Copyright © 2001, American Society for Microbiology. All Rights Reserved.
DNA Microarray Analysis of Chimpanzee Liver during Acute
Resolving Hepatitis C Virus Infection
CATHERINE B. BIGGER,1KATHLEEN M. BRASKY,2ANDROBERT E. LANFORD1*
Department of Virology and Immunology1and Department of Laboratory Animal Medicine,2Southwest Regional
Primate Research Center, Southwest Foundation for Biomedical Research, San Antonio, Texas 78227 Received 21 February 2001/Accepted 25 April 2001
Hepatitis C virus (HCV) poses a worldwide health problem in that the majority of individuals exposed to HCV become chronically infected and are predisposed for developing significant liver disease. DNA microarray technology provides an opportunity to survey transcription modulation in the context of an infectious disease and is a particularly attractive approach in characterizing HCV-host interactions, since the mechanisms underlying viral persistence and disease progression are not understood and are difficult to study. Here, we describe the changes in liver gene expression during the course of an acute-resolving HCV infection in a chimpanzee. Clearance of viremia in this animal occurred between weeks 6 and 8, while clearance of residual infected hepatocytes did not occur until 14 weeks postinfection. The most notable changes in gene expression occurred in numerous interferon response genes (including all three classical interferon antiviral pathways) that increased dramatically, some as early as day 2 postinfection. The data suggest a biphasic mechanism of viral clearance dependent on both the innate and adaptive immune responses and provide insight into the response of the liver to a hepatotropic viral infection.
Approximately 3% of the worldwide population is persis-tently infected with hepatitis C virus (HCV) (3). Although chronic HCV infections are often clinically silent for decades, an estimated 20% of persistently infected individuals will de-velop serious liver disease, including cirrhosis and liver cancer (2, 3). Currently no vaccine is available, and antiviral therapy is limited to alpha interferon (IFN-␣) and ribavirin, which afford viral clearance in a minority of cases (1). HCV persists despite the presence of specific humoral and cellular immune re-sponses, and little is understood with regard to the factors leading to viral clearance or persistence, yet early events in the acute infection probably influence the outcome (12, 16, 17, 23, 39). These events are difficult to examine in human popula-tions, since the time of infection is rarely known and access to serial liver samples is constrained. Most immunological studies of HCV infection understandably involve chronically infected patients and thus are directed more at disease manifestations than at events leading to viral persistence. The chimpanzee model is particularly attractive for the analysis of the factors involved in viral clearance, since greater than 60% of inocu-lated animals rapidly clear the infection (6, 7, 36). These same factors may lead to rapid viral clearance in humans; however, the identification and analysis of individuals at the time of exposure is difficult. We have chosen to use DNA microarrays as a method to probe the liver for changes in gene expression associated with resolution of an HCV infection in a chimpan-zee.
DNA microarrays provide a powerful technique for explor-ing the myriad of changes in gene expression characteristic of various physiological and pathological conditions. Microarray technology has been used in a variety of systems, including
studies in whole organisms (40, 60). To date, microarray stud-ies involving viral infections (e.g., human immunodeficiency virus, human cytomegalovirus, and human papillomavirus) (13, 28, 63) have been restricted to tissue culture models. These systems provide a controlled environment for analysis of virus-host interactions, although they occur in the absence of the immune response. In this study, we anticipated four types of changes in gene expression within the liver as a consequence of HCV infection: (i) changes due to the endogenous antiviral response of the liver (e.g., induction of IFN-␣ and IFN-␥
response genes), (ii) changes due to the innate and adaptive immune response to the infection (e.g., activation and infiltra-tion of NK cells, macrophages, and lymphocytes), (iii) changes due to the hepatocyte response to the cytokines expressed by these cells, and (iv) changes due to interactions between the hepatocyte and HCV proteins. Ultimately, we expect that some of these changes may become predictive of viral clear-ance or persistence.
MATERIALS AND METHODS
Chimpanzee.The chimpanzee was housed at the Southwest Regional Primate Research Center at the Southwest Foundation for Biomedical Research. The animal was cared for by members of the Department of Laboratory Animal Medicine in accordance withGuide for the Care and Use of Laboratory Animals (16a), and all protocols were approved by the Institutional Animal Care and Use Committee. Chimpanzee 4x0271 was inoculated intravenously with chimpanzee serum containing 1.5⫻107genome equivalents (ge) of HCV genotype 1a, H77 strain (6, 8, 50). Serum and liver needle biopsies were taken through the course of infection. Liver biopsies were obtained early in the morning under fasting conditions to avoid postprandial changes in liver metabolism.
ALT, antibody, and TaqMan analyses.Total RNA prepared from liver biop-sies was used to perform microarray analyses and to monitor viral RNA levels by quantitative reverse transcriptase-PCR (RT-PCR) (37). Serum samples were taken to monitor viral RNA levels, changes in serum alanine transaminases (ALT), and antibody response to HCV proteins (ELISA [enzyme-linked immu-nosorbent assay] Testing System 3.0; Ortho Diagnostic Systems, Raritan, N.J.). HCV RNA was quantified by a real-time, 5⬘exonuclease RT-PCR (TaqMan) assay using an ABI 7700 sequence detector (PE Biosystems, Foster City, Calif.). The primers and probe were derived from the conserved region of the 5⬘
non-* Corresponding author. Mailing address: 7620 NW Loop 410, San Antonio, TX 78227. Phone: (210) 258-9445. Fax: (210) 670-3329. E-mail: [email protected].
7059
on November 9, 2019 by guest
http://jvi.asm.org/
coding region and were selected using the Primer Express software designed for this purpose (PE Biosystems). The forward primer was from nucleotides 149 to 167 (5⬘-TGCGGAACCGGTGAGTACA-3⬘), the reverse primer was from nu-cleotides 210 to 191 (5⬘-CGGGTTTATCCAAGAAAGGA-3⬘), and the probe was from nucleotides 189 to 169 (5⬘-CCGGTCGTCCTGGCAATTCCG-3⬘). The fluorogenic probe was labeled with 6-carboxyfluorescein and 6-carboxytet-ramethylrhodamine and was obtained from Synthegen (Houston, Tex.).
Microarray analysis.All RNA and DNA preparations were made according to Affymetrix (Santa Clara, Calif.) protocols, and hybridizations and data analyses were also performed using Affymetrix protocols, equipment, and software. Briefly, liver punch biopsies from days 0, 2, and 7 and weeks 2, 4, 6, 8, and 14 were homogenized and processed for total RNA using TRIZOL (Life Technol-ogies, Gaithersburg, Md.). These time points were chosen to analyze changes in liver gene expression during the very early stages of viral infection and during peaks in viremia and ALT. Total liver RNA was further purified using an RNeasy total RNA isolation kit (Qiagen, Valencia, Calif.); cDNA synthesis reactions were carried out with 20g of total RNA, using Superscript Choice system (Life Technologies) with an oligo (dT) primer containing a T7 RNA polymerase promoter (Research Genetics, Huntsville, Ala.). Following second-strand syn-thesis, the reaction mixture was extracted with phenol-chloroform-isoamyl alco-hol, and double-stranded cDNA was ethanol precipitated. The DNA was resus-pended in RNase-free water and used to synthesize biotinylated cRNA using a BioArray high-yield RNA transcription labeling kit (ENZO Diagnostics, Farm-ingdale, N.Y.). Biotinylated cRNA was purified using RNeasy affinity columns
(Qiagen). Fragmentation and hybridizations to Affymetrix HumanFL microar-rays were performed by Research Genetics. Data analysis was facilitated by the GeneChip expression analysis sequence information database (Affymetrix).
RESULTS AND DISCUSSION
To initiate this study, a young adult male chimpanzee, not previously exposed to infectious agents, was inoculated with the genotype 1a, H77 strain of HCV (1.50⫻107ge) (6, 7, 49).
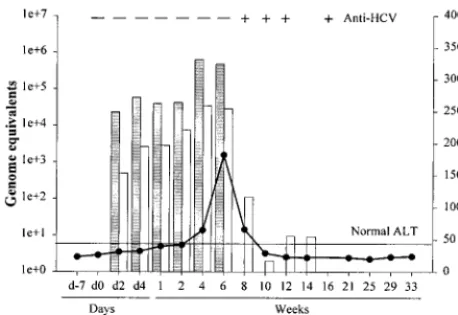
The animal experienced a rapid viral clearance profile (Fig. 1). Viremia was first detected by day 2 postinoculation (p.i.) at 2⫻
104 ge/ml of serum, peak serum titers occurred at week 4
(4.5⫻ 105 ge/ml), and viremia was no longer detectable by
week 8. A similar profile was observed for viral RNA in the liver; a 3-log decline in liver viral RNA levels occurred between weeks 6 and 8, concomitant with the loss of viremia at week 8; however, residual viral RNA persisted in the liver until week 14 p.i. Seroconversion for anti-HCV antibodies occurred be-tween weeks 6 and 8, as well. Serum ALT levels rose sharply between weeks 4 and 6 and declined between weeks 6 and 8 coincident with viral clearance and seroconversion (Fig. 1). The delay in complete clearance of viral RNA from the liver suggests that different mechanisms may be operative between inhibition of replication and subsequent clearance of viremia and the clearance of residual infected hepatocytes.
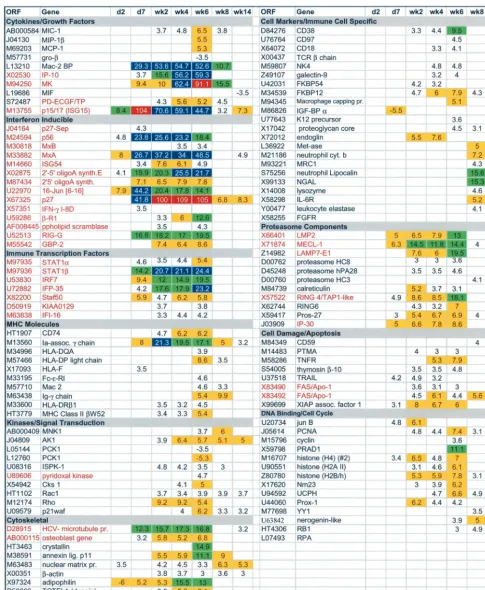
[image:2.612.59.288.74.231.2]Microarray analyses were performed using the Affymetrix HumanFL DNA microarrays which contain oligonucleotides representing approximately 7,000 human genes. Transcripts from approximately one-third (2,300) of the genes on the mi-croarray were detected at each time point and are probably well representative of the total gene expression of the liver. Samples from days 2 and 7 and weeks 2, 4, 6, 8, and 14 were compared to day 0 samples, and only those genes that had a fold change (FC) in expression ofⱖ3.5 were included in this analysis (the baseline cutoff for Affymetrix HumanFL microar-rays is actually a twofold change). Several important aspects of the data were immediately apparent. First, relatively few changes (14 genes) were observed on day 2 (Table 1; seven are listed in Fig. 2), indicating that changes in liver gene expression unrelated to HCV did not present a problem with the assay. This was of concern due to the potential of the mammalian liver to undergo dramatic changes in gene expression in re-sponse to the environment (e.g., postprandial metabolic chang-es). Second, most of the genes with altered expression were detected in more than one sample (Fig. 2), again arguing against spurious changes. FC ofⱖ3.5 were observed for 327 FIG. 1. HCV infection profile of the chimpanzee used in DNA
microarray studies. Chimpanzee 4x0271 was inoculated with HCV genotype 1a, H77 strain, and serial serum and liver biopsy samples were obtained during the course of infection. Serum ALT values were measured as an indication of liver damage (F; horizontal line indicates upper limit of normal). Quantitative RT-PCR (TaqMan) values for viral RNA in the serum or liver are given as genome equivalents per milliliter of serum (shaded bars) or micrograms of total liver RNA (unshaded bars), respectively. Seroconversion for anti-HCV antibodies was monitored by third-generation ELISA (⫹and⫺). d, day.
TABLE 1. FC in liver gene expression during acute HCV infectiona
FC Day 2 Day 7 Wk 2 Wk 4 Wk 6 Wk 8 Wk 14
⫺3.5 to⫺4.9 0 15 9 19 32 10 22
⫺5.0 to⫺9.9 2 6 1 4 20 3 17
⫺10 to⫺20 0 2 2 1 1 2 5
⬎⫺20 0 0 0 1 0 1 1
3.5 to 4.9 9 22 58 64 65 35 41
5.0 to 9.9 3 19 31 29 28 28 26
10 to 20 0 4 9 10 19 6 0
⬎20 0 6 7 10 14 1 1
Total 14 74 117 138 179 86 113
aIn comparison to a day 0 sample taken before HCV infection. The number of genes within a given FC range are indicated in the columns below each time point.
The total number of genes that changed in expression levels at each time point is also listed.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.53.552.601.710.2]FIG. 2. Microarray analysis of changes in liver gene expression during acute HCV infection. FC of 3.5 or greater compared to the preinocu-lation values for a subset of genes are shown in rows from days (d) 2 and 7 and weeks 2, 4, 6, 8, and 14. Genes are listed by open reading frame (ORF) (GenBank accession number or TIGR database HT number) and name and are grouped into functional classes (titles in gray). IFN-inducible genes (22) are in red. Yellow boxes, FC of 5.0 to 9.9; green boxes, FC of 10.0 to 20.0; blue boxes, FC of⬎20; red boxes, FC of⬎90.0. Genes represented on the array multiple times (e.g., FAS/APO-1) reflect probe sets obtained from different accession numbers.
on November 9, 2019 by guest
http://jvi.asm.org/
genes (222 prior to clearance of viremia); of these, 162 were altered in expression in more than one time point. The major-ity of genes with FC ofⱖ3.5 at a single time point occurred at weeks 6 (32%) and 14 (47%); however, many of these single time point changes at the 3.5-fold cutoff level exhibited FC between 3.0 and 3.4 in adjacent time points.
Of the 327 genes that experienced FC of ⱖ3.5, 139 genes exhibited FC of 5 to 10, 37 genes exhibited FC of 10 to 20, 14 genes exhibited FC ofⱖ20, and 3 genes exhibited FC ofⱖ90 (Table 1; Fig. 2). Last, clearance of viremia at week 8 was accompanied by a decline to baseline levels for the majority of genes with altered expression levels, especially the IFN re-sponse genes (Fig. 2, p15/17 and MK [Midkine]). For evalua-tion purposes, these genes were divided into funcevalua-tional group-ings, and 116 are depicted in Fig. 2 (a complete data set can be viewed at www.sfbr.org/sfbr/department/virology/hepatitis.html).
IFN response.Thirty-three of the genes that exhibited a fold
change in expression by microarray analysis are known IFN response genes (22) (red type in Fig. 2), and 5 of the 14 genes exhibiting changes in expression at day 2 were IFN-inducible genes. Most changes at this early time point probably reflect a response to IFN-␣/ secreted by infected hepatocytes. Al-though changes in expression of the IFN genes themselves were not detected, if a relatively small percentage of hepato-cytes were infected (see below), IFN induction may be below the level of detection. However, even low levels of IFN can result in substantial amplification of IFN response genes in adjacent, uninfected cells. For example, two of the genes (p15/17 [22] and p27 [21]) with FC ofⱖ90 are IFN-␣response genes. MK, a third gene up-regulated⬎90-fold, is a retinoic acid response gene that plays a role in inflammation and an-giogenesis (55). MK probably is regulated also by IFN-␣, given that considerable cross-talk exits between the IFN and retinoic acid pathways (14). MK and p15/17 encode cytokines that are chemotatic for neutrophils and enhance the proliferation and cytolytic activity of NK cells, respectively (21, 55).
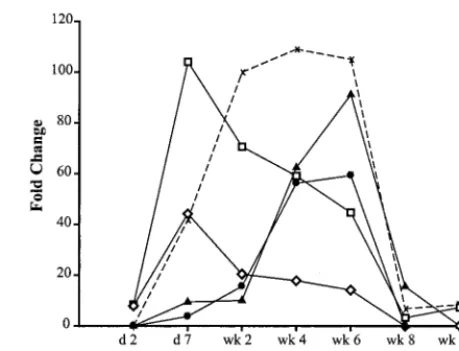
Interestingly, the expression patterns of IFN response genes were temporally distinct. Three patterns in expression of IFN response genes could be discerned (Fig. 3): genes whose ex-pression levels peaked early (day 7 for p15/17 and 16-Jun) then declined; genes whose expression peaked late (week 6 for IFN-inducible protein 10 [IP-10] and MK); and genes whose expression peaked early and was sustained until clearance of viremia (p27). This differential regulation of IFN response genes suggests that different regulatory pathways and/or in-volvement of different cell types and IFN types (IFN-␣/ or IFN-␥) are operative over time. The mechanisms regulating these distinct expression patterns are not understood, but ev-idence suggests that the IFN-␣/antiviral response is certainly complex and constitutes more than the classically defined an-tiviral pathways of 2⬘,5⬘-oligoadenylate synthetase (OAS), dou-ble-stranded RNA-dependent protein kinase R (PKR), and the Mx genes. Indeed, numerous IFN-␣ genes have been de-scribed, and the roles of many of these in antiviral responses have yet to be defined (22).
Cytokines and growth factors.Other cytokine or
immuno-modulatory genes that were upregulated in liver tissue (Fig. 2) included macrophage inhibitory cytokine 1 (MIC-1), macro-phage inflammatory protein 1 beta (MIP-1), macrophage chemoattractant protein 1 (MCP-1), IP-10, platelet derived
endothelial cell growth factor/thymidine phosphorylase (PD-ECGF/TP), and Mac-2-binding protein (Mac2BP). Generally, these gene products have been shown to be chemotactic and/or stimulatory to various immune cells. For example, MIC-1 is a distant member of the transforming growth factor superfam-ily of genes that may serve as an autocrine inhibitor of mac-rophage activation (11). The chemokines MCP-1, MIP-1, and IP-10 are important in recruitment and stimulation of mono-cytes, macrophages, dendritic cells, NK cells, and T lympho-cytes (5, 53). PD-ECGF/TP is functionally diverse, promoting chemotaxis of endothelial cells, angiogenesis, and regulation of steady-state levels of thymidine within cells, and is IFN induc-ible (43). Mac-2BP is a multifunctional protein important in cell adhesion and interaction with extracellular matrix pro-teins, stimulation of NK cell activity, and production of inter-leukin-1 (IL-1) and IL-6 by monocytes (52). Assigning specific functions for these cytokines with regard to HCV infection requires further investigation.
Gene regulation and cell replication.Many genes encoding
DNA-binding proteins and transcription factors changed in expression throughout the infection. Specific increases in mR-NAs encoding proliferating cell nuclear antigen (PCNA), his-tones, and cyclin genes (Fig. 2) indicated the presence of pro-liferative changes in the liver, which may result from liver regeneration to replace damaged hepatocytes and other liver-specific cells. These increases coincided with peak ALT levels (Fig. 1).
[image:4.612.313.543.71.246.2]Genes encoding transcription factors involved in generating the IFN response were increased in expression, including sig-nal transducer and activator of transcription 1 (STAT1) and IFN regulatory factor 7 (IRF-7) (Fig. 2). STAT1 is a member of a family of latent, cytosolic transcription factors activated and/or up-regulated by a variety of stimuli (prolactin, MK, IFN-␣/, and IFN-␥) (20, 41, 51). Upon activation, STAT1 forms hetero- or homodimers, translocates to the nucleus, and FIG. 3. IFN-inducible genes are expressed in distinct temporal pat-terns. Many of the IFN-inducible genes have overlapping patterns of expression, although several exhibit distinct temporal changes in peak expression levels. At least three distinct patterns were observed: genes with an early maximum expression level (䊐[ISG15] and{[16-Jun]), genes with a late peak in expression level (F[IP-10] andŒ[MK]), and genes with an intermediate pattern in which maximum levels were reached early and were sustained (X [p27]). d, day.
on November 9, 2019 by guest
http://jvi.asm.org/
mediates transcription of various genes, including IFN re-sponse genes (e.g., IRF-1) (41, 54). STAT1 encodes two splice variants, STAT1␣ and STAT1. The latter is a dominant-negative form of the protein that binds DNA but cannot acti-vate transcription (41, 54). In this study STAT1 mRNA in-creases were greater than for STAT1␣. This disparity may reflect the in vivo levels of the transcripts in hepatocytes fol-lowing IFN activation, or STAT1may be important in regu-lating STAT1␣activity following activation and induction by IFNs or other cytokines (e.g., MK) (51). IRF-7 is a multifunc-tional gene product that is transcripmultifunc-tionally activated in virus-infected cells and is a key player in transactivating downstream IFN-␣ genes (44, 45). Ironically, IRF-7 also functions as a transcriptional repressor of IRF-1 in Epstein-Barr virus-in-fected cells (62).
Other up-regulated immune transcription factors in this study included stimulated trans-acting factor of 50 kDa (STAF50) and IFN-inducible protein 16 (IFI-16). STAF50 is the human counterpart to the mouse Rpt-1 (regulatory pro-tein, T-lymphocyte 1) gene that has been shown to negatively regulate the IL-2 receptor gene (57). IFI-16 encodes an IFN-␥
response gene that also can function as a repressor of tran-scription (33). The roles of these gene products during the acute stages of HCV infection remain to be determined, espe-cially since several of these genes function as transcriptional repressors. Two central players in activating downstream IFN response genes, p48 of IFN-stimulated gene factor 3 (ISGF3) (complex of STAT1␣-STAT2-p48) and IRF-1 (44, 47, 54) did not change in expression levels throughout the study beyond baseline levels (twofold change). The IRF-1 expression profile seen by microarray analysis was confirmed by quantitative RT-PCR (TaqMan) analysis (data not shown). That IRF-1 is re-quired for activation of NK cells and that the liver contains a significant population of resident NK cells raises an interesting question as to whether HCV partially avoids aspects of the IFN response by circumventing up-regulation of ISGF3 and IRF-1 by as yet unknown mechanisms (9, 24). The likelihood also exists that the IFN response to HCV infection in the liver does not require expression of these specific gene products. Small increases in IRF-1 or p48 not considered significant by mi-croarray analysis still may be biologically relevant in the im-mune response to this virus. The fact that the IFN-␣/ re-sponse was demonstrable by increases in IFN-␣/-inducible genes and not by increases in the IFN-␣/ genes themselves supports this supposition.
Immune cell markers. Several immune cell markers (e.g.,
CD18 and CD38) (Fig. 2) were detected at the later time points, possibly indicating an immune cell infiltrate. However, increases in T-cell-specific markers (e.g., CD3, CD4, and CD8) remained essentially unchanged, with the exception of an in-crease in the T-cell receptor (TCR)chain at week 14 (Fig. 2). Genes encoding major histocompatibility complex (MHC) class II and proteasome components were also up-regulated during the later time points of infection, further suggesting an immune cell infiltrate, although liver-specific endothelial cells also express MHC class II (35). Since the liver participates in clearance of activated T cells from the circulation (19), small increases due to HCV-specific lymphocytes might not be de-tected above the baseline. The apparent lack of a measurable lymphocytic infiltrate by microarray analysis is, however,
con-sistent with our histopathological findings from numerous HCV-infected chimpanzees. The majority of HCV-infected animals do not exhibit significant inflammatory lesions during the acute phase of infection, and the histopathology typically involves hepatocyte swelling with disruption of the sinusoidal spaces that returns to normal following viral clearance. Occa-sionally, isolated lymphocytic infiltration in portal areas is noted. The hepatocyte swelling appears to involve all hepato-cytes, suggesting that the observed histological changes are in response to IFN or other cytokines, since our calculations suggest that not all hepatocytes are infected during the acute phase (see below). Importantly, the apparent minimal T-cell response in this animal occurred in the presence of high levels of IP-10 (FC of⬎50 for weeks 4 to 6), which is chemoattrac-tant for liver-infiltrating lymphocytes in hepatitis. Other immu-nomodulatory genes (e.g., immunophilins FKBP54 and FKBP12, lipocalin) exhibited peak expression levels between weeks 4 and 8 (Fig. 2) and probably reflect activation of NK cells, Kupffer cells, and other polymorphonuclear cells.
Apoptosis. Genes involved in apoptosis in other systems
(e.g., tumor necrosis factor [TNF]-related apoptosis-inducing ligand (TRAIL), TNF receptor [TNFR], and FAS/APO-1) (15, 59, 61) were up-regulated as early as day 7. Apoptosis of infected hepatocytes may be one mechanism of viral clearance, and increases in some apoptotic markers were concurrent with the rise in serum ALT values, an indicator of hepatocyte dam-age. However, increases in apoptotic markers were present prior to a significant rise in serum ALT values and persisted beyond viral clearance. Since biochemical data do not always correlate with histological data, other mechanisms of cell death are likely involved during HCV infection.
Genes associated with HCV infection. Changes were
ob-served in expression levels of several genes with specific rele-vance to HCV infection, including adipophilin, HCV microtu-bule aggregate protein, and Mac-2BP (31, 34, 52). Adipophilin associates with lipid droplets, is increased in pathological con-ditions such as alcoholic liver cirrhosis, and has been impli-cated as a marker for steatosis (31). The involvement of adi-pophilin in the generation of steatosis, a hallmark of HCV infection (48), requires further characterization. An increase in the HCV-associated microtubule aggregate protein was an an-ticipated outcome of HCV infection (34) and served as a pos-itive internal control during the study. Both adipophilin and HCV-associated microtubule aggregate protein mRNA levels returned to baseline levels following viral clearance.
Mac-2BP is one of the 14 genes with an FC of⬎20 (Table 1; Fig. 2). Mac-2BP mRNA levels were 30-fold higher at day 7, steadily increased through week 6 (FC of⬎50), and returned to baseline levels by week 14. Increased levels of Mac-2BP have been demonstrated in hepatocellular carcinoma and HCV-infected patients and may be a marker for IFN-␣ unre-sponsiveness in HCV chronically infected patients (4, 18). The role of Mac-2BP in HCV infection is unclear, although ele-vated levels of Mac-2BP mRNA did not correlate with the development of chronicity, as the animal in this study cleared the infection.
Classical IFN-induced antiviral response. Three classical
IFN response pathways (PKR, Mx proteins, and OAS) are generally associated with antiviral activity (54). Gale et al. and Taylor et al. demonstrated that HCV-encoded NS5A and E2,
on November 9, 2019 by guest
http://jvi.asm.org/
respectively, bind to and inhibit PKR function and proposed that modulation of PKR activity may be one mechanism by which the virus escapes the IFN response (27, 56). Therefore, the lack of an increase in PKR mRNA by microarray analysis, in the context of increases in the other two IFN response pathways (Fig. 2, MxA and OAS), was particularly notable. We sought to confirm these data using quantitative, real-time (TaqMan) RT-PCR. By TaqMan analysis, PKR mRNA in-creased fivefold by week 1, remained elevated through week 6, and returned to baseline levels by week 8 (data not shown). This finding was confirmed using serial liver samples from several other chimpanzees during the acute phase of infection. TaqMan analysis confirmed the microarray expression data for two other genes, adipophilin and IRF-1. The explanation for the disparity with PKR may lie in the choice of probes used in TaqMan versus microarray analysis. The TaqMan probe and primers for PKR mapped within the coding sequence of the gene, whereas all of the probe sets for PKR used in microarray analysis are encoded in the 3⬘untranslated region of the gene. Given that chimpanzee and human DNAs are highly homolo-gous, sequence discrepancies are probably more likely in non-coding regions. Ultimately, virus replication in this animal was significantly curtailed despite possible NS5A/E2 modulation of PKR and presumably without a large specific immune cell infiltrate.
Resolution of infection. Inherent to interpretation of the
data from this experiment is the observation that the chimpan-zee cleared viremia by week 8 and that peak serum ALT levels did not coincide with the decline in viral RNA levels in the serum or liver. These results suggest that viral clearance was not associated with extensive hepatocellular death. This is con-sistent with our observations that some animals clear viral infection without a significant rise in serum ALT values (6, 36). Noncytolytic mechanisms of viral RNA clearance have been demonstrated for hepatitis B virus; in this case, however, the clearance is associated with elevations in expression of IFN-␥
and TNF-␣and a measurable increase in T-cell markers in the liver (16, 30).
Viral clearance in this study was not associated with mea-surable increases in markers for lymphocytic infiltrates (CD3, CD8, and CD4). Additionally, IFN-␥and TNF-␣mRNA levels in the liver remained essentially unchanged by microarray analysis, although small increases in mRNA levels were de-tected by TaqMan assay at week 6 (twofold for IFN-␥ and fourfold for TNF-␣). The significance of these changes is ques-tionable, since a twofold variation was observed between two baseline samples taken 1 week apart prior to inoculation. Even if the elevations in IFN-␥and TNF-␣ that were observed by TaqMan at week 6 were accurate, the biological significance of such small increases is not known.
Viral clearance must also be interpreted in context of the percentage of hepatocytes infected. Peak viral RNA levels in the liver approached 1 ge per hepatocyte (105ge/g of liver
RNA) (Fig. 1). If the average infected cell contains at least 10 ge (1 to 2 copies of negative strand RNA and 9 to 10 copies of positive-strand RNA), then no more than 10% of hepatocytes were infected. The factors that limited infection to a subset of hepatocytes may be the same factors involved in viral clear-ance. Hepatocytes may respond to IFN-␣ and subsequently other cytokines by creating an environment incompatible with
viral replication. Such a scenario would rapidly proceed to a situation in which limited available replication space existed within the liver as an increasing percentage of the uninfected hepatocytes became exposed to IFN-␣. The spread of the in-fection would be limited, and a decrease in viremia would ensue. However, it is unlikely that this mechanism alone would result in complete viral clearance in the absence of an adaptive immune response.
This hypothesis would explain the loss of viremia at 8 weeks p.i., prior to the elimination of viral RNA from the liver. Many IFN response proteins were up-regulated, some of which have known antiviral effects (e.g., OAS, PKR, Mx proteins, and STAT1 as an IFN response transcriptional activator) and are probably important in controlling virus spread and replication. The final phase of viral clearance may be mechanistically dif-ferent and may involve localized cytotoxic T-lymphocyte-de-pendent or cytokine-mediated killing of the residual infected cells. Detection of small increases in HCV-specific CD4⫹and
CD8⫹T cells above the resident population of T cells in the
liver may not be possible by microarray analysis, since the liver is involved in the clearance of activated T cells from the cir-culation (19). However, an early cytotoxic T-lymphocyte re-sponse to multiple HCV antigens has been associated with viral clearance in HCV-infected chimpanzees (17). The data in this report do not minimize the role of HCV-specific T cells in viral clearance. However, the data do demonstrate that an early and extensive increase in IFN response genes is associ-ated with the clearance of viremia during acute infection.
Several lines of evidence support the importance of IFN-␣/
in viral clearance. More recent therapies using pegylated
IFN-␣2b with ribavirin provide a high level of sustained viral clear-ance (29), suggesting that the low response rate of earlier therapies was, in part, due to the inability to maintain IFN levels. HCV infection may also result in down-regulation of the IFN response over time, making chronic infections particularly difficult to treat. Indeed, the use of traditional IFN-␣ mono-therapy resulted in virtually 100% viral clearance rate when applied to individuals during the acute phase of infection (E. Jaekel, M. Cornberg, J. Mayer, J. N. Koerbel, H. Wedemeyer, A. Schueler, M. Zankel, C. Trautwein, and M. P. Manns, Abstr. 51st Annu. Meet. Am. Assoc. Study Liver Dis. abstr. 634, 2000). Other studies have demonstrated that the IFN-␣/
receptor is down-regulated in HCV chronically infected pa-tients (25, 26, 46). The importance of the host in IFN respon-siveness was also demonstrated when the virus from an IFN-unresponsive patient became responsive to IFN when transmitted to another individual (58). Finally, the sensitivity of HCV replication to IFN has been demonstrated in vitro using HCV replicons (10, 42).
The association of a vigorous IFN-␣/response with clear-ance of viremia in conjunction with the apparent biphasic pat-tern of viral clearance observed in this chimpanzee suggests that both the innate and adaptive immune responses play crit-ical roles in elimination of HCV during the acute phase of infection. This biphasic mechanism of viral clearance is sup-ported by the kinetics of viral decline observed during IFN treatment of HCV infection. The early, rapid phase of viral decline is hypothesized to involve inhibition of viral replica-tion, while the second, slower phase of decline is thought to result from the destruction of infected hepatocytes (32, 38).
on November 9, 2019 by guest
http://jvi.asm.org/
Clearly, the use of DNA microarray technology to examine the changes in gene expression during the acute phase infection of HCV has provided new insight into the role of IFN in viral clearance and many opportunities for future studies on the myriad other changes observed.
ACKNOWLEDGMENTS We thank Bernadette Guerra for TaqMan analyses.
This work was supported by grants U19 AI40035 and P51 RR13986 from the National Institutes of Health.
REFERENCES
1.Ahmed, A., and E. B. Keeffe.1999. Treatment strategies for chronic hepatitis C: update since the 1997 National Institutes of Health Consensus Develop-ment Conference. J. Gastroenterol. Hepatol.14:S12–S18.
2.Alter, H., and L. Seeff.2000. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis.
20:17–35.
3.Anonymous.1997. Hepatitis C: global prevalence. Wkly. Epidemiol. Rec.
72:341–344.
4.Artini, M., C. Natoli, N. Tinari, A. Costanzo, R. Marinelli, C. Balsano, P. Porcari, D. Angelucci, M. D’Egidio, M. Levrero, and S. Iacobelli.1996. Elevated serum levels of 90K/MAC-2 BP predict unresponsiveness to alpha-interferon therapy in chronic HCV hepatitis patients. J. Hepatol.25:212– 217.
5.Baggiolini, M., B. Dewald, and B. Moser.1997. Human chemokines: an update. Annu. Rev. Immunol.15:675–705.
6.Bassett, S. E., K. M. Brasky, and R. E. Lanford.1998. Analysis of hepatitis C virus-inoculated chimpanzees reveals unexpected clinical profiles. J. Virol.
72:2589–2599.
7.Bassett, S. E., D. L. Thomas, K. Brasky, and R. E. Lanford.1999. Viral persistence, antibody to E1 and E2, and hypervariable region 1 sequence stability in hepatitis C virus-inoculated chimpanzees. J. Virol.73:1118–1126. 8.Bassett, S. E., D. L. Thomas, K. M. Brasky, and R. E. Lanford.1999. Viral persistence, antibody to E1 and E2, and hypervariable region 1 sequence stability in hepatitis C virus-inoculated chimpanzees. J. Virol.73:1118–1126. 9.Biron, C. A., K. B. Nguyen, G. C. Pien, L. P. Cousens, and T. P. Salazar-Mather.1999. Natural killer cells in antiviral defense: function and regula-tion by innate cytokines. Annu. Rev. Immunol.17:189–220.
10. Blight, K. J., A. A. Kolykhalov, and C. M. Rice.2000. Efficient initiation of HCV RNA replication in cell culture. Science290:1972–1975.
11. Bootcov, M. R., A. R. Bauskin, S. M. Valenzuela, A. G. Moore, M. Bansal, X. Y. He, H. P. Zhang, M. Donnellan, S. Mahler, K. Pryor, B. J. Walsh, R. C. Nicholson, W. D. Fairlie, S. B. Por, J. M. Robbins, and S. N. Breit.1997. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-superfamily. Proc. Natl. Acad. Sci. USA94:11514–11519. 12. Cerny, A., and F. V. Chisari.1999. Pathogenesis of chronic hepatitis C:
immunological features of hepatic injury and viral persistence. Hepatology
30:595–601.
13. Chang, Y. E., and L. A. Laimins.2000. Microarray analysis identifies inter-feron-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol.74:4174–4182.
14. Chelbi-Alix, M. K., and L. Pelicano.1999. Retinoic acid and interferon signaling cross talk in normal and RA-resistant APL cells. Leukemia13:
1167–1174.
15. Cheng, J., T. Zhou, C. Liu, J. P. Shapiro, M. J. Brauer, M. C. Kiefer, P. J. Barr, and J. D. Mountz.1994. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science263:1759–1762.
16. Chisari, F. V.1997. Cytotoxic T cells and viral hepatitis. J. Clin. Investig.
99:1472–1477.
16a.Committee on Care and Use of Laboratory Animals.1996. Guide for the care and use of laboratory animals. Institute of Laboratory Animal Re-sources, National Research Council. Washington, D.C.
17. Cooper, S., A. L. Erickson, E. J. Adams, J. Kansopon, A. J. Weiner, D. Y. Chien, M. Houghton, P. Parham, and C. M. Walker.1999. Analysis of a successful immune response against hepatitis C virus. Immunity10:439–449. 18. Correale, M., V. Giannuzzi, P. A. Iacovazzi, M. A. Valenza, S. Lanzillotta, I. Abbate, M. Quaranta, M. L. Caruso, S. Elba, and O. G. Manghisi.1999. Serum 90K/MAC-2BP glycoprotein levels in hepatocellular carcinoma and cirrhosis. Anticancer Res.19:3469–3472.
19. Crispe, I. N., T. Dao, K. Klugewitz, W. Z. Mehal, and D. P. Metz.2000. The liver as a site of T-cell apoptosis: graveyard, or killing field? Immunol. Rev.
174:47–62.
20. Darnell, J. E. J.1997. STATs and gene regulation. Science277:1630–1635. 21. D’Cunha, J., E. J. Knight, A. L. Haas, R. L. Truitt, and E. C. Borden.1996. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl. Acad. Sci. USA93:211–215.
22. Der, S. D., A. Zhou, B. R. Williams, and R. H. Silverman.1998. Identification
of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA95:15623–15628. 23. Diepolder, H. M., R. Zachoval, R. M. Hoffmann, M. C. Jung, T. Gerlach, and
G. R. Pape.1996. The role of hepatitis C virus specific CD4⫹T lymphocytes
in acute and chronic hepatitis C. J. Mol. Med.74:583–588.
24. Duncan, G. S., H. W. Mittrucker, D. Kagi, T. Matsuyama, and T. W. Mak.
1996. The transcription factor interferon regulatory factor-1 is essential for natural killer cell function in vivo. J. Exp. Med.184:2043–2048.
25. Fukuda, R., N. Ishimura, S. Ishihara, A. Tokuda, S. Satoh, S. Sakai, S. Akagi, M. Watanabe, and S. Fukumoto.1996. Expression of interferon-alpha receptor mRNA in the liver in chronic liver diseases associated with hepatitis C virus: relation to effectiveness of interferon therapy. J. Gastroenterol.
31:806–811.
26. Fukuda, R., N. Ishimura, Y. Kushiyama, N. Moriyama, S. Ishihara, S. Nagasawa, T. Miyake, M. Niigaki, S. Satoh, S. Sakai, S. Akagi, M. Wa-tanabe, and S. Fukumoto.1997. Effectiveness of interferon-alpha therapy in chronic hepatitis C is associated with the amount of interferon-alpha recep-tor mRNA in the liver. J. Hepatol.26:455–461.
27. Gale, M., Jr., C. M. Blakely, B. Kwieciszewski, S. L. Tan, M. Dossett, N. M. Tang, M. J. Korth, S. J. Polyak, D. R. Gretch, and M. G. Katze.1998. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol. Cell. Biol.18:5208–5218. 28. Geiss, G. K., R. E. Bumgarner, M. C. An, M. B. Agy, A. B. van’t Wout, E. Hammersmark, V. S. Carter, D. Upchurch, J. I. Mullins, and M. G. Katze.
2000. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology266:8–16.
29. Glue, P., R. Rouzier-Panis, C. Raffanel, R. Sabo, S. K. Gupta, M. Salfi, S. Jacobs, and R. P. Clement.2000. A dose-ranging study of pegylated inter-feron alfa-2b and ribavirin in chronic hepatitis C. The Hepatitis C Interven-tion Therapy Group. Hepatology32:647–653.
30. Guidotti, L. G., R. Rochford, J. Chung, M. Shapiro, R. Purcell, and F. V. Chisari.1999. Viral clearance without destruction of infected cells during acute HBV infection. Science284:825–829.
31. Heid, H. W., R. Moll, I. Schwetlick, H. R. Rackwitz, and T. W. Keenan.1998. Adipophilin is a specific marker of lipid accumulation in diverse cell types and diseases. Cell Tissue Res.294:309–321.
32. Herrmann, E., A. U. Neumann, J. M. Schmidt, and S. Zeuzem.2000. Hep-atitis C virus kinetics. Antiviral Ther.5:85–90.
33. Johnstone, R. W., J. A. Kerry, and J. A. Trapani.1998. The human inter-feron-inducible protein, IFI 16, is a repressor of transcription. J. Biol. Chem.
273:17172–17177.
34. Kitamura, A., K. Takahashi, A. Okajima, and N. Kitamura.1994. Induction of the human gene for p44, a hepatitis-C-associated microtubular aggregate protein, by interferon-alpha/beta. Eur. J. Biochem.224:877–883. 35. Knolle, P. A., and G. Gerken.2000. Local control of the immune response in
the liver. Immunol. Rev.174:21–34.
36. Lanford, R. E., C. Bigger, S. Bassett, and G. R. Klimpel.2001. The chim-panzee model of hepatitis C virus infections. ILAR J.42:117–126. 37. Lanford, R. E., H. Lee, D. Chavez, B. Guerra, and K. Brasky.2001.
Infec-tious cDNA clone of the hepatitis C virus genotype 1 prototype sequence. J. Gen. Virol.82:1291–1297.
38. Layden, T. J.1999. Principles of interferon induction therapy. Am. J. Med.
107:71S–73S.
39. Lechner, F., D. K. Wong, P. R. Dunbar, R. Chapman, R. T. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B. D. Walker.
2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med.191:1499–1512.
40. Lee, C. K., R. G. Klopp, R. Weindruch, and T. A. Prolla. 1999. Gene expression profile of aging and its retardation by caloric restriction. Science
285:1390–1393.
41. Leonard, W. J., and J. J. O’Shea.1998. Jaks and STATs: biological impli-cations. Annu. Rev. Immunol.16:293–322.
42. Lohmann, V., F. Ko¨rner, J. O. Koch, U. Herian, L. Theilmann, and R. Bartenschlager.1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science285:110–113.
43. Makower, D., S. Wadler, H. Haynes, and E. L. Schwartz.1997. Interferon induces thymidine phosphorylase/platelet-derived endothelial cell growth factor expression in vivo. Clin. Cancer Res.3:923–929.
44. Mamane, Y., C. Heylbroeck, P. Genin, M. Algarte, M. J. Servant, C. LePage, C. DeLuca, H. Kwon, R. Lin, and J. Hiscott.1999. Interferon regulatory factors: the next generation. Gene237:1–14.
45. Marie, I., J. E. Durbin, and D. E. Levy.1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon reg-ulatory factor-7. EMBO J.17:6660–6669.
46. Mathai, J., K. Shimoda, B. F. Banner, M. Mori, H. L. Bonkovsky, and G. F. Barnard.1999. IFN-alpha receptor mRNA expression in a United States sample with predominantly genotype 1a/I chronic hepatitis C liver biopsies correlates with response to IFN therapy. J. Interferon Cytokine Res.19:
1011–1018.
47. Matsumoto, M., N. Tanaka, H. Harada, T. Kimura, T. Yokochi, M. Kita-gawa, C. Schindler, and T. Taniguchi.1999. Activation of the transcription factor ISGF3 by interferon-gamma. Biol. Chem.380:699–703.
on November 9, 2019 by guest
http://jvi.asm.org/
48.Moriya, K., H. Yotsuyanagi, Y. Shintani, H. Fujie, K. Ishibashi, Y. Mat-suura, T. Miyamura, and K. Koike.1997. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol.78:1527–1531. 49.Ogata, N., H. J. Alter, R. H. Miller, and R. H. Purcell.1991. Nucleotide
sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA88:3392–3396.
50. Ogata, N., H. J. Alter, R. H. Miller, and R. H. Purcell.1991. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA88:3392–3396.
51. Ratovitski, E. A., P. T. Kotzbauer, J. Milbrandt, C. J. Lowenstein, and C. R. Burrow.1998. Midkine induces tumor cell proliferation and binds to a high affinity signaling receptor associated with JAK tyrosine kinases. J. Biol. Chem.273:3654–3660.
52. Sasaki, T., C. Brakebusch, J. Engel, and R. Timpl.1998. Mac-2 binding protein is a cell-adhesive protein of the extracellular matrix which self-assembles into ring-like structures and binds1 integrins, collagens and fibronectin. EMBO J.17:1606–1613.
53. Scapini, P., J. A. Lapinet-Vera, S. Gasperini, F. Calzetti, F. Bazzoni, and M. A. Cassatella.2000. The neutrophil as a cellular source of chemokines. Immunol. Rev.177:195–203.
54. Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber.1998. How cells respond to interferons. Annu. Rev. Biochem.
67:227–264.
55. Takada, T., T. Kinkori, H. Muramatsu, A. Hayakawa, S. Torii, and T. Muramatsu.1997. Midkine, a retinoic acid-inducible heparin-binding cyto-kine, is a novel regulator of intracellular calcium in human neutrophils. Biochem. Biophys. Res. Commun.241:756–761.
56. Taylor, D. R., S. T. Shi, P. R. Romano, G. N. Barber, and M. M. Lai.1999. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 pro-tein. Science285:107–110.
57. Tissot, C., and N. Mechti.1995. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J. Biol. Chem.270:14891–14898.
58. Toyoda, H., H. Sakamoto, T. Mizuno, Y. Horiguchi, and H. Nakano.2000. Eradication of hepatitis C virus 1b by interferon in a health care worker with acute hepatitis following needlestick transmission from a patient with chronic hepatitis C unresponsive to interferon. Scand. J. Gastroenterol.
35:1117–1120.
59. Wallach, D., E. E. Varfolomeev, N. L. Malinin, Y. V. Goltsev, A. V. Kovalenko, and M. P. Boldin.1999. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol.17:331–367.
60. White, K. P., S. A. Rifkin, P. Hurban, and D. S. Hogness.1999. Microarray analysis of Drosophila development during metamorphosis. Science286:
2179–2184.
61. Wiley, S. R., K. Schooley, P. J. Smolak, W. S. Din, C. P. Huang, J. K. Nicholl, G. R. Sutherland, T. D. Smith, C. Rauch, and C. A. Smith.1995. Identifi-cation and characterization of a new member of the TNF family that induces apoptosis. Immunity3:673–682.
62. Zhang, L., and J. S. Pagano.1997. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol.17:5748–5757. 63. Zhu, H., J. P. Cong, G. Mamtora, T. Gingeras, and T. Shenk.1998. Cellular
gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc. Natl. Acad. Sci. USA95:14470–14475.