0022-538X/83/060944-12$02.00/0
Copyright © 1983,AmericanSociety forMicrobiology
Reciprocal
Productive
and
Restrictive Virus-Cell Interactions
of Immunosuppressive
and Prototype Strains
of Minute Virus
of Mice
PETERTATTERSALL* ANDJESSICABRATTON
DepartmentofHumanGenetics, Yale UniversitySchoolofMedicine, New Haven, Connecticut 06510
Received 26 January1983/Accepted24February1983
Viral and cellular factors responsible for parvovirustargetcellspecificityhave beenexamined fortwoserologically indistinguishable strains of the minute virus of mice which infectmousecellsof dissimilar differentiated phenotype. Both the prototype strain and theimmunosuppressive straingrowin and formplaques on monolayersof simian virus 40-transformed human fibroblast's, afinding that has allowed the
comparison
of several aspects of their virus-host cell interactions. Although closely related by antigenic and genomic criteria, both the prototype strain and theimmunosuppressive
strain arerestricted for lytic growth in each other's murine hostcell, thatis,
inTcells andfibroblasts,
respectively. The host rangeof each virus variantappears tobespecified byagenetic determinant that is stably inherited in the absence of selection. Inthe restrictive virus-host interac-tionlytic growth is limitedto asmallor,insomecases,undetectable subset ofthe host cellpopulation,
and the majority of the infected cells remain viable,continuing
to grow at the normal rate withoutexpressing
viral antigens. Thesusceptible host cell phenotype is dominant inTlymphocyte x fibroblast fusion hybrids, implying that different cell types express different developmentally regulated virus helper functions that can only be
exploited
by the virus variant thatcarries theappropriate strain-specific
determinant.The parvovirus
family
consists of alarge
number of
physically
andchemically
similar viruses that infect many animalspecies
(36). These agents are small,nonenveloped,
icosahe-dral virionsapproximately
20 nm indiameter,
containing
a5-kilobase,
single-stranded
DNAgenome (36). The vertebrate
parvoviruses
aredivided intotwo
subgroups
onthebasis of their requirementforhelper
viruses. Membersofthe adeno-associated virus subgroup are defective anddependentirely
uponadenovirusorherpes-virus coinfection for their own
replication.
In contrast, members of the autonomous parvovi-rus subgroup arecapable ofproductive replica-tion withouttheaidofahelper
virus both in vivo and in vitro.Numerous studiesover the past 20 years on
the
pathogenicity
ofautonomousparvoviruses
have shown thattheyare
predominantly
terato-genic agents. In
general,
they cause fetal and neonatal abnormalities by destroyingspecific
cell
populations
that arerapidly
proliferating
during the normal course ofdevelopment (re-viewed in references 12 and 19). These same tissues are generally resistant in the mature animal; consequently, few oftheviruses cause clinical disease in the adult. However, some of
these tissues can be rendered susceptible to virus infection by inducing them toundergo an abnormal
proliferative
response. For example,the
induction
of mitotic activity in the liver bypartial hepatectomy (28), by carbon tetrachlo-ride toxicity (12), and byparasite infection (13) resultsinparvovirus hepatitis,wherevirus repli-cation is localized in theregenerating portion of theliver.Asimilarpreferential attackon regen-erating tissue is seenin parvovirus infection of healingosseouswounds (2).
Subsequent
analysis
of the autonomous par-vovirus growth cycle in cultured cells has pro-vided a rationale for the predilection of these viruses for dividing cells observed in vivo. These studieshave shownthatvirusreplication is dependent upon cellular functions expressed transiently during the S-phase of the cell cycle (27, 30, 33, 37). Sincethevirusescannotinduce resting cells to enter the S-phase (33), it is therefore not surprising that viral replication is restricted to dividing cell populations both in vitro (33) andinvivo (15).Althoughproliferative activity appears to be a prerequisite for target organs, it is clear that not all tissues thatturn over rapidly are necessarilysubject
to virus-induced damage (15). Although most adulttis-944
on November 10, 2019 by guest
http://jvi.asm.org/
sues are mitotically quiescent compared with those of the fetus and neonate, many, such as gut epithelium and the lymphopoietic system, contain large numbers of cycling cells. One might expect these cells, which are essential for the host organism's well being and survival, to be targetsfor parvovirus attack inthe adult. The sparing of these adult tissues by the majority of autonomous parvoviruses is underlined by the existence of a small subset of parvoviruses, namely, the feline panleukopenia-mink enteritis-canine parvovirus group and the Aleutian dis-ease virus ofmink, which frequently causefatal disease in adult animals. The disease involves extensive destruction ofgut epithelium and re-ticuloendothelial cells (10, 21, 26).
Studiesonthereplication ofautonomous par-voviruses in vitro, particularly with the minute virus of mice(MVM) (38), have provided signifi-cant support for the hypothesis that lytic virus growth is modulated by developmentally regu-lated components operating in the host at the cellularlevel. Mohanty and Bachmann(23) have reported thatactively dividing cells of the early mouseembryoareresistant tokillingby MVM. Murine embryonal carcinoma cells, the stem cells of teratocarcinoma, are resistant to the prototype strain MVM(p), as are many of their differentiated derivatives (22, 34). However, when these cells are induced to differentiate in vitrothey give rise to at least one differentiated cell type, resembling a fibroblast, which sup-portslytic MVM(p) replication(34). These stud-iessuggest thatcell cycling, although necessary, isnotsufficient for thelytic replicationof parvo-viruses, and that the differentiated state of the host cellis ofparamount importance.
That differences in pathogenic potential exist not only between virus serotypes, but also be-tween virus strains ofthe same serotype (4-6, 11, 18, 20) suggests that a particular tissue tropism might not be an invariant property of each virus. Theisolation ofan additional strain of minute virus ofmice, MVM(i),as an immuno-suppressiveagentfromamurine lymphoma indi-cates that a mutable genetic component of the virusmayplay a role in determining the type of differentiated cell the virus can lytically infect. MVM(i)suppresses anumber ofTcell-mediated functions as measured in vitro, whereas MVM(p) does not (6), despite their genomes being closelyrelatedby restriction endonuclease mapping(20).
We describe here a single infectivity assay system for both MVM(p) and MVM(i) which has allowed us to compare directly the interactions ofboth virus strains with host cells of lympho-cyte and fibroblast origin. The results of this study suggest that both the viral component determiningtarget cellspecificity and the
devel-opmentally regulated host factors with which it interacts can be dissected invitro.
MATERIALS ANDMETHODS
Cell lines and culture conditions. A9 ouabrl1 cells
werederivedfromtheoriginal HGPRT- L-cell line A9
(16)byselection for clones resistantto10'- Mouabain
afternitrosoguanidinemutagenesis. A9clone 8Eisan
MVM(p) resistant derivative of A9 which does not
carrythereceptorforMVM(p)onitssurface(14). S49
1TB2 isathymidine kinase-negativemutant ofthe
T-cell lymphoma line S49 (3). EL4-sti is an adherent
variantoftheT-celllymphoma lineEL4(8). RPC5.4is
animmunoglobulin G2a-secreting myeloma line (24).
C127 isafibroblast line derivedby Lowy etal. (17).
Hyb 1/11 is a clonal hybrid line made by fusing A9
ouabrll with EL4-sti essentially by the method of
O'Malley and Davidson (25) followedby selection in
medium containing
hypoxanthine-aminopterin-thymi-dine (HAT) (32) and 10-3 M ouabain. Hyb 2/40 isa
clonalhybrid cell line resulting from asimilar fusion
between A9 ouabrll and S49 1TB2, followed by
selectionin mediumcontainingHAT.324Kcellsarea
clone of simian virus40-transformed human newborn
kidney cells (29). These cellscontain simian virus40 T
antigens as detected by immunofluorescent staining
withmonoclonalantibodies of theseriesgenerated by
Harlowetal. (9), but donotproduce infectious simian
virus40eitherspontaneouslyor uponfusion with
CV-1 monkey cells (unpublished results). Lymphocyte
cultures weremaintained inAutopowmonolayer
me-dium(FlowLaboratories, Inc.) with nonessential
ami-no acids and 10% heat-inactivated fetal calf serum.
324K, C127, and hybrid cell lines werecultivated in
thesame medium containing5%serum,and A9lines
were maintained in suspension culture in Autopow
spinner medium with nonessential amino acids and 5%
serum. Forcomparative infections all cell lineswere
cultured inmonolayer medium with nonessential
ami-noacids and10%serum.
Virus stocks and infectivity assay. The original
cloned stock of MVM, previously designated
MVM(T), has been described elsewhere (33). This
strain is nowdesignated MVM(p)-forprototype-to
avoidconfusion with thepreexisting nomenclature of
T- and B-lymphocyte populations. Culture
superna-tant containing the uncloned immunosuppressive
strain of MVM (4), now designated MVM(i), was
generouslyprovided byB.Hirt. This viruswascloned
by terminal dilutionassayinEL4lymphocyte cultures
as described below. Both viruses were assayed by
plaque titrationon324Kcellmonolayers. Briefly,
60-mm plastic tissue culture dishes were inoculated, at
25% confluency, with 0.2-ml samples of virus diluted
in monolayer mediumcontaining 1% fetal calfserum
and buffered at pH 7.3 with 25 mM HEPES
(N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic
acid)-NaOH. After adsorption for 60 min at 37°C, the
monolayers were overlaid with 8 mlof medium
con-taining0.6% agarose(Seakem;type ME), 0.2%
tryp-tose phosphate, 5% serum, and buffered 15 mM
HEPES-10 mM
N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid adjusted to pH 7.8 with
NaOH. After incubation for 6 days, plaques were visualized by stainingwithneutralred.
Virusproduction andpurification. High-titer stocks of [3H]thymidine-labeled virus were produced in 46,
on November 10, 2019 by guest
http://jvi.asm.org/
946 AND
monolayers of324Kcells byinfectionat 10PFU per
cell followed by incubation in medium containing
[methyl-3H]thymidine (1 ,uCi/ml;Sx10-6M).After48
to72 h,cellswerescraped into themedium, collected
bylow-speedcentrifugation, washed with
phosphate-buffered saline, andfinally suspended in asmall
vol-ume of TE8.7 (50 mMTris,0.5 mM EDTA, pH 8.7)
(35). The cellswere lysedby three cycles offreezing
and thawing, and cell debris was removed by
low-speedcentrifugation. Afterfurtherclearing(5 minin
anEppendorfmicrofuge),thesupernatantwaslayered
on an11.5-ml10to30% glycerol gradientin TE8.7 and
centrifuged for 2.5hat35,000rpmand5°CinanSW41
rotor.The peak of 110Sinfectious virionswaslocated
by fractionating thegradient from the bottom of the
tubefollowed by liquid scintillation countingtolocate
the peak of labeled full virions. Peakfractions were
pooled and assayed for infectivity by the 324K cell assay.Allcomparisonsof thebiological activity of the
twoMVM strains reported herewereperformed with
such glycerol gradient-purified virions, unless
other-wise indicated in the legend to thefigure. High-titer
stocksofunlabeled MVM(p) and MVM(i) were also prepared in spinnercultures ofA9 ouabrl1 and S49
1TB2, respectively. Infected cells wereharvested by
centrifugation, andaTE8.7lysatewaspreparedasfor
324K cells. After the high-speed clearing spin, the
supernatant was assayedfor infectivity by the 324K
assay, anddilutions wereusedtoinfect cellswithout furtherpurification.
RESULTS
Isolation and assay of MVM(i). The original stock of MVM(i) was derived from the T-cell lymphomaEL4,which had been maintainedas a transplantable tumor in C57/BL mice (4). The virus was isolated by growing these cells in culture until cytopathic effect (CPE) was ob-served, which was manifest by massive clump-ing of the cells followed by cessationofgrowth andloss ofviability asmeasuredbytrypanblue
staining. The uncloned stock of virus used to
initiate these studies was aculture supernatant
from such infected cells obtained from B. Hirt
(Lausanne, Switzerland). Initial stocks were
prepared by inoculating this supernatant into
cultures ofrapidlydividing, virus-free,EL4cells
and allowing CPE to develop. The virus was harvested from these cellsbyfreezingand
thaw-ing in TE8.7. Suchan extractwasthenassayed by inoculatingdilutions into replicate EL4
cul-tures, which were then scored for CPE after
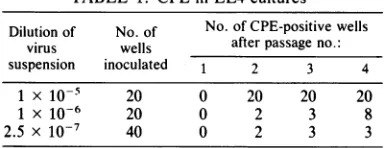
each offour passages as described in Table 1.
Theinfected cells fromoneof thethreepositive cultures atthe highest dilution were extracted, and the virus was used to derive a high-titer stock in EL4 cells. Since less than 10% of the
cultures at this dilution developed CPE, we assumethatthisstockisclonal.Thisassumption
hasbeen confirmedbythefindingthatthis virus
uniformly lacks an HindIII restriction site at map unit 79, which ispresentin approximately 80% of the genomes in the uncloned stock and
TABLE 1. CPE in EL4culturesa
Dilution of No.of No. ofCPE-positivewells virus wells afterpassage no.:
suspension inoculated 1 2 3 4
1x10-5 20 0 20 20 20
1 x 10-6 20 0 2 3 8
2.5 x 10-7 40 0 2 3 3
aReplicate 2-ml cultures of EL4-sti inoculated with
dilutions ofanuncloned stock ofMVM(i)were
subcul-tured every 5 to 7 days by transferring 0.1 ml of
mediumplusunattached cellsto2 ml offresh medium.
uniformly present in virus subsequently grown up from another of these three CPE-positive cultures (datanotshown).
Using
the initial cloned stock, we havecom-paredthe abilityofthis virus and the prototype strainMVM(p) togrowlytically in a numberof celllines. Althoughwe have not found amouse
cell line that supports extensive lytic infection bybothviruses, wefound that twosimian virus 40-transformed humannewbornkidneycelllines NB-E and 324K would support both viruses and indeed afford equivalent plaque assay systems forboth. Figure 1 shows that both
MVM(p)
and MVM(i) formplaques on324K cellmonolayers with single-hit kinetics. The low virus input required for plaque formation and the kinetics observed indicate that a single particle of each virus is capable offorming a plaque in these cells.MVM(p)
is 5- to 10-fold moreefficient,per particle,atforming plaqueson324Kmonolayers thanMVM(i), and this assay forMVM(p)
infec-tivity is about 20-fold more sensitive than the previously described assay on A9 cell monolay-ers (33). MVM(i), however, is at least 107-fold more efficient at plaque formation on 324K monolayers than on A9 monolayers.Wehave used the 324K cellassay to examine the sensitivity of both viruses to neutralization by a high-titer antiserum raised in rabbits against
MVM(p).
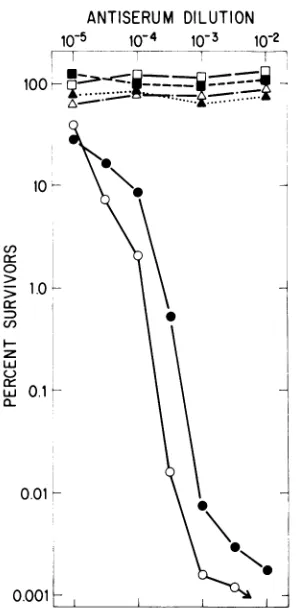
BothMVM(p)
and MVM(i) areneu-tralized with similar kinetics by this antiserum (Fig. 2). The serumappears to have a higher titer against
MVM(p),
which may reflect minor dif-ferences in the amino acid sequence of the capsid polypeptides between the two viruses; however, it is clear that they are antigenically very closely related. This closeness is empha-sized by the fact that this antiserum has no neutralizing activity againstH-1,anotherrodent parvovirus (Fig.2),althoughH-1, MVM(p),and MVM(i) shareantigenic determinantsdetectableby
cross-immunoprecipitation
oftheir structuralpolypeptides (S. F. Cotmore and P. Tattersall, manuscriptin preparation).
Reciprocal susceptibility of host cells to both virus strains. The screening ofmurine cell lines
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.488.257.449.79.153.2]2.0
U)
r-lL
LL
z
LLU
0c ct
-J
0
-J
1.0
05K
0
0 0.5 1.0
LOG1ORELATIVE DILUTION
FIG. 1. Dose-response curve for MVM(p) and MVM(i) on 324K cell monolayers. Serial twofold
dilutions ofgradient-purifiedMVM(p) virions (0) and MVM(i) virions (0) were assayed for PFU with the assay described in the text. Error bars indicate the samplestandarddeviationfor each virus dose.
for theirabilitytosupportthelytic replication of
MVM(p) and MVM(i) has revealed a general pattern. Undifferentiated teratocarcinoma cells
andanumberof differentiated cell lines appear
to be refractory to infection by both virus
strains. Of the T-cell lymphomas examined, all
wereinfected by MVM(i), butnotMVM(p), and fibroblastic cell lines, including L cell deriva-tives, were infectable with MVM(p), but not
MVM(i). On the assumption that these
interac-tions reflect differences in thetissue tropism of
the virus strains invivo, we haveexamined the
course of infection of both viruses in two such cell lines, the L-cell derivative A9ouabr1l and theT-celllymphomaline S491TB2.
Infections of these two cell types by both
MVM strainsatvariousinput multiplicitieswere
monitored for the appearance of nuclear viral
capsid antigen at 26 h after infection (Fig. 3).
Since autonomous parvoviruses require a host
cellfunction that isexpressedtransiently during
the Sphaseof the cellcycle(36),the timecourse
ofappearance of viralantigen early after
infec-tion reflects the asynchrony of the cell
popula-tion. However, by26 hpostinfection the
major-ity of cells in which the input virus has
established an infectious cycle are expressing
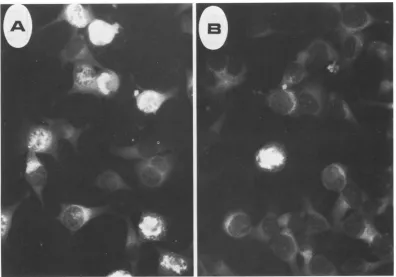
highlevelsofcapsid antigen. Ascanbeseen,A9
and S49cells showconsiderablesusceptibilityto
infectionbyMVM(p)andMVM(i), respectively,
asmeasuredby this assay, throughout the range ofmultiplicities examined. However, the recip-rocal infections show much less efficient virus takeover of the cell.Thus, in S49 cultures infect-ed with MVM(p) all nuclei, out of the several thousandexamined,wereindistinguishablefrom those of uninfected cells, as were the great majority of nuclei in A9 cultures infected with MVM(i). Significantly, in the small fraction of antigen-positive nuclei in MVM(i)-infected A9 cultures, each nucleus appeared to express a level of viral antigen equivalent to that of posi-tive nuclei in the reciprocal infection (Fig. 4),
ANTISERUM DILUTION
10-5
10-4
10-3
10-2
10012i- -S
°>101
a\
0.0- K.
U)~\
LUJ
C-_
Lu 0.1
0.001
FIG. 2. Neutralization curves for MVM(p),
MVM(i), and H-1 with nonimmune andanti-MVM(p)
rabbitsera.Ahigh-titer stock of each viruswasmixed
with dilutions of either nonimmune rabbit serum (nr serum)or ahyperimmune anti-MVM(p) serumraised inrabbitsbyrepeated subcutaneousinjectionof
puri-fied MVM(p)emptycapsids (apserum). Symbols: O,
MVM(p) + nr; E, MVM(i) + nr; A, H-1 + nr; 0,
MVM(p) +ap;*,MVM(i) +ap;A,H-1 + oxp.After
incubation for1 h at37°C, the virus wasdiluted and
assayedforsurvivingvirus(PFU) asdescribed in the
text, except that the indicator monolayers were washedafter adsorption to remove any residual anti-serum before theoverlay with agar-containing medi-um.
46,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.488.65.215.62.284.2] [image:4.488.272.420.232.538.2]BRATTON
100
w
LLJ
z
wi
10-0
Q-z
.Z
w
(! I
z
0.1
o0.052
3 10 30 100
INPUT MULTIPLICITY
FIG. 3. Infectionof A9 ouabrll and S49 1TB2 host
cellswithincreasing multiplicities of virus. Replicate
culturesof A9 ouabrll and S49 1TB2 cellsat2 x 105
cellspermlwereinfectedat3, 10,30,and100 PFUper
cell with glycerol gradient-purified MVM(p) or
MVM(i) virions. Cellswerefixed inacetone26 h after
infection and stained by indirect fluorescence for
capsid antigen. The primary antibodywas the same
hyperimmune rabbitantiserum raisedagainstMVM(p)
capsids described in the legend to Fig. 2, and the
secondary antibody was fluorescein-conjugated goat
anti-rabbit immunoglobulin. Symbols: 0,
MVM(p)-infectedA9ouabrll;*, MVM(p)-infected S491TB2;
*, MVM(i)-infected S49 1TB2; 0, MVM(i)-infected
A9 ouabrll.
indicating that these cells might comprise a subpopulation of cells thatare lytically infecta-bleatthisinputmultiplicity. Thus,inthe major-ity of each cell type, each virus showed a reciprocal difference in its ability to establish lytic infection.
Productive and restrictive virus-host cell
inter-actions. To explore the kinetics of these virus
infections and their longer-term consequences
for eachhost cellculture,wemonitored various parametersof both virus andcellgrowthduring infection. Cultures wereinfected at10 PFU per
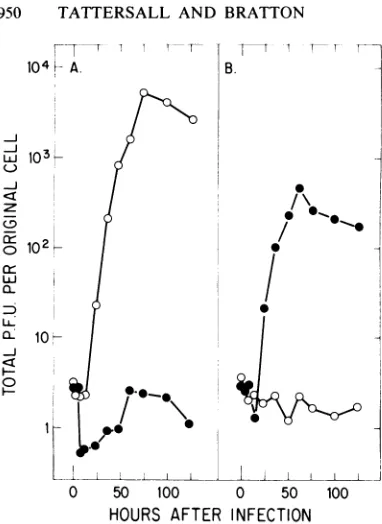
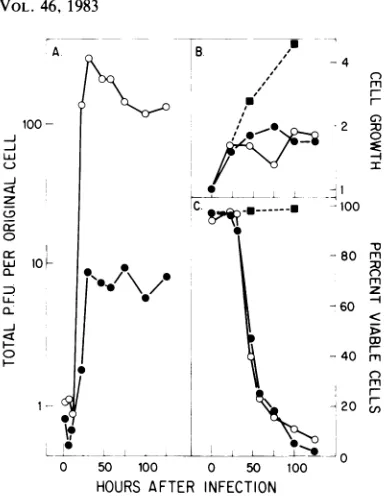
cell and followed for a time span sufficient for several cycles of virus replication, release, and reinfection. Figure 5 shows the accumulation of each virus, interms ofinfectivity, in each host
cell type under these conditions. In Fig. 5, the
total virus produced, both extracellular and cell
associated, is normalized tothenumberof cells
originally infected.The virustiterrisestoapeak value at approximately 60 h postinfection,
ex-cept in the infection of S49 with MVM(p), in
whichnoincreaseoverearly titerswasdetected. However, the peak titer for MVM(p)was 1,000-fold higherthan that forMVM(i)ininfected A9 cultures.Reciprocally,thepeaktiter reached
by
MVM(i) in S49 cells rose more than 200 times higher than thelevelofMVM(p) observed in a
parallel infectionof these cells.
When we examined the parameters of cell growthandviability,asmeasuredby dye exclu-sion with trypan blue, an even more dramatic difference was found between the two types of infection. The infections of A9 with MVM(p) and of S49 with MVM(i) resulted in a rapid cessation of cell growth paralleled by asimilarly rapid decline in cell viability, both startingabout 40 h postinfection, at atime when exponential virus production was underway in these cells (Fig. 6). In this type of virus-host cell interac-tion, which we term a productive infection, the great majority of the cells in the culture are ultimately involved in virus productionfollowed by cell death. In such host cell-virus combina-tions, a lowered initial multiplicity ofinfection merelyincreases the number of cyclesof infec-tion and reinfection that occur before the maxi-mum yield of virus is achieved, which again results in the death ofessentially every cell in the culture. On the other hand, the reciprocal infections of A9 with MVM(i) and S49 with MVM(p) do not appear to affect the rate of growth of the cells or their viability. Virus production and cell death appear toberestricted to a small [or, in the case of S49cells infected withMVM(p),undetectable]subset of the whole population which, in the case ofMVM(i) infect-ed A9 cultures, appears to behave as if it was composedofnormalproductivehost cells. How-ever, themajority of the culture remains refrac-tory to the infection and continues to divide without apparently being affected. We have termed this type ofvirus-host interaction a re-strictive infection, to indicate its self-limiting nature.
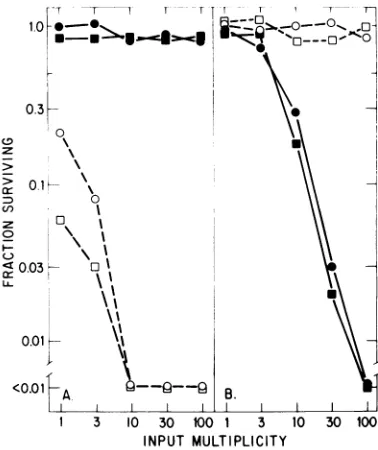
Effect ofmultiplicityofinfectiononproductive and restrictive interactions. We have used the assay for cell killing to investigate further the restrictive and productive interactions of both viruses with other cell lines. In these experi-ments, rapidlygrowing cells were infected at a variety of inputmultiplicities,and the numberof trypanblue-excludingcells wasdeterminedafter 5 or6days of growth andcomparedwiththatin control, uninfected cultures. Figure 7 summa-rizes the results of suchassays withA9-8E,RPC 5.4, C127, and EL4-sti in addition to the A9 ouabrll and S49 1TB2 cell lines. The long-term consequences of these infections reflect the con-clusions drawn above. Restrictive infections ini-tiated at high multiplicities do not show very significantcell
death,
evenwhen examined after _//
_
,~~~/
o0
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.488.76.224.54.286.2]FIG. 4. MVMcapsid antigenexpression. (A) Average field ofA9ouabrll cells infectedat10PFU percell
with MVM(p) as described in the legend to Fig. 3. (B) Selectedfield fromaparallel A9ouabrllcultureinfectedat
10PFU percell withMVM(i), showingarelatively rareMVMcapsidantigen-expressingcell.
timeforseveralcyclesofgrowth has elapsed. In thecase ofMVM(i) infection ofA9ouabrll at
100PFU percell, where almost5% ofthecells
are viral antigen positive at 26 h (Fig. 3), the long-term effect on the culture appears to be littlemorethan theremoval of this smallcohort of cells from thepopulation.However, the infec-tion ofS49 1TB2 cultures with this virus at 3 PFU percell, whileinducing approximately the same fraction of infected cellsinthe firstcycle of virusgrowth, ledtoalmost90% destruction of theculture by 5days. It should be emphasized here that these results represent a single time point inanumberof growthcurves,and that this was chosen as being logistically optimal for examining the viral life cycle. Further
subcul-ture of infected restrictive host cultures does not, ingeneral, show anychange in the viability
orgrowthrateof theculture,exceptininstances where apersistent infection is established,
lead-inginsome cases to theevolution of host-range
mutants (D. Ron and J.Tal, personal
communi-cation; P. Tattersall, E. M. Gardiner, and J. Bratton, unpublished results). On the other hand, further subculture of infected productive host cells leads ultimately to the destruction of essentially every cell in the population, even
when starting multiplicities of less than 0.001 PFU per cell are used. In all ofthe fibroblast lines we have examined to date, infection with MVM(p)at any
multiplicity
results in theeven-tual death ofmorethan 99.9% of the cells and reveals a preexisting subpopulation ofcells
re-sistant to extremely high multiplicities of the virus (unpublished data).
The fibroblast line C127 and the T-cell lym-phoma EL4-sti show productive and restrictive interactions withthetwovirusstrains, similarto
those shown by A9 ouabrll and S49 1TB2, respectively (Fig.7). Interestingly,the A9 deriv-ative A9-8E, which had been selected for the absence of the
MVM(p)-specific
cell surface receptor(14), and theB-cell myeloma line RPC5.4 were completely resistant to both virus
strains, at all multiplicities. As we have shown elsewhere,the resistance of theseparticularcell lines is duetolackofreceptorsfor both viruses
(31).
Stabilityof the viralphenotype. The availabil-ityofa hostcell such as324K, which supports the replication of both MVM(p) and MVM(i), has allowedus to testthestabilityof the strain-specific phenotype ofeachvirusduring subclon-ing and growth in the absence of selection.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.488.45.441.74.351.2]BRATTON 104k A.
/
o
LLj 103 IS
I
-j
(3
z-J
z
o
12
0cr
D.
LLJ
JI
rLlo
!
0
F--l.
l'
T--T I-T1T r
B.
S
/ .
I~~~~
0 50 100 0 50 100
HOURS AFTER INFECTION
FIG. 5. Kinetics of virus production during
infec-tion. Replicatecultures of(A)A9ouabrl1 and(B)S49
1TB2 cellsat2 x 105cellsperml wereinfectedat10
PFUpercellwithglycerol gradient-purified MVM(p)
(0) or MVM(i) (0) virions. At various times after
infection, samples were withdrawn and assayed for
extracellular virus and cell-associated virus by the
324K cell infectivity assay as described in the text.
The resultsareshownasthesumof thesetwo
determi-nations, normalized to the number of cellsoriginally
infected. Cultures weremaintained between 2 x 105
and 8x 105cellsperml for A9ouabrl1 and between 2
x 105 and 16 x 105 cells per ml for S49 1TB2 by
dilution,wherenecessary,with fresh medium.
Single, well-isolated plaques of each virus strain were picked from 324K monolayers andgrown upthroughtwopassagesin 324K cultures. Titers
of these stocks were then determined with the 324Kplaqueassay,andthe cell killing ability of
thestockswasmeasured,as afunction of multi-plicity, by the phenotypic assay described above. The results obtained with two indepen-dent subclones of each of the two virus strains are shown in Fig. 8. Virus stocks grown from single MVM(p) plaques maintain the MVM(p) phenotype of being abletokill A9, but notS49, cells, and MVM(i) derived subclones maintain theiroriginal, reciprocal phenotypeeventhough they have been cloned and grown in cells that wouldsupportviruses of either phenotype. This
suggests that the MVMgenome carriesa stable genetic determinantspecifying the differentiated celltypein which the viruscangrow.The stocks growntohigh titer by multiplepassagesin 324K
cellsappear to be slowerthan theparent stocks in their kinetics ofcell killing. This might be accounted for by the recent finding (M. Merch-linsky, personal communication)that high-mul-tiplicitypassageofMVM in 324K cells leads to the rapid accumulation of defective genomes such as those described byFaustand Ward (7). Two otherindependent subclonesof each virus strainhave beensimilarlytested and werefound togive similarresults(datanot shown).
Restrictive host phenotype is recessive in somat-ic cell hybrids. To examine the nature of the block to virus replication in the majority of individuals in a restrictive host cell population, we constructed a numberof A9 cellx T lympho-mahybrids and tested themfor susceptibilityto both viruses. Thegrowth cycle ofMVM(p)and MVM(i) in hyb2/40,anA9ouabrll x S491TB2 hybrid, initially infected at 10 PFU per cell is showninFig.9.Bothvirusesgrow inthis hybrid line, reachingpeaktiters somewhatearlierthan the infections described in Fig. 5 and 6. The peaktiters per original cell of both viruses are some20-fold lowerhere than in the correspond-ing productive infection ofeachof the parents of this hybrid cell line. The productive nature of theinteraction of this cell line with both MVM strains is most convincingly demonstrated by the effect of each virus on cell growth and viability. (Fig. 9B and C). The more rapid kinet-icsof virus production, cessation ofcellgrowth, and cell killing here compared with those ob-served forparental cell infections suggests that the hybrid line is more sensitive to infection at 10 PFUpercellthan eitherof theparents. There is lessthan a2-fold increasein cell numberafter infection of the hybrid (Fig. 9B), whereas A9 infected with MVM(p) and S49 infected with MVM(i) showa5- and10-fold increase, respec-tively, in cell number before growth ceases. When the differences in cell growth after infec-tion are taken into account, the numbers of infectious virions produced perinfected cell at the time ofpeak titer are comparable between two sets of infections. Taken with the growth inhibition and cellkilling elicited by both viruses in hyb 2/40, we conclude that the block to infection exhibited by themajorityof cells in a
restrictive population behaves as a recessive trait in somatic cell hybrids.
It has notbeenpossible as yet to demonstrate plaque formation in monolayers of hyb 2/40, probably because this cell line does not grow well under agar. However, we have examined plaqueformationbyboth viruses in a somewhat more robust hybrid, hyb 1/11, constructed by fusingA9ouabrll toEL4-sti.Thecomparison of twoindependentlygrown stocksofMVM(p) and MVM(i) for plaquing efficiency on 324K, A9 ouabrll, and hyb 1/11 monolayers is
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.488.55.246.58.320.2]HOST RANGE VARIANTS OF MVM 951
50
3 0 cr 10
-J
(-) LL
5-100*
w 80
L,J
m 60
H 40 -z LL x 20 CL
C.
0OL
0
D.
*-50 100 0 50 100
HOURS AFTER INFECTION
1000
C-)
m
r-G:
C~)
I 0
i
-v
m
z
D
m
m
Cl)
FIG. 6. Cellgrowth and viabilityduringinfection. Cellgrowthduringtheinfections of (A)A9ouabrl1 and (B)
S491TB2cultureswith 10PFUofMVM(p)(0)orMVM(i)(0)per cellasdescribed inthelegendtoFig.5.Cell
growthisexpressedasthe totalcell numberateachtimepointdividedby theoriginal number of cells present at
thestartof infection. Cellviabilityduringthesameinfections of(C)A9ouabrll and(D) S491TB2is expressed as
the percentage of cells inthe culture which exclude trypan blue.
rized in Table 2. This shows that MVM(p) plaques with approximately equal efficiency on
both A9 ouabrll and hyb 1/11 monolayers, al-thoughwith about1/20 ofthe titerdemonstrable
on 324K monolayers.
MVM(i),
on the other hand, plaques on 324K at least107-fold
moreefficiently than on A9 ouabrll; indeed, no
plaques were detected in either ofthese assays
onA9ouabrll monolayersatthelowestMVM(i) dilutions tested. However, MVM(i) does form plaques on hyb 1/11 monolayers at about 1/100
ofits
efficiency
on324K cells. Given the5- to10-fold difference in efficiency between MVM(p) and MVM(i) plaque formation per virion on
324K monolayers mentioned above(Fig. 1),this shows that, per virion, MVM(p) and MVM(i) plaque at thesame efficiency on hyb 1/11 cells. This confirms the conclusion drawn from the growth of both viruses shown in Fig. 9, that susceptibility to each virus strain is expressed codominantly in somatic cell hybrids.
DISCUSSION
Inanattempt toestablishanin vitrocorrelate for the strain-specific tissue tropisms exhibited by members of the autonomous parvovirus group(4-6, 11,12, 15, 18-20)wehave examined the virus-host interactions of two variants of MVM which infect cells of dissimilar differenti-ated phenotypes. The prototype strain, MVM(p), was
initially
isolated in whole mouseembryo fibroblast cultures and subsequently cloned by plaque
purification
in A9 cells (33). This virus has been the object of considerable study over the past 10 years, and many of its basic biochemical properties have been well characterized (38). The immunosuppressive strain, MVM(i), wits first demonstrated as acontaminant of invivo-passagedEL4T-cell lym-phoma cells (4), and wereporthere its isolation and cloning by limitdilution in EL4 cultures in vitro (Table 1). This virus grows well in a
VOL.46,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.488.122.361.69.367.2]1.0
z
>
0.3-=D
cn
z £
0.1-s
0
0.03-
\
<0.03 A.
1 3 10 30 lOO 1 3 10 30 100 INPUTMULTIPLICITY
FIG. 7. Cellkillingasfunction ofinputmultiplicity.
Replicate cultures ofA9ouabrll(0),A9-8E(V), C127
(O), RPC 5.4(V),S491TB2(0),and EL4-sti(A)at104
per ml were infected by the addition of an equal
volume ofvirus togive variousmultiplicitiesof
infec-tion. (A)Cells were infected with dilutions ofa
high-titer clearedlysateof MVM(p) grown in A9 ouabrll.
(B) Cells were infected witha similarpreparation of
MVM(i) growninS491TB2.All cultureswerediluted
on day 2 or 3 sufficiently to keep the uninfected
controls in exponential growth, and the number of
trypan blue-excluding cells was determinedon day5
for RPC 5.4 and S49 1TB2 and on day 6 for the
remainder. The surviving fraction is that number
de-termined for each infected culture divided by that
determined for uninfected control cultures. Allpoints
represent the averages ofduplicate determinations.
number of T-cell tumor lines, especially S49 1TB2, the major host cell usedin thisstudy.
The in vitro immunosuppressive activity of MVM(i) appears to be due to its ability to kill cytotoxic T lymphocyte precursors thatare
re-sponding toantigenorallogenic cellsbyentering the S-phaseasthefirst step in clonalexpansion (6). These respondingcells do not appear to be susceptible to MVM(p) infection, although it was not possible in that study to be sure that equal infectious doses of each variant were compared (6, 20). Neither virus appears to sup-press B-cell functiondirectly(6). None of the
B-cell lines we haveexamined to datecarry specif-ic cell surface receptors for MVM, offering a probable explanation for this finding (31; P. Tattersall, unpublished results). We show in an accompanying paper (31), however, that the susceptibility of T cells to MVM(i), but not MVM(p), is not mediated at the cellsurface, but
is due to a requirement for different intracellular factorsfor the growth of each virus.
The MVM(i) clone described here has been physically characterized and compared with the prototype strain by McMaster and colleagues (20), who found that the sedimentation coeffi-cients, buoyant densities, and structural poly-peptidesofMVM(p) and MVM(i) virionsappear
identical. The genomesofthe two variants have been compared by a number of biochemical techniques, including length measurement on alkalinegels, restrictionmapping,and heterodu-plexformation. The genome of MVM(i) is some 60 nucleotides shorter than that ofMVM(p), and this apparent deletion has been located at ap-proximately 92 map units, close to the 5' end of viral DNA (20; P. Tattersall, unpublished re-sults). So far,well over 100restriction siteshave been mapped on both genomes, and approxi-mately80%of them arecoincident,from the left hand(3') end in both genomes (20; E. M. Gardi-nerand P. Tattersall, unpublished results).
We have shown here that both ofthese vari-antswillgrowin, and form plaques on, monolay-ers of the simian virus 40-transformed human newbornkidney fibroblast cell line324K(29). It
-r 1 r T Ir r
1.0 _0 -, O s@S0
0.3
> 01.
enC
\0 C0.03 UO
LL.
0.01
<0.01AIB
[image:9.488.54.243.59.273.2]1 3 10 30 100 1 3 10 30 100 INPUT MULTIPLICITY
FIG. 8. Stability ofvirus phenotype. Independent
subclones ofMVM(p)andMVM(i)wereisolatedand
expanded, titers were determined in 324K cells as
described inthe text, andtheirphenotypewas
deter-mined by the cellkilling assay described in the legend
toFig. 7 for(A) A9ouabrl1 and(B) S49 1TB2cells.
Symbols: 0, MVM(p) subclone 1; 0, MVM(p)
sub-clone3;*, MVM(i) subclone 2; *, MVM(i) subclone
3. I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.488.260.449.368.593.2]HOST RANGE VARIANTS OF MVM 953
-J
La
-j
0
CL
LL
0
100
-
lOF-0 50 100 0 50 100
HOURS AFTER INFECTION
FIG. 9. Virus growth in hybrid cells. Cultures
hyb 2/40, a cloned A9 ouabrll x S49 1TB2 hybr
wereinfected at 10 PFU percell with glycerolgra
ent-purifiedvirions. (A)Total PFU weredetermined
described in the legendtoFig.5. (B) Cell growtha
(C) cellviabilityweredeterminedasdescribed in I
legend toFig. 6. Symbols: 0, MVM(p) infection;
MVM(i) infection; *, growthandviabilitydetermir
foraparallelculture of mock-infectedhyb2/40 cel
is notclearwhy either variant shouldgrowat
in human cells. We have recently tested t
ability of these twomurine virus strains togr innormal human fibroblast lines and have fou thatthey both undergo abortive infection,rest ing in the expression of viral antigens, but detectable viral DNA synthesis or infectic progenyvirusproduction (S. F. Cotmore and Tattersall, unpublished results). Weare curre ly examining the role, ifany,of simian virus transformation in productive MVM infection human cells.
The 324K assay has allowed us to exami several biological parameters of infection w these two MVM variants, with the assurar thatwe arecomparing the effects of equal inf
tivity inputs. The two strains are serologica very closely related, perhaps indistinguishab with heterologous antisera. Despite their hi degree of physical, antigenic, and genomic rel edness, MVM(p) and MVM(i) are reciproca restricted in their abilitytogrowin cell lineso lymphocyte and fibroblast origin, respective We suggest the term allotropic variant to ( scribe virus strains of the same serotype whi exhibit such reciprocal interactions with h cells of the same species, but of dissimi
differentiated types. This viral phenotype is a
stable characteristic of each strain even when plaque purified and amplified in 324K cells, which are permissive foreither allotropic vari-ant. We have shown that these
phenotypes
are> also stable through plaque purification and
am-plification in hyb 1/11 cells, indicating that the
target cell specificity of each variant is not
dependent upon the phenotype of the cell in which it is grown, and therefore is unlikely to
m resemble the host-controlled restriction and
m modification systems of bacteriophages and
z their hosts (1). We take these resultsas
demon-< strating that thetwovirus variants each carrya D different genetic determinant, which wecall the r allotropic determinant, which specifies the
pro-, ductive host cell type for that virus. In support
r of this hypothesis, wehave been able toisolate,
at lowfrequency, stablemutantsof each variant which have an extended host range and can infect cells of both lymphocyte and fibroblast origin (P. Tattersall and E. M. Gardiner, unpub-lished results).
of The existence of thesetwo
allotropic
variants rid,obviously
posesthequestion
of theirorigin.
Doidi- theyexist in natureasseparatefieldstrainsoras
as complexfield strainscomprisingseveraldistinct
tnd allotropic variants? Perhaps theyarise from
pan-the tropic field strains by mutation followed by * selection in the laboratory, either bypassage in
ied differentiated tumors in the whole animal orby
ls. direct isolation in cultures of differentiated cells.
Theavailability of less stringent host cells, such
all as the 324K cell line described
here,
shouldthe allow the isolation and
study
of field strains inow the absence of such selection.
ind In this paper we have also shown that the
Ilt- cellular
component
with which theallotropic
no determinant of the virusactsbehaves inadomi-)us
IP.
nt- TABLE 2. Plaquetiters of virus stockson different 40 indicator cell lines"
Iof
ine ith ice ec-illy
le, igh
lat-Llly
ffT
ly.
de-ich
ost
ilar
PFUof indicatorcelllineper ml Virus Virus
stock strain 324K A9 hyb1/11
ouabrllhyI 11
1 MVM(p) 1 x 109 5.8 x 107 1.8 x 107
2 MVM(p) 1.5 x 108 8.5 x 106 8.3 x 106
3 MVM(i) 1.7 x 108 <250 1.3 x 106
4 MVM(i) 4.7 x 10X <25 4.5 x 106
'Two independently grown and glycerol gradient
purified stocks of each of MVM(p) and MVM(i),
produced in 324K cells, were assayed for PFU on
324K, A9ouabrll,andhyb1/11 (A9 ouabrll x
EL4-sti) cell monolayers as described in the text, except
that the hyb 1/11 assays were stained on day 10.
IncreasingthelengthofA9ouabrl1 assay incubations
does not lead to anincrease in the observed titer of
MVM(p)stocks or to theappearance of plaques due to
MVM(i).
VOL.46, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:10.488.45.238.59.307.2] [image:10.488.250.445.512.589.2]954 TATTERSALL AND BRATTON
nant fashion in L cell x Tcell hybrids. Indeed,
hybrids between A9 and EL4 such as hyb 1/11
give plaques with approximately equal
parti-cle/infectivity ratios for both viruses, whereas
MVM(p) is over 105-fold more efficient than
MVM(i) in producing plaques on monolayers of
the A9 parent. A simple interpretation of these
results is that thesetwodissimilar differentiated celltypesexpressdifferent developmentally
reg-ulated helper functions which are exploited by therespective virus variant. That thishost factor
isdevelopmentallyregulated, rather thana
func-tion of our selection of cell lines derived from different mouse strains, is supported by recent
experiments with BALB/c 3T3 fibroblasts.
These cellsare syngeneic with the S491TB2
T-cell line used here andarerestrictivefor MVM(i)
butproductiveforMVM(p). BALB/c 3T3 cells,
although restrictive for MVM(i), are, however,
somewhatmoresusceptibletoMVM(i) killingat
high multiplicities of infectionthan the fibroblast
cell lines examinedhere, although they maintain
a 10-to 100-fold greater sensitivityto MVM(p)
compared with MVM(i) over a broad range of
multiplicities (P. Tattersall and J. Bratton,
un-published results). This implies that there may
be germ line-transmitted differences between
mouse strains in susceptibility, at the cellular
level, to MVMper se.
It is of considerable importancetothe
under-standing ofparvovirus tissue tropism to
deter-mine the biochemicalnature of the
developmen-tally regulated host cell factororfactors that are
involvedin MVM replicationand thestepsin the
virus growth cycle at which they act. In an
accompanying paper (31) we demonstrate that, unlikethe majority of examplesof specific tissue
tropism reported for members of other virus
groups, the restriction ofMVM replication
de-scribed here does notoccuratthe level of viral
cell-surface receptors, but is mediated by
intra-cellular host factors.
Threefactors, therefore, appear to act at the
cellular leveltoconfercompetenceas ahostfor
MVM. First, the cell must be of the correct
species; second, the cell must be traversing the
cell cycle; and third,the cellmusthavea
partic-ular differentiated phenotype. The last factor
appears to be variable, and in this case the outcome of the interaction depends upon a
ge-netic locus within the viral chromosome for
which there exist, at present, two alleles.
ACKNOWLEDGMENTS
Ric Armentrout,Peter Beard, PeterHowley, SolonRhode, andNancy Ruddlekindlyprovided cell lines. We thank Susan Cotmore,Bernhard Hirt, Solon Rhode, and Barbara Spalholz forvaluable discussions.
Thisworkwassupported byaSwebilius Cancer Award and
by Public Health Service grantCA29303 from the National Cancer Institute.
LITERATURE CITED
1. Arber, W., and S. Linn. 1968. DNA modification and restriction. Annu.Rev. Biochem. 38:467-500.
2. Baer, P. N.,G.E. Garrington, and L.Kilham. 1971. Effect of age and H-1 virus on healing fractures in hamsters. J. Gerontol. 26:373-377.
3. Baxter, J. D., A. W. Harris, G.M. Tomkins, and M. Cohn. 1971.Glucocorticoid receptors inlymphoma cells inculture: relationshipto glucocorticoid killing activity. Science 171:189-191.
4. Bonnard, G. D., E. K. Manders, D. A. Campbell, Jr., R. B. Herberman,and M. J. Collins, Jr. 1976. Immuno-suppressive activity of a subline of the mouse EL-4 lymphoma. Evidenceforminutevirus of mice causing the inhibition.J. Exp. Med. 143:187-205.
5. Campbell, D. A., Jr., S. P. Staal, E. K. Manders, G. D. Bonnard, R. K. Oldham, L. A. Salzman, and R. B. Her-berman. 1977. Inhibition of in vitro lymphoproliferative responsesbyinivivopassagedrat 13762 mammary adeno-carcinoma cells. II. Evidence that Kilham rat virus is responsible for the inhibitory effect. Cell. Immunol. 33:378-391.
6. Engers, H. D., J. A. Louis,R. H. Zubler, and B. Hirt. 1981. Inhibitionof Tcell-mediated functionsby MVM(i), aparvovirus closely relatedto minute virus of mice. J. Immunol. 127:2280-2285.
7. Faust,E. A.,andD. C.Ward. 1979.Incomplete genomes of theparvovirusminutevirusofmice: selective conser-vation ofgenome termini, including the origin of DNA replication.J. Virol. 32:276-292.
8. Gorer,P. A.1950. Studiesin antibody response of mice to tumorinoculation.Br. J.Cancer4:372-379.
9. Harlow, E., L. V. Crawford, D. C. Pim, and N. M. Williamson. 1981. Monoclonal antibodies specificfor simi-anvirus40tumor antigens.J.Virol.39:861-869. 10. Kahn,D. E.1978.Pathogenesisof felinepanleukopenia.J.
Am. Vet.Med. Assoc. 173:628-630.
11. Kilham,L.,andG. Margolis. 1966. Spontaneous hepatitis andcerebellarhypoplasiainsucklingrats due to congeni-talinfectionswith ratvirus. Am. J.Pathol.49:457-475. 12. Kilham, L., andG. Margolis. 1975. Problems ofhuman
concern arising from animal models ofintrauterine and neonatal infectionsdue to viruses:areview. I. Introduc-tionandvirologic studies. Prog. Med. Virol.20:113-143. 13. Kilham, L., G. Margolis, and E. D. Colby. 1970.
En-hancedproliferation ofH-1 virus inliversofrats infected withCvstericusfasciolaris. J.Infect.Dis. 121:648-652. 14. Linser, P., H. Bruning, and R. W. Armentrout. 1977.
Specific binding sites for a parvovirus, minute virus of mice,oncultured mouse cells.J. Virol. 24:211-221. 15. Lipton,H. L., and R. T.Johnson. 1972. Thepathogenesis
of Rat Virus infections in the newborn hamster. Lab. Invest. 27:508-513.
16. Littlefield, J. W. 1964. Three degrees ofguanylic acid-inosinic acid pyrophosphorylase deficiency in mouse fi-broblasts. Nature (London)203:1142-1144.
17. Lowy,D. R., E.Rands,and E. M.Scolnick. 1978. Helper-independent transformationbyunintegratedHarvey sar-comavirus DNA.J.Virol.26:291-298.
18. Lum, G. S. 1970. Serological studies of rat viruses in relationtotumors. Oncology24:335-343.
19. Margolis,G., and L. Kilham. 1975. Problemsof human concern arising from animal models ofintrauterine and neonatal infectionsdue toviruses:areview.II. Pathologic studies. Prog. Med. Virol.20:144-179.
20. McMaster,G. K., P. Beard, H. D.Engers, andB. Hirt. 1981.Characterizationof animmunosuppressive parvovi-rus relatedto theminute virusofmice. J. Virol. 38:317-326.
21. Meunier, P. C., L. T.Glickman,M. J. Appel, and S. J. Shih. 1981. Canine parvovirus in a commercial kennel: epidemiologic and pathologic findings. Cornell Vet. 71:96-110.
22. Miller, R. A., D. C. Ward, and F. H. Ruddle. 1977. Embryonal carcinoma cells (and their somatic cell
on November 10, 2019 by guest
http://jvi.asm.org/
brids) areresistant to infection by the murineparvovirus MVM, which does infect otherteratocarcinoma-derived celllines. J. Cell.Physiol. 91:393-402.
23. Mohanty, S. B., and P. A. Bachmann. 1974. Susceptibility of fertilized mouse eggs to minute virus of mice. Infect. Immun. 9:762-763.
24. Mohit, B., and K. Fan. 1971. Hybrid cell line from a clonedimmunoglobulin-producing mouse myeloma and a nonproducing mouselymphoma. Science 171:75-77. 25. O'Maliey, K. A., and R. L. Davidson. 1977. A new
dimensioninsuspension fusion techniques with polyethyl-ene glycol.Somat. Cell Genet. 3:441-448.
26. Porter, D. D.,and H. J. Cho. 1980. Aleutian disease of mink: a modelfor persistent infection, p. 233-256. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology, vol 16. Plenum Publishing Corp., New York. 27. Rhode,S.L.,III. 1973.Replication process of the
parvo-virus H-1.I.Kinetics in a parasynchronous cell system. J. Virol.11:856-861.
28. Ruffolo, P. R., G. Margolis, and L. Kilham. 1966. The induction of hepatitis by prior partial hepatectomy in resistant adult rats injected with H-1 virus. Am. J. Pathol. 49:795-824.
29. Shein, H., and J. F. Enders. 1962. Multiplication and cytopathogenicity of Simian vacuolatingvirus 40 in cul-tures of human tissues. Proc. Soc. Exp. Biol. Med. 109:495-500.
30. Siegl, G., and M. Gautschi. 1973. The multiplication of parvovirus LuIII in a synchronized culture system. I. Optimum conditions for virus replication. Arch. Gesamte
Virusforsch. 40:105-118.
31. Spalholz, B. A., and P. Tattersall. 1983. Interaction of minute virus of mice with differentiated cells: strain-dependent target cellspecificity is mediated by intracellu-lar factors. J. Virol. 46:937-943.
32. Szybalski, W., E. H. Szybalska, and G. Ragni. 1962. Genetic studieswithhumancell lines. Natl. CancerInst. Monogr. 7:75-88.
33. Tattersall, P. 1972. Replication oftheparvovirusMVM.I. Dependence of virusmultiplication and plaque formation oncellgrowth. J. Virol.10:586-590.
34. Tattersall,P.1978.Susceptibilitytominutevirus of mice as afunction of host-cell differentiation, p. 131-149. In D.C. Ward and P. Tattersall (ed.), Replication of mam-malian parvoviruses. Cold Spring Harbor Laboratory, ColdSpring Harbor,N.Y.
35. Tattersall, P., P. J. Cawte, A. J. Shatkin, and D. C. Ward. 1976. Three structuralpolypeptides coded for by minute virus of mice, a parvovirus. J. Virol. 20:273-289. 36. Tattersall, P., and D. C. Ward. 1978. Theparvoviruses: an
introduction, p. 3-12. In D.C. Ward and P. Tattersall (ed.), Replication of mammalian parvoviruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. 37. Tennant,R.W.,K. R.Layman, and R. E. Hand, Jr. 1969.
Effect of cellphysiological state on infection byratvirus. J. Virol.4:872-878.
38. Ward,D.C.,and P.J.Tattersall.1982. Minute virus of mice, p. 313-334.InH. L.Foster,J. D.Small, and J. G. Fox (ed.), The mouse in biomedical research, vol 2. Academic Press, Inc.,NewYork.
VOL.46,1983