay of total RNA purified from 293T cells transfected with pIMVCBGH constructs bearing either the wild-type (WT, lane 5) or mutant (5X, lane 6) NP1 gene, as described in the text, using the 2A3D probe. The probe alone is shown in lane 4. The sizes of the probe (243 nt) and the protected fragments (206 nt, 164 nt, and 105 nt) are shown on the left. Bands reflecting RNA species polyadenylated at (pA)d (RT), RNAs spliced at the second intron acceptor but not at the third intron donor (2Aspl3DUnspl), and RNAs spliced at the second intron acceptor and also the third intron donor (2Aspl3Dspl), as described more fully in the text, are indicated on the right. (D) 293T cells transfected with pIMVCBGH constructs bearing either the wild-type (WT; lane 2) or mutant (5X; lane 3) NP1 gene were harvested 48 h posttransfection. The samples were analyzed by immunoblotting with NP1 and tubulin antibodies.
on November 7, 2019 by guest
http://jvi.asm.org/
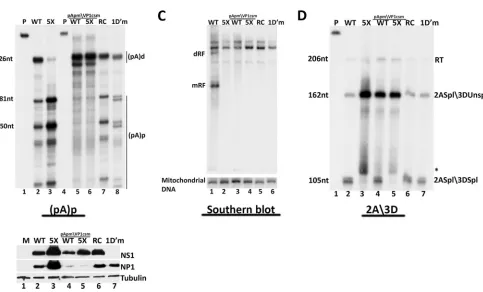
[image:6.585.70.515.67.329.2]generated by constructs containing the pIMVC pApmVP1csm mutation, in either the NP1 wild-type or NP1 mutant back-ground, failed to polyadenylate at (pA)p and rather extended downstream to be processed at the distal polyadenylation site (pA)d (Fig. 5B, lanes 5 and 6). Importantly, the NP1-replete con-struct (bearing the wild-type NP1 gene) which contained this (pA)p mutation showed a marked reduction in replication follow-ing transfection of WRD cells (Fig. 5C, lane 3). This result implied
that internal polyadenylation of MVC transcripts was necessary for efficient genome replication.
We found that the levels of NP1 were substantially reduced in the pIMVCpApmVP1 mutant, while the levels of NS1 were more modestly affected (Fig. 5E, lanes 4 and 5). Since NP1 and NS1 have previously been shown to have essential roles in MVC genome replication (8), reduced replication of pIMVC pApmVP1csm could be explained by the significant loss of NP1 and the reduction FIG 4The S⁄R domain of NP1 is required for its effect on splicing but not on polyadenylation. (A) Alignment of nt 2547 to 2740 of wild-type MVC NP1 (WT), NP1SRsm (SR silent mutant as described in text) and NP1 SR mutants (SRm1, SRm2, SRm3, SRm4, and SRm5) showing the RS dipeptide repeats (under-lined).The alanine (A) codon substitutions are shown in italics for the mutants (NP1 SRm). (B) RNase protection of total RNA extracted from 293T cells 48 h following transfection with pIMVC WT (lane 2), pIMVC 5XNP1 (lane 3), pIMVC NP1 SR mutants (lanes 4 to 8), and pIMVC NP1 SRsm (lane 9) (the mutant is described in the text), using the 2A3D probe. The sizes of the probe (243 nt) and protected RNAs (206 nt, 164 nt, and 105 nt) are shown on the left. Bands reflecting RNA species polyadenylated at (pA)d (RT), RNAs spliced at the second intron acceptor but not at the third intron donor (2Aspl3DUnspl), and RNAs spliced at the second intron acceptor and also the third intron donor (2Aspl3Dspl), as described more fully in the text, are indicated on the right. (C) RNase protections of total RNA isolated from transfections described for panel B, as indicated, using the (pA)p proximal polyadenylation probe. The sizes of the probe (267 nt) and protected RNAs (226 nt, 180 nt, 150 nt) are shown on the left. RNAs polyadenylated at the distal [(pA)d] and proximal [(pA)p] polyadenylation sites are indicated on the right. (D) 293T cells transfected with constructs described for panel B were harvested 48 h posttransfection. The samples were analyzed by immunoblotting with NP1 and tubulin antibodies.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.58.528.64.502.2]of NS1. But what was the primary cause of the reduction of NP1 and NS1 in this mutant? The reduction in MVC protein levels seen inFig. 5Ecould have merely been the consequence of decreased genome template accumulation, which was expected as the con-sequence of a primary loss of NP1 and NS1; however, this was not the case. Transfection of a nonreplicating NS1 mutant in an oth-erwise wild-type background, as well as transfection of a replica-tion-defective construct lacking the left-end viral hairpin (1D=m, and RepCap, respectively) (Fig. 5E, lanes 6 and 7), resulted in the generation of significant amounts of both NP1 (Fig. 5E, lanes 6 and 7) and capsid proteins (data not shown). The nonreplicating RepCap construct also generated substantial amounts of NS1 (Fig. 5E, lane 6). These results indicated that reduced NP1 expression
from pIMVCpApmVP1csm was not due merely to the paucity of transcription templates. Rather, they suggested that internally polyadenylated mRNAs generated from proximal polyadenyla-tion at (pA)p were likely the primary generators of NP1 and NS1 from the MVC clone, and it was the lack of their production which was the primary deficiency which rendered the construct replica-tion defective. Addireplica-tionally, we showed previously that mutareplica-tions of NP1 led to increased expression of that protein (7), as well as NS1 (Fig. 5E, compare lane 3 to lane 2). This is consistent with the model in which increased NP1 (and to a lesser extent NS1) was due to the lack of the ability of mutant NP1 to suppress (pA)p, resulting in the accumulation of NP1-encoding internally poly-adenylated transcripts (7). Also consistent with this notion, the FIG 5The MVC proximal polyadenylation element is essential for viral genome replication. (A) Alignment of the MVC proximal polyadenylation region (nt 3111 to 3326) in pIMVC WT and the pIMVC (pA)pmVP1csm mutant. The third-nucleotide substitutions in the VP1 reading frame are shown in italics. (B) RNase protection assays of total RNA extracted from 293T cells transfected with pIMVC WT (lane 2), pIMVC 5XNP1 (lane 3), pIMVC (pA)pmVP1csm bearing either a wild-type (WT, lane 5) or mutant (5X, lane 6) NP1 gene, MVC RepCap (RC, lane 7), or pIMVCNS1 (1D=m, lane 8) using either wild-type (lanes 2, 3, 7, and 8) or homologous mutant (lanes 5, 6) proximal polyadenylation (pA)p probes. The wild-type and homologous probes are shown in lanes 1 and 4, respectively. Bands reflecting RNAs using the proximal polyadenylation site [(pA)p] or the distal polyadenylation site [(pA)d] are indicated on the right. The sizes of the protected fragments (226 nt, 181 nt, and 150 nt) are shown on the left. (C) Southern blot showing total DNA extracted 72 h after transfection of 45% confluent canine WRD cells transfected with pIMVC WT (lane 1), pIMVC 5XNP1 (lane 2), pIMVC (pA)pmVP1csm bearing a wild-type (WT; lane 3), or mutant (5X; lane 4) NP1 gene, MVC RepCap (RC; lane 5), or pIMVC NS1 mutant (1D=m; lane 6). Equivalent amounts of total DNA were loaded in each lane, as determined with a Nanodrop spectrophotometer, and confirmed by hybridization to mitochondrial DNA using a mitochondrial DNA-specific probe. Positions of the viral DNA replication intermediates monomer (mRF) and dimer (dRF) replicative forms are shown on the left. (D) RNase protection of total RNA isolated from the transfections described for panel B, using the 2A3D probe. The probe alone is shown in lane 1. The sizes of the protected RNA fragments (206 nt, 162 nt, and 105 nt) are indicated on the left. Bands reflecting RNA species polyadenylated at (pA)d (RT), RNAs spliced at the second intron acceptor but not at the third intron donor (2Aspl3DUnspl), and RNAs spliced at the second intron acceptor and also the third intron donor (2Aspl3Dspl), as described more fully in the text, are indicated on the right. The asterisk indicates a nonspecific unidentified fragment. (E) Protein samples taken from the transfections described for panels B and D above were subjected to immunoblotting using antibodies directed against NS1, NP1, and tubulin.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.55.538.114.406.2]previously described pIMVCBGH construct, which generated in-ternally polyadenylated RNAs exclusively (Fig. 3C), produced abundant amounts of NP1 (Fig. 3D, lanes 2 and 3) and NS1 (data not shown).
Splicing of the 3D⁄3A intron was also impaired in the pIMVC pApmVP1csm background, not only in the presence of a mutant NP1 gene, as expected (Fig. 5D, compare lane 5 to lane 2), but also in the presence of the wild-type NP1 gene (Fig. 5D, compare lane 4 to 2). We show above (Fig. 1) that splicing of the 3D⁄3A intron requires NP1. Because levels of NP1 were reduced (likely due to the loss of internally polyadenylated RNA) in this construct, the reduction in splicing was consistent with results demonstrating that 3D⁄3A excision depends upon NP1. The nonreplicating NS1 mutant and Rep⁄Cap control constructs, which had a functional (pA)p, generated higher levels of NP1 (5E, lanes 6 and 7) and so were able to efficiently excise the 3D⁄3A intron to wild-type levels (Fig. 5D, lanes 6 and 7).
In summary, our results demonstrate that NP1, itself primarily made from an internally polyadenylated transcript, is necessary for both read-through of the internal (pA)p signal and splicing of the 3D⁄3A intron. Both events are necessary to allow proper initi-ation at the VP1 AUG required for accessing the capsid gene in-formation. This suggests the following model ofBocaparvovirus
gene expression. Initially, internally polyadenylated RNAs encode NP1 and NS1. As the concentration of NP1 increases during in-fection, internal polyadenylation at (pA)p is suppressed and splic-ing of the upstream 3A⁄3D intron is enhanced, generatsplic-ing mRNAs competent for encoding the capsid proteins. Some internal poly-adenylation continues to provide a source of NP1 and NS1. Thus, both polyadenylation at (pA)p and its read-through must be finely tuned throughout infection to maintain the proper levels of the nonstructural and capsid proteins for efficient infection.
DISCUSSION
Although parvoviruses have only subtle early-to-late expression shifts, they all regulate access to their capsid genes, and different parvoviruses do so in different ways. Members of the genera
Pro-toparvovirusandDependoparvovirusfeature transcriptional
trans-activation of their capsid gene promoters by their nonstructural proteins for this purpose (14,15,39). For parvoviruses that have an internal polyadenylation site, such as AAV5, B19, and GPV, this site lies within an intron whose excision allows extension of the spliced RNA into the capsid gene (16, 18, 40–42). For the
Bocaparvoviruses, however, these potent motifs are retained in the
capsid protein-encoding cytoplasmic mRNA. Thus, it was unclear
howBocaparvovirusesgovern access to their capsid gene
informa-tion.
We recently showed that NP1 is required for accumulation in the cytoplasm of partially processed full-length MVC RNA retain-ing the internal (pA)p site (7). The NP1 requirement was shown to be independent of viral replication (7). We have shown here that splicing of the 3D⁄3A intron immediately upstream of (pA)p, nec-essary in the capsid-encoding mRNA for access to the VP1 AUG, was also enhanced by NP1. Further, NP1’s role in internal poly-adenylation at (pA)p could be independent of its role in upstream splicing: NP1 was still required for read-through of (pA)p when the 3D⁄3A intron was debilitated, and the requirement of NP1 for efficient 3D⁄3A excision remained when (pA)p was replaced by a heterologous polyadenylation site. Consistent with these results, we have also found that mutations which disrupt the S⁄R-like
do-main within the unordered amino terminus of NP1 inhibited its role in splicing but not polyadenylation. Finally, we demonstrated that the internal polyadenylation site (pA)p was essential for rep-lication of the MVC infectious clone and that NP1 and to a lesser extent NS1 were only poorly expressed when the endogenous (pA)p signal in the infectious MVC clone was altered to prevent internal polyadenylation. NP1, itself made primarily from an in-ternally polyadenylated transcript, is thus an RNA processing fac-tor necessary for the accumulation of RNAs that both extend through the internal (pA)p signal into the capsid coding gene and splice the 3D⁄3A intron, which allows proper initiation at the VP1 AUG essential for accessing MVC capsid gene information.
NP1 is common to all members of theBocaparvovirusesbut has little homology to proteins of other parvoviruses and no substan-tial homology to any protein in the databases. Interestingly, it has recently been shown to complement early events controlled in minute virus of mice (MVM) infection by the MVM protein NS2 (43). NP1 contains multiple Arg⁄Ser di-repeats in the N-terminal intrinsically disordered portion of the protein. These motifs are characteristic of the SR class of RNA processing proteins involved primarily in splicing, but also other aspects of the pathways of mRNA maturation. Mutations of the S⁄R repeats in the unordered region of NP1 had the discriminatory phenotype of debilitating NP1’s role in splicing but not its role in alternative polyadenyla-tion. This result is consistent with our mutational results regard-ing the 3D⁄3A intron and (pA)p, which indicate that NP1 can act on each element independently. How NP1 can function separately in these steps of RNA processing is under investigation, which should be aided by these mutants.
Our results also indicate that NP1’s activity in alternative poly-adenylation is not a general effect on all polypoly-adenylation sites. When the MVC (pA)p region was replaced by the bovine growth hormone (bGH) polyadenylation site it was not affected by NP1. This suggested that there might be some specificity to the effect of NP1, perhaps mediated by sequences outside the core AAUAAA and G⁄U-rich motifs within (pA)p. This is under investigation. The role of NP1 in enhancing splicing is also restricted in function. NP1 enhances the 3D⁄3A intron quite strongly but has only limited action on other MVC introns. Interestingly, when an artificial donor site was introduced downstream of the authentic 3D site, the new donor site was used exclusively, and excision of the intron was dependent upon NP1. This suggested that NP1’s effect is tied to the third intron acceptor, from which it helps identify the prox-imal upstream donor. For reasons that are not yet clear, in the double-donor constructs, use of the (strongly consensus) wild-type donor was not regained in the absence of NP1. Whether NP1 has effects on RNA processing of cellular genes is also currently being investigated.
NP1’s effect on MVC internal polyadenylation is different from the effect on polyadenylation in the parvoviruses AAV5 and B19, which feature direct competition between splicing and poly-adenylation (16,40). Recently, the HPV-16 E2 protein was re-ported to regulate the usage of the proximal polyadenylationcis -acting elements in the middle of its genome by modulating the recruitment of the polyadenylation machinery to this site (44,45). In addition, viral proteins such as herpes simplex virus 1 (HSV-1) ICP27 (46, 47), the Kaposi sarcoma-associated herpesvirus (KSHV) ORF57 protein (48–50), and the human cytomegalovirus protein UL69 (51,52) have been shown to modulate the process-ing of HSV, KSHV, and HCMV transcripts, respectively. Because
on November 7, 2019 by guest
http://jvi.asm.org/
of its specificity, NP1’s function and how it regulates RNA pro-cessing may be different from these examples. However, a com-mon feature found in HSV-1 ICP27, KSHV ORF57, and HCMV UL69 is an intrinsically disordered region that contains motifs that mediate protein-protein or RNA-protein interaction (53). MVC NP1 has a similar predicted disordered N terminus embed-ded with SR repeats, which may yet suggest some commonality of function. In other systems, posttranslation modification of these regions, including phosphorylation of resident S⁄R residues, have been shown to be important for their function (38, 53–56). Whether such modifications of NP1 influence its function is cur-rently being investigated.
How does alternative polyadenylation at (pA)p factor into the virus life cycle? Our results indicated that loss of (pA)p resulted in a dramatic inhibition of viral replication. Examination of the pro-tein production from these mutants indicated that NP1 levels were significantly depleted, while NS1 was modestly depleted. NP1 and NS1 are known to be required for replication; however, the reduction in protein levels resulted not from a decrease in template accumulation but rather from a block to expression, which then itself likely led to a reduction in replication. Together, these results strongly suggest that within the MVC plasmid clone, primarily NP1 and to a lesser extent NS1 are generated by the internally polyadenylated RNAs, which in turn suggests the fol-lowing model for the differential control ofBocaparvovirusgene expression.
We suggest that perhaps at the earliest times in infection, NP1 and NS1 are primarily produced from internally polyadenylated RNA. As NS1- and NP1-dependent replication ensues and the concentration of NP1 increases, internal polyadenylation at (pA)p is suppressed and splicing of the 3D⁄3A intron is enhanced. This leads to the accumulation of the capsid mRNAs, which then can read through into the capsid gene and are spliced so that the VP1 AUG can be properly accessed. Some internal polyadenylation continues to provide a source of NP1 and NS1. Thus, both poly-adenylation at (pA)p and its read-through need to be finely tuned throughout infection to maintain the proper levels of the non-structural and capsid proteins for efficient infection. NP1 is the first parvovirus protein to be implicated in RNA processing. While a temporal shift in the MVC RNA profile has not yet been de-tected, it seems likely that NP1 mediates yet another way that parvoviruses have evolved to govern essential access to its capsid-protein gene, namely, at the RNA level, by regulating the use of both the essential splicing of an intron and the suppression of an internal polyadenylation site.
ACKNOWLEDGMENTS
We thank Jianming Qiu, University of Kansas Medical Center, for cloned HBoV, other valuable reagents, and information prior to publication and Lisa Burger for excellent technical assistance.
FUNDING INFORMATION
HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) provided funding to David J. Pintel under grant number AI 046458.
REFERENCES
1.Carmichael LE, Schlafer DH, Hashimoto A.1994. Minute virus of ca-nines (MVC, canine parvovirus type-1): pathogenicity for pups and sero-prevalence estimate. J Vet Diagn Invest6:165–174.http://dx.doi.org/10 .1177/104063879400600206.
2.Harrison LR, Styer EL, Pursell AR, Carmichael LE, Nietfeld JC.1992. Fatal
disease in nursing puppies associated with minute virus of canines. J Vet Diagn Invest4:19 –22.http://dx.doi.org/10.1177/104063879200400105. 3.Decaro N, Carmichael LE, Buonavoglia C. 2012. Viral reproductive
pathogens of dogs and cats. Vet Clin North Am Small Anim Pract42:583– 598, vii.http://dx.doi.org/10.1016/j.cvsm.2012.01.006.
4.Cotmore S, Agbandje-McKenna M, Chiorini J, Mukha D, Pintel D, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P, Gatherer D, Davison A.2014. The family Parvoviridae. Arch Virol159:1239 –1247.http://dx .doi.org/10.1007/s00705-013-1914-1.
5.Cotmore SF, Tattersall P.2014. Parvoviruses: small does not mean simple. Annu Rev Virol1:517–537.http://dx.doi.org/10.1146/annurev -virology-031413-085444.
6.Qiu J, Pintel D.2008. Processing of adeno-associated virus RNA. Front Biosci13:3101–3115.http://dx.doi.org/10.2741/2912.
7.Sukhu L, Fasina O, Burger L, Rai A, Qiu J, Pintel DJ.2013. Character-ization of the nonstructural proteins of the bocavirus minute virus of canines. J Virol87:1098 –1104.http://dx.doi.org/10.1128/JVI.02627-12. 8.Sun Y, Chen AY, Cheng F, Guan W, Johnson FB, Qiu J.2009. Molecular
characterization of infectious clones of the minute virus of canines reveals unique features of bocaviruses. J Virol83:3956 –3967.http://dx.doi.org/10 .1128/JVI.02569-08.
9.Lederman M, Patton JT, Stout ER, Bates RC. 1984. Virally coded noncapsid protein associated with bovine parvovirus infection. J Virol 49:315–318.
10. Lederman M, Cotmore SF, Stout ER, Bates RC. 1987. Detection of bovine parvovirus proteins homologous to the nonstructural NS-1 pro-teins of other autonomous parvoviruses. J Virol61:3612–3616. 11. Lederman M, Bates RC, Stout ER.1983. In vitro and in vivo studies of
bovine parvovirus proteins. J Virol48:10 –17.
12. Schwartz D, Green B, Carmichael LE, Parrish CR. 2002. The canine minute virus (minute virus of canines) is a distinct parvovirus that is most similar to bovine parvovirus. Virology302:219 –223.http://dx.doi.org/10 .1006/viro.2002.1674.
13. Green MR, Roeder RG.1980. Transcripts of the adeno-associated virus genome: mapping of the major RNAs. J Virol36:79 –92.
14. Christensen J, Cotmore SF, Tattersall P. 1995. Minute virus of mice transcriptional activator protein NS1 binds directly to the transactivation region of the viral P38 promoter in a strictly ATP-dependent manner. J Virol69:5422–5430.
15. Lorson C, Burger LR, Mouw M, Pintel DJ.1996. Efficient transactivation of the minute virus of mice P38 promoter requires upstream binding of NS1. J Virol70:834 – 842.
16. Guan W, Huang Q, Cheng F, Qiu J.2011. Internal polyadenylation of the parvovirus B19 precursor mRNA is regulated by alternative splicing. J Biol Chem286:24793–24805.http://dx.doi.org/10.1074/jbc.M111.227439. 17. Qiu J, Nayak R, Tullis GE, Pintel DJ. 2002. Characterization of the
transcription profile of adeno-associated virus type 5 reveals a number of unique features compared to previously characterized adeno-associated viruses. J Virol76:12435–12447. http://dx.doi.org/10.1128/JVI.76.24 .12435-12447.2002.
18. Yoto Y, Qiu J, Pintel DJ.2006. Identification and characterization of two internal cleavage and polyadenylation sites of parvovirus B19 RNA. J Virol 80:1604 –1609.http://dx.doi.org/10.1128/JVI.80.3.1604-1609.2006. 19. Venkatesh LK, Fasina O, Pintel DJ.2012. RNAse mapping and
quanti-tation of RNA isoforms. Methods Mol Biol883:121–129.http://dx.doi.org /10.1007/978-1-61779-839-9_9.
20. Miller CL, Pintel DJ.2002. Interaction between parvovirus NS2 protein and nuclear export factor Crm1 is important for viral egress from the nucleus of murine cells. J Virol76:3257–3266.http://dx.doi.org/10.1128 /JVI.76.7.3257-3266.2002.
21. Bentley DL.2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet15:163–175.http://dx.doi.org/10.1038/nrg3662. 22. Catania F, Lynch M.2013. A simple model to explain evolutionary trends of eukaryotic gene architecture and expression: how competition between splicing and cleavage/polyadenylation factors may affect gene expression and splice-site recognition in eukaryotes. Bioessays35:561–570.http://dx .doi.org/10.1002/bies.201200127.
23. Martinson HG.2011. An active role for splicing in 3=-end formation. Wiley Interdiscip Rev RNA2:459 – 470.http://dx.doi.org/10.1002/wrna.68. 24. Dye MJ, Proudfoot NJ.1999. Terminal exon definition occurs
cotrans-criptionally and promotes termination of RNA polymerase II. Mol Cell 3:371–378.http://dx.doi.org/10.1016/S1097-2765(00)80464-5. 25. Niwa M, MacDonald CC, Berget SM.1992. Are vertebrate exons scanned
on November 7, 2019 by guest
http://jvi.asm.org/
during splice-site selection? Nature 360:277–280. http://dx.doi.org/10 .1038/360277a0.
26. Davidson L, West S.2013. Splicing-coupled 3=end formation requires a terminal splice acceptor site, but not intron excision. Nucleic Acids Res 41:7101–7114.http://dx.doi.org/10.1093/nar/gkt446.
27. Cooke C, Hans H, Alwine JC.1999. Utilization of splicing elements and polyadenylation signal elements in the coupling of polyadenylation and last-intron removal. Mol Cell Biol19:4971– 4979.http://dx.doi.org/10 .1128/MCB.19.7.4971.
28. Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT.2013. Scalable web services for the PSIPRED protein analysis workbench. Nu-cleic Acids Res41:W349 –W357.http://dx.doi.org/10.1093/nar/gkt381. 29. Bradley T, Cook ME, Blanchette M.2015. SR proteins control a complex
network of RNA-processing events. RNA21:75–92.http://dx.doi.org/10 .1261/rna.043893.113.
30. Long JC, Caceres JF.2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J417:15–27.http://dx.doi .org/10.1042/BJ20081501.
31. Lai MC, Peng TY, Tarn WY.2009. Functional interplay between viral and cellular SR proteins in control of post-transcriptional gene regulation. FEBS J 276:1517–1526.http://dx.doi.org/10.1111/j.1742-4658.2009.06894.x. 32. Matlin AJ, Clark F, Smith CW.2005. Understanding alternative splicing:
towards a cellular code. Nat Rev Mol Cell Biol6:386 –398.http://dx.doi .org/10.1038/nrm1645.
33. Blencowe BJ, Bowman JA, McCracken S, Rosonina E.1999. SR-related proteins and the processing of messenger RNA precursors. Biochem Cell Biol77:277–291.http://dx.doi.org/10.1139/o99-048.
34. Wang Y, Ma M, Xiao X, Wang Z.2012. Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat Struct Mol Biol19:1044 –1052.http://dx.doi.org/10.1038/nsmb.2377. 35. McCullough AJ, Berget SM.1997. G triplets located throughout a class of
small vertebrate introns enforce intron borders and regulate splice site selection. Mol Cell Biol17:4562– 4571.http://dx.doi.org/10.1128/MCB .17.8.4562.
36. Chou MY, Rooke N, Turck CW, Black DL.1999. hnRNP H is a com-ponent of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol19:69 –77.http://dx.doi.org/10.1128 /MCB.19.1.69.
37. Zhou Z, Fu XD.2013. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma122:191–207.http://dx.doi.org/10 .1007/s00412-013-0407-z.
38. Misteli T, Caceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL.1998. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J Cell Biol143:297–307.http: //dx.doi.org/10.1083/jcb.143.2.297.
39. Trempe JP, Carter BJ.1988. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J Virol62:3356 – 3363.
40. Qiu J, Pintel DJ.2004. Alternative polyadenylation of adeno-associated virus type 5 RNA within an internal intron is governed by the distance between the promoter and the intron and is inhibited by U1 small nuclear RNP binding to the intervening donor. J Biol Chem279:14889 –14898.
http://dx.doi.org/10.1074/jbc.M312734200.
41. Qiu J, Cheng F, Pintel D.2007. Distance-dependent processing of adeno-associated virus type 5 RNA is controlled by 5=exon definition. J Virol 81:7974 –7984.http://dx.doi.org/10.1128/JVI.00714-07.
42. Qiu J, Cheng F, Yoto Y, Zadori Z, Pintel D. 2005. The expression
strategy of goose parvovirus exhibits features of both the Dependovirus and Parvovirus genera. J Virol79:11035–11044.http://dx.doi.org/10.1128 /JVI.79.17.11035-11044.2005.
43. Mihaylov IS, Cotmore SF, Tattersall P.2014. Complementation for an essential ancillary non-structural protein function across parvovirus gen-era. Virology 468 – 470:226 –237. http://dx.doi.org/10.1016/j.virol.2014 .07.043.
44. Johansson C, Somberg M, Li X, Backstrom Winquist E, Fay J, Ryan F, Pim D, Banks L, Schwartz S.2012. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J31:3212–3227.http://dx.doi.org/10.1038/emboj.2012.147. 45. Johansson C, Schwartz S.2013. Regulation of human papillomavirus
gene expression by splicing and polyadenylation. Nat Rev Microbiol11: 239 –251.http://dx.doi.org/10.1038/nrmicro2984.
46. Sandri-Goldin RM.2011. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol6:1261– 1277.http://dx.doi.org/10.2217/fmb.11.119.
47. Hann LE, Cook WJ, Uprichard SL, Knipe DM, Coen DM.1998. The role of herpes simplex virus ICP27 in the regulation of UL24 gene expression by differential polyadenylation. J Virol72:7709 –7714.
48. Schumann S, Jackson BR, Baquero-Perez B, Whitehouse A. 2013. Kaposi’s sarcoma-associated herpesvirus ORF57 protein: exploiting all stages of viral mRNA processing. Viruses5:1901–1923.http://dx.doi.org /10.3390/v5081901.
49. Majerciak V, Zheng ZM.2009. Kaposi’s sarcoma-associated herpesvirus ORF57 in viral RNA processing. Front Biosci (Landmark Ed)14:1516 – 1528.
50. Sahin BB, Patel D, Conrad NK.2010. Kaposi’s sarcoma-associated her-pesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog6:e1000799.http://dx .doi.org/10.1371/journal.ppat.1000799.
51. Toth Z, Lischka P, Stamminger T.2006. RNA-binding of the human cytomegalovirus transactivator protein UL69, mediated by arginine-rich motifs, is not required for nuclear export of unspliced RNA. Nucleic Acids Res34:1237–1249.http://dx.doi.org/10.1093/nar/gkl007.
52. Toth Z, Stamminger T.2008. The human cytomegalovirus regulatory protein UL69 and its effect on mRNA export. Front Biosci13:2939 –2949.
http://dx.doi.org/10.2741/2899.
53. Majerciak V, Pripuzova N, Chan C, Temkin N, Specht SI, Zheng ZM. 2015. Stability of structured Kaposi sarcoma-associated herpesvirus ORF57 protein is regulated by protein phosphorylation and ho-modimerization. J Virol 89:3256 –3274. http://dx.doi.org/10.1128/JVI .03721-14.
54. Xiang S, Gapsys V, Kim HY, Bessonov S, Hsiao HH, Mohlmann S, Klaukien V, Ficner R, Becker S, Urlaub H, Luhrmann R, de Groot B, Zweckstetter M.2013. Phosphorylation drives a dynamic switch in serine/ arginine-rich proteins. Structure 21:2162–2174. http://dx.doi.org/10 .1016/j.str.2013.09.014.
55. Wang JT, Smith J, Chen BC, Schmidt H, Rasoloson D, Paix A, Lambrus BG, Calidas D, Betzig E, Seydoux G.2014. Regulation of RNA granule dynamics by phosphorylation of serine-rich, intrinsically disordered pro-teins in C. elegans. eLife3:e04591.http://dx.doi.org/10.1016/j.virol.2014 .07.043.
56. Wright PE, Dyson HJ.2015. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol16:18 –29.http://dx.doi .org/10.1038/nrm3920.