Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Alternative Splicing of the Latency-Related Transcript of
Bovine Herpesvirus 1 Yields RNAs Containing
Unique Open Reading Frames
LAXMINARAYANA R. DEVIREDDY
ANDCLINTON JONES*
Center for Biotechnology, Department of Veterinary and Biomedical Sciences,
University of Nebraska—Lincoln, Lincoln, Nebraska 68583-0905
Received 2 April 1998/Accepted 27 May 1998
The latency-related transcript (LRT) of bovine herpesvirus 1 (BHV-1) is the only abundant viral RNA
detected during latency. A previous study (A. Hossain, L. M. Schang, and C. Jones, J. Virol. 69:5345–5352,
1995) concluded that splicing of polyadenylated [poly(A)
1] and splicing of nonpolyadenylated [poly(A)
2] LRT
are different. In this study, splice junction sites of LRT were identified. In trigeminal ganglia of acutely infected
calves (1, 7, or 15 days postinfection [p.i.]) or in latently infected calves (60 days p.i.), alternative splicing of
poly(A)
1LRT occurred. Productive viral gene expression in trigeminal ganglia is readily detected from 2 to 7
days p.i. but not at 15 days p.i. (L. M. Schang and C. Jones, J. Virol. 71:6786–6795, 1997), suggesting that
certain aspects of a lytic infection occur in neurons and that these factors influence LRT splicing. Splicing of
poly(A)
2LRT was also detected in transfected COS-7 cells or infected MDBK cells. DNA sequence analysis of
spliced LRT cDNAs, poly(A)
1or poly(A)
2, revealed nonconsensus splice signals at exon/intron and intron/
exon boundaries. The GC-AG splicing signal utilized by the herpes simplex virus type 1 latency-associated
transcript in latently infected mice is also used by LRT in latently infected calves. Taken together, these results
led us to hypothesize that (i) poly(A)
1LRT is spliced in trigeminal ganglia by neuron-specific factors, (ii) viral
or virus-induced factors participate in splicing, and (iii) alternative splicing of LRT may result in protein
isoforms which have novel biological properties.
All members of the alphaherpesvirus subfamily establish and
maintain a latent infection in the peripheral nervous system of
their natural hosts. Bovine herpesvirus 1 (BHV-1), a member
of the alphaherpesvirus subfamily, is an important pathogen of
cattle and establishes latent infection in sensory ganglia of
infected cattle (reviewed in references 57 and 58). Since
neu-rons are terminally differentiated cells, it may not be necessary
for the virus to replicate in these cells to maintain latency.
Viral gene expression in latently infected neurons is restricted
to the latency-related transcript (LRT). By using in situ
hy-bridization, LRT was detected in trigeminal ganglia (TG) of
BHV-1-infected rabbits (55, 56) or cattle (41). These studies
mapped the approximate 59
and 39
ends of LRT and estimated
its length to be 1.15 kb. LRT is also expressed during the late
stages of productively infected bovine cells (56). A 41-kDa
protein is encoded by the LR (latency-related) gene in
tran-siently transfected cells or infected bovine cells (35). LR gene
products inhibit entry of cells into S phase, suggesting that the
LR gene regulates some aspect of latency (65).
The latency-associated transcript (LAT) of herpes simplex
virus type 1 (HSV-1) has been the subject of intense scrutiny
(reviewed in references 4, 9 24, 34, and 80). It is not known if
HSV-1 LAT encodes a protein even though LAT is associated
with polysomes (28). LAT is a stable 2.0-kb intron (22, 40, 59,
83), and the 1.5- or 1.45-kb transcript is derived from the 2.0-kb
LAT by further splicing (71). The splicing event that generates
the 1.5-kb LAT utilizes a novel splice donor that is GC instead
of GT (71, 74), and this splicing event requires neuron-specific
splicing factors (44). Polyadenylation of the spliced 1.5-kb LAT
is controversial (18, 50, 52, 70, 79). Disruption of splice donor
or acceptor sites prevents synthesis of the 2-kb LAT in
pro-ductively infected nonneuronal cells but not in latently infected
neurons (3).
Although cis-acting sequences that regulate neuron-specific
transcription of the BHV-1 LR gene have been studied (10, 11,
17, 37), processing of LRT has not been well characterized. A
previous study concluded that LRT is spliced, but splice
junc-tions were not identified (35). In this study, LRT splicing
pat-terns in TG of infected calves were compared to those of
productively infected bovine cells or COS-7 cells transfected
with a plasmid that expresses LR gene products. We have
identified three alternatively spliced poly(A)
1LRT isoforms at
7, 15, or 60 days postinfection (p.i.). A spliced poly(A)
2LRT
was detected at 1 day p.i., suggesting LRT is expressed early in
TG. LRT was spliced in the poly(A)
2RNA fraction after
bovine cells were infected or after COS-7 cells were
trans-fected with a plasmid containing the LR gene. It is
hypothe-sized that poly(A)
1LRT is alternatively spliced in TG and that
these spliced variants have the potential to encode novel
pro-teins.
MATERIALS AND METHODS
Virus, plasmids, and cells.MDBK (Madin-Darby bovine kidney) cells or
COS-7 cells (American Type Culture Collection, Rockville, Md.) were grown in Earle’s modified Eagle’s medium supplemented with 10% fetal calf serum. The Cooper strain of BHV-1 was obtained from the National Veterinary Services Laboratory, Animal and Plant Health Inspection Services (Ames, Iowa). MDBK cells were infected with 5 PFU of BHV-1 per cell, and RNA was extracted 24 h p.i.
Plasmid pcDNA1/LRT was constructed by inserting a 2-kb HindIII-SalI frag-ment which contains the LR gene (35) (Fig. 1) into the mammalian expression vector pcDNA1/Amp (Invitrogen). A SalI site was inserted into the unique XbaI site of pcDNA1/Amp prior to insertion of the LR gene. COS-7 cells were
* Corresponding author. Mailing address: Center for Biotechnology,
Dept. of Veterinary and Biomedical Sciences, University of
Nebras-ka—Lincoln, Fair St. at East Campus Loop, Lincoln, NE 68583-0905.
Phone: (402) 472-1890. Fax: (402) 472-9690. E-mail: [email protected]
.edu.
7294
on November 9, 2019 by guest
http://jvi.asm.org/
transfected with pcDNA1/LRT by calcium phosphate precipitation (19), and total RNA was prepared 48 h after transfection.
Infection of cattle and preparation of tissue samples.BHV-1-free calves were
divided into six groups of two each and then infected with 108.8 50% tissue culture infective doses intranasally and intraocularly as described previously (66). Two animals were used as controls. TG were collected 1, 2, 4, 7, 15, or 60 days p.i. (two calves per time point) or from two uninfected calves. TG were frozen in an ethanol-dry ice bath and stored at2120°C (66). Some of the RNA samples from the cattle described by Schang and Jones (66) were used for these studies.
Preparation of RNA.Total RNA was extracted from TG as described
previ-ously (35). Total RNA from infected or transfected cells was extracted by using the RNAgents Total RNA Isolation system (catalog no. Z5110; Promega) ac-cording to the manufacturer’s instructions. RNA concentrations were measured in a spectrophotometer at 260 nm.
Preparation of poly(A)1and poly(A)2LRT RNA by oligo(dT)
chromatogra-phy.Poly(A)1RNA was prepared by using an mRNA purification kit (catalog
no. 27-9258; Pharmacia) according to manufacturer’s instructions. Total RNA was passed through an oligo(dT)-cellulose column twice, and the column was washed with a buffer supplied in the kit. Bound RNA was recovered by incubat-ing the column with 250ml of elution buffer at 65°C, and RNA was subsequently precipitated with ethanol. RNA in the flowthrough fraction of the oligo(dT) column [designated poly(A2)] was recovered and precipitated with ethanol.
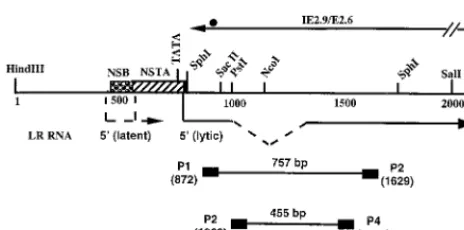
Primers for RT-PCR.Figure 1 shows the design of primers used for reverse
transcriptase (RT)-mediated PCR (RT-PCR) and location of the hybridization probe. LRT was detected by using primers P1 (same sense as LRT 59-AGGCT GGGGGTCGCAAATACACGGC-39) and P2 (antisense to LRT 59-GGCCCG CCGGAGAAGAAGGACAGAGT-39), which amplify a 757-bp fragment. With primers P3 (same sense as LRT 59-CCCCAGGAGGCTTTCTCGCACC-39) and primer P4 (antisense to LRT 59 -CACAGTGATAGACCTGACGGCGAACG-39), spliced LRT was amplified. The probe used for detection of LRT cDNA was from nucleotides (nt) 1092 to 1117 (59-GCGCACCGAAATGGAAGTGGCCG CC-39). The numbering system for the LR gene was described previously (41) and is based on the Cooper strain of BHV-1. The 39termini of primers P1, P2, P3, and P4 correspond to positions 872, 1629, 1068, and 1523, respectively. The primers were designed based on the following criteria: (i) the oligonucleotides are located adjacent to an AT-rich region, (ii) the oligonucleotides have a GC content greater than 50%, (iii) there is no significant similarity to other viral genes, and (iv) PCR product size is more than 300 bp.
RT-PCR and sequence analysis of cDNAs.The method for RT-PCR was
described previously (14). Prior to cDNA synthesis, RNA was treated with 1 U of DNase I (GIBCO-BRL) to eliminate contaminating DNA (20). Five hundred nanograms of DNase I-treated poly(A)1or 3mg of poly(A)2RNA was
dena-tured at 65°C for 7.5 min, incubated at room temperature with 1mg of oligo(dT) or random primers (Invitrogen) for 15 min, and then incubated on ice. This mixture was incubated with 200 U of Moloney murine leukemia virus RT (GIBCO-BRL) in the presence of 20 U of RNase inhibitor (Promega) according
to the manufacturer’s instructions for synthesis of cDNA. RT was inactivated by heating at 95°C for 5 min. Amplification of cDNA was conducted with 2.5 U of
Taq DNA polymerase and 100mM deoxynucleoside triphosphates in a 50-ml reaction. Forty cycles of amplification were carried out with primers P1 and P2 (200 ng of each) in the presence of 10% glycerol to improve denaturation of GC-rich DNA and to enhance the extension through secondary structures (68) on a DNA thermal cycler (Hybaid). The following conditions were used for amplification: 1 min at 94°C (denaturation), 2 min at 55°C (annealing), 2 min at 72°C (polymerization), and 7 min at 72°C to complete the extension. The PCR products were then reamplified with primers P3 and P4 (200 ng of each) under the same conditions. To avoid contamination, PCR was performed in a separate room, gloves were changed frequently, all reagents were used exclusively for these studies, and numerous other precautions were taken to avoid contamina-tion (32). Amplified products were purified either by polyacrylamide gel elec-trophoresis or by selective precipitation (62). Briefly, 0.1 volume of 103STE (1 M NaCl, 200 mM Tris-HCl [pH 7.5], 100 mM EDTA) was added to PCR products, followed by addition of equal amounts of 4 M ammonium acetate, and precipitated with 2.5 volumes of ethanol at room temperature. Purified PCR products were cloned into pCR-Script vector (Stratagene) according to the manufacturer’s instructions. Both strands of the inserts were sequenced by the dideoxynucleotide chain termination method using the Fidelity DNA sequencing system (catalog no. 57600; Oncor), which is designed for sequencing GC-rich DNA. As a positive control, BHV-1 DNA was used. Negative controls included RNA from TG of uninfected calves, infected MDBK cells, or mock-transfected COS-7 cells.
Southern blot analysis.PCR products were separated on 2% agarose gels and
transferred onto Hybond N1membrane (Amersham) by capillary transfer ac-cording to the protocol of the manufacturer. Hybridization was done acac-cording to the manufacturer’s instructions. The probe was prepared by end labeling with T4 polynucleotide kinase (New England Biolabs) and [g-32P]ATP (Amersham).
RESULTS
Amplification of LRT splice junction sites by RT-PCR.
A
previous study (35) demonstrated that splicing of LRT
oc-curred, but splice junction sites were not identified. To further
study splicing of LRT, RT-PCR was conducted because this
approach has been used successfully to identify alternative
splicing of other primary transcripts (15, 23, 54). To this end,
total RNA from TG of infected calves was used. Poly(A)
1RNA was purified by oligo(dT) chromatography. No attempts
were made to prove how efficient the purification procedure
was because unnecessary manipulation of TG RNA increases
the probability of degradation. To avoid amplification of
con-taminating viral DNA, total RNA was treated with DNase I.
Single-stranded cDNA was synthesized by using an oligo(dT)
primer, RT, and conditions which allow for optimal
amplifica-tion of LRT. The resulting cDNA was then amplified in a
nested PCR using the primers shown in Fig. 1. The rationale
for using nested PCR is that (i) primers P1 and P2 are adjacent
to the transcription start sites (11) and the poly(A) signals (41),
(ii) primers P3 and P4 flank the region which was spliced (35),
and (iii) this strategy enables detection of small amounts of
LRT. Although these primers will amplify the IE2.9/E2.6
mRNA, this region of the RNA is not spliced (82), and thus the
amplified product migrates with the same mobility as genomic
DNA (data not shown).
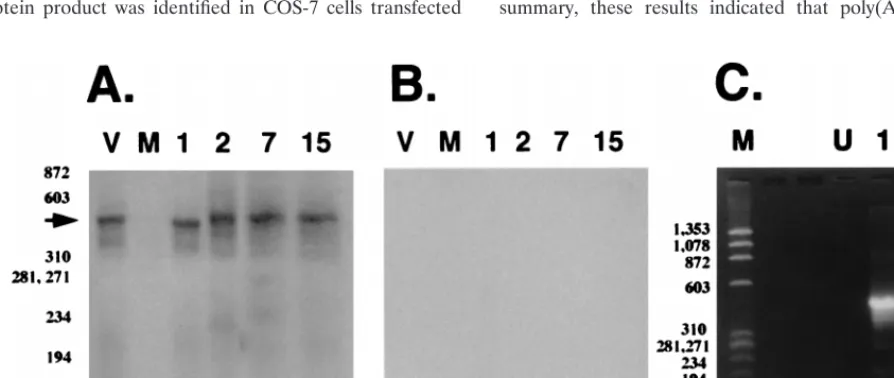
Poly(A)
1LRT was detected in bovine TG at 7, 15, or 60
days p.i. (Fig. 2A). A previous study demonstrated that
infec-tious virus was detected in ocular swabs at 2, 4, or 7 days p.i.
but not 15 or 60 days p.i. (66). Alternative splicing apparently
occurred in TG during acute infection because amplified
prod-ucts detected at 7 or 15 days p.i. were smaller than amplified
LR DNA or LRT cDNA at 60 days p.i. Amplified products
were not detected when RT was excluded from the cDNA
synthesis (Fig. 2B) or when RNA was prepared from TG of an
uninfected calf (Fig. 2A, lane U).
The flowthrough from the oligo(dT) column [poly(A)
2RNA] was subjected to cDNA synthesis using random primers
and nested PCR to detect LRT. A 455-bp PCR product was
detected by Southern blot analysis using the LRT-specific
probe described in Fig. 1 (Fig. 3A, lanes 2, 7, and 15).
Ampli-FIG. 1. Schematic of the LR promoter, locations of 59termini of LR
tran-scripts, and partial restriction enzyme map of the LR gene. The 59ends of LRT were mapped by RACE (rapid amplification of cDNA ends) PCR or primer extension (11, 35). DNA sequences within the LR promoter which are bound by neuron-specific proteins (NSB) were identified by electrophoretic mobility shift assays and exonuclease III footprinting (17). DNA sequences within the LR promoter which cis activate a minimal tk promoter in neuronal cells are desig-nated as a neuron-specific transcriptional activator (NSTA) (10). Splicing of LR RNA occurs in LRT; this is designated by the dashed lines (35). The transcripts (IE2.9/E2.6) antisense to LRT are indicated by a solid black line, and the circle at the 39end of the transcript is the position of the stop codons for the protein encoded by IE2.9/E2.6. The primers P1, P2, P3, and P4 used for amplification of LRT are indicated by small black rectangles. The number below each primer indicates the position of the 59terminus. The predicted sizes of the PCR products that can be amplified by these primers are also indicated. The small hatched rectangle indicates the probe used in Southern blot analysis to detect the PCR product. Except for the boxes depicting P1, P2, P3, P4, and the probe, the line map is drawn to scale. Plasmid pcDNA1/LRT contains the 2-kb HindIII-SalI fragment cloned into pcDNA1/Amp as described in Materials and Methods.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.53.285.68.183.2]fied products were not detected when RT was omitted from the
cDNA synthesis reaction (Fig. 3B). Poly(A)
2LRT was also
detected at 1 day p.i. (Fig. 3A, lane 1), and the PCR product
appeared to be slightly smaller than genomic viral DNA.
Fur-thermore, poly(A)
2LRT from latently infected animals (60
days p.i.) migrated as a 455-bp amplified product (Fig. 3C, lane
1), and the specificity of the amplified product was confirmed
by Southern blot hybridization (data not shown). As expected,
amplified products were not observed when RT was left out of
the cDNA synthesis reaction (Fig. 3C, lane 2). RNA from TG
of a second set of infected calves yielded similar bands (data
not shown). In summary, these results demonstrated that (i)
alternative splicing of poly(A)
1LRT occurred in TG during
acute infection, (ii) splicing of poly(A)
2LRT apparently
oc-curred at 1 day p.i., and (iii) splicing of poly(A)
2LRT in TG
at 2, 7, 15, or 60 days p.i. was not readily detected.
Analysis of LRT synthesized in transfected or infected cells.
A protein product was identified in COS-7 cells transfected
with the LR gene or after MDBK cells were infected (35, 65).
In this study, splicing of LRT was investigated after COS-7
cells were transfected with a plasmid expressing LR gene
prod-ucts or after MDBK cells were infected with BHV-1. Total
RNA was extracted 48 h after transfection or 24 h after
infec-tion. Following DNase I treatment, poly(A)
1RNA was used as
a template to synthesize single-stranded cDNA with an
oli-go(dT) primer, and LRT cDNA was amplified. Although
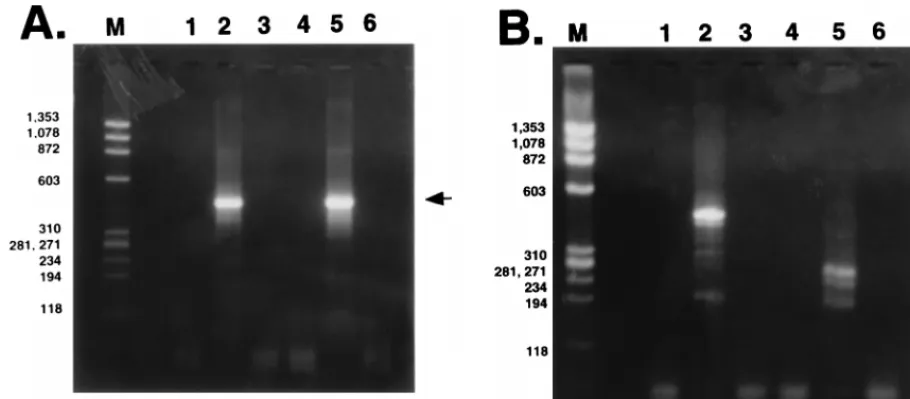
poly(A)
1LRT was detected in transfected or infected cells,
spliced poly(A)
1LRT was not detected, as judged by the size
of the PCR product (Fig. 4A, lane 2 or 5). A previous study
detected small amounts of spliced poly(A)
1LRT in infected
MDBK cells (35). Although the results in Fig. 4A appear to be
at odds with that conclusion, the RNA used for this study was
purified by oligo(dT) chromatography. Thus, we hypothesize
that either the spliced poly(A)
1LRT was degraded during
purification, the poly(A) tail was too short to be stably bound
on an oligo(dT) column, or the use of a strand-specific primer
in the previous study (35) allowed detection of small amounts
of spliced poly(A)
1LRT in infected MDBK cells.
[image:3.612.65.277.69.166.2]When cDNA synthesis of poly(A)
2RNA was primed with a
random primer and LRT cDNA was amplified by nested PCR,
bands smaller than amplified BHV-1 DNA (455 bp) were
de-tected (Fig. 4B, lanes 2 and 5). poly(A)
2RNA which was
prepared from productively infected cells yielded amplified
products migrating as 280-, 240-, or 200-bp fragments (Fig. 4B,
lane 5). In contrast, poly(A)
2RNA in transiently transfected
COS-7 cells contained bands migrating as 455-, 300-, or 200-bp
fragments (Fig. 4B, lane 2). All of the amplified products
hybridized to the LR-specific probe described in Fig. 1 (data
not shown). No PCR products were observed when RT was left
out of the cDNA synthesis reaction (Fig. 4A and B, lanes 3 and
6). Unspliced poly(A)
2LRT was also detected in transfected
cells because a 455-bp band was amplified (Fig. 4B, lane 2).
Plasmid pcDNA1/LRT does not contain the IE2.9/E2.6 gene,
demonstrating that LRT can be spliced in the absence of any
other known viral gene and a subset of LRT was not spliced. In
summary, these results indicated that poly(A)
2LRT was
FIG. 2. Detection of splicing in poly(A)1LR RNA in TG of infected cattle.
The cDNA reaction containing RT was subsequently amplified by using the LRT-specific primers in the nested PCR (A). Lane V, BHV-1 genomic DNA used as an unspliced target; lane U, RNA from TG of an uninfected calf. Numbers above the lanes indicate the days p.i. at which RNA was prepared from TG.f174 DNA which was digested with HaeIII was used as a molecular weight marker, and the positions of the bands are listed as base pairs. RNA samples were incubated in the standard RT reaction, but RT was omitted and then the nested PCR was performed (B). The lanes are labeled as in panel A. PCR products were detected by Southern blot analysis as described in Materials and Methods.
FIG. 3. Analysis of poly(A)2LR RNA. (A) Southern blot analysis of PCR-amplified products derived from the reaction performed with RT. BHV-1 genomic DNA
was used as a positive control (lane V). Lane M, RNA from an uninfected calf. Numbers above the lanes indicate RNA prepared from TG which were obtained at different days p.i. The arrow indicates the position of LRT. (B) Southern blot analysis of PCR products derived from the reaction performed without RT. Water was used as a negative control (lane V). The remainder of the lanes are labeled as described for panel A.f174 DNA digested with HaeIII was used as a marker, and the positions of the bands are designated in base pairs. (C) Amplification of poly(A)2LR RNA from latently infected calves (60 days p.i.). Lane M,f174 DNA digested
with HaeIII; lane U, RNA prepared from TG of an uninfected calf; lane 1, reaction with RT and LRT cDNA subsequently amplified; lane 2, reaction without RT followed by PCR to amplify LRT cDNA.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.77.524.477.666.2]spliced in MDBK cells after infection or when COS-7 cells
were transfected with pcDNA1/LRT.
Sequencing of cloned fragments spanning LRT splice sites.
Although it was possible to sequence the PCR products
di-rectly, they were cloned into the pCR-Script vector prior to
DNA sequencing. This approach was used in an attempt to
identify minor splice site variants and to enhance selection of
full-length products. Purified PCR products shown in Fig. 2A,
3A and C, and 4B were cloned into pCR-Script vector. A
fragment migrating as a 200-bp fragment was the only band
less than 455 bp which was able to be cloned from transfected
COS-7 cells or infected MDBK cells (Fig. 4B, lanes 2 and 5).
Prior to DNA sequencing, plasmids were analyzed by
restric-tion enzyme digesrestric-tion to verify that the inserts were similar in
size to the amplified products. Less than 2% of the plasmids
had different-size inserts, suggesting that most of the PCR
products were not deleted during cloning. DNA sequence of
these variants did not match the published LRT gene
se-quence, indicating they were rearranged during cloning or
were not bona fide LRT cDNAs. In contrast, fragments
mi-grating at the expected position of the amplified product
yielded DNA sequence which matched the LR gene sequence
with an interruption in the middle. The 455-bp PCR product
matched the known sequence of the LR gene but did not
contain an interruption and thus was not spliced. Figure 5
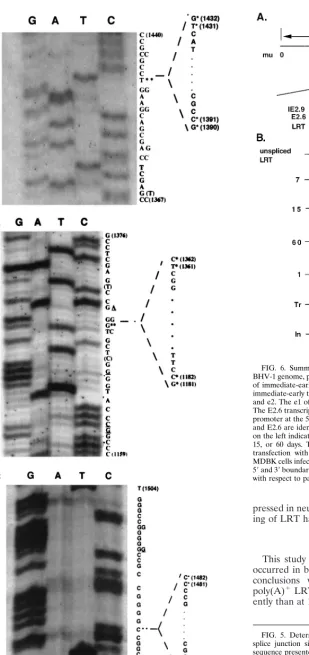
shows representative examples of the DNA sequence spanning
splice junction sites at 1, 7, or 15 days p.i. from TG. At least 10
independent clones were sequenced for each time point, and
they yielded the same sequence (locations of splice sites are
summarized in Fig. 6).
Regardless of whether LRT was poly(A)
1or poly(A)
2,
splice sites did not match consensus 59
or 39
splice sites (Table
1). The 59
splice sites of poly(A)
1LRT at 60 days p.i. were GC,
and they match the 59
splice site of HSV-1 LAT (71, 74), duck
a-globin, or bovine aspartyl protease (reviewed in references
36 and 47). The 59
GC splice site was also identified at the
second exon/intron border in transiently transfected COS-7
cells. The remainder of the 59
splice sites were CG, and to date
no transcript has been identified with this splice donor. Except
for the 39
TC splice site identified at 7 days p.i., the remainder
of the 39
splice sites have been described for other mRNAs.
The 39
TG splice site identified at 1 day p.i. or in transfected
cells (second 39
splice site) is present in the 39
splice acceptor
site of human or Drosophila melanogaster
a
subunit of guanine
nucleotide-binding protein (39, 53). The 39
CC splice site
ob-served at 15 days p.i. in transfected COS-7 cells (first 39
splice
site) or infected MDBK cells was described for yeast HAC1
mRNA (67). HAC1 and D. melanogaster
a
subunit of guanine
nucleotide-binding protein RNAs are alternatively spliced (39,
67). Finally, the 39
AG splice site present at 60 days p.i.
matches HSV-1 LAT (71, 74). In summary, these studies
indi-cated that (i) in TG of latently infected calves, the 59
and 39
splice sites of LRT match HSV-1 LAT; and (ii) most of the
nonconsensus splice sites are utilized by other mRNAs.
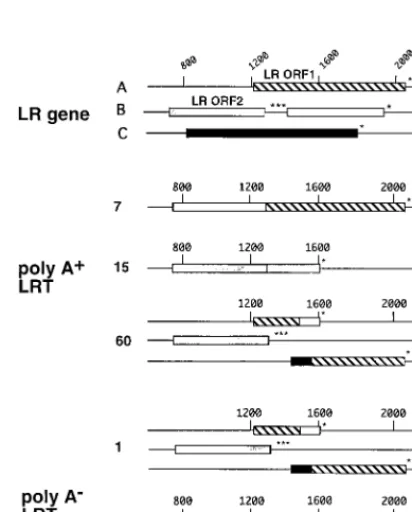
Effects of splicing on LR ORFs.
The LR gene contains two
open reading frames (ORFs) and two reading frames without
an initiating methionine. One reading frame without an
initi-ating methionine is contained after the three in-frame stop
codons of ORF 2, and the other is in reading frame C. Each
spliced LRT isoform was examined to determine the effect of
splicing on the ORFs. A fusion between ORF 2 and ORF 1 was
generated by splicing at 7 days p.i. in infected MDBK cells or
transfected COS-7 cells (Fig. 7). However, these putative
pro-teins would not be identical because the splice junction signals
are different. At 15 days p.i., the stop codons at the 39
end of
ORF 2 were removed and fused to the reading frame which is
in frame with ORF 2 (Fig. 7). At 1 day or 60 days p.i., the
ORFs were organized in a similar fashion (Fig. 7).
Interest-ingly, at 1 or 60 days p.i., a new ORF which is a fusion between
reading frame C and ORF 1 is generated (Fig. 7). When ORF
2 is fused to ORF 1 (7 days p.i., infected or transfected cells)
or to reading frame B (15 days p.i.), the predicted molecular
masses of these proteins were 35 to 45 kDa. This finding agrees
with previous conclusions that the P2 antibody directed against
the amino terminus of LR ORF 2 recognizes a 40-kDa protein
(35, 65). Although we do not know if these proteins are
ex-FIG. 4. Analysis of LRT expressed in transfected COS-7 or infected MDBK cells. (A) PCR amplification of poly(A)1LRT from transfected or infected cells. LaneM,f174 DNA digested with HaeIII; lane 1, RNA prepared from untransfected COS-7 cells; lanes 2 and 3, reactions with (lane 2) and without (lane 3) RT, using poly(A)1LRT prepared from COS-7 cells transfected with pcDNA1/LRT; lane 4, RNA prepared from uninfected MDBK cells; lanes 5 and 6, reactions with (lane 5)
and without (lane 6) RT, using poly(A)1LRT prepared from MDBK cells which were infected for 24 h. The arrow indicates the position of LRT. (B) PCR amplification
of poly(A)2LRT from transfected or infected cells. Lanes are as in panel A. The top arrow indicates the position of unspliced LRT, and the bottom arrows indicate
the positions of spliced LRT. Sizes are indicated in base pairs.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.72.527.72.271.2]pressed in neurons, these studies suggest that alternative
splic-ing of LRT has the potential to generate novel proteins.
DISCUSSION
[image:5.612.74.383.64.719.2]This study demonstrated that alternative splicing of LRT
occurred in bovine TG compared to nonneural cells. Several
conclusions were drawn from the sequencing data: (i)
poly(A)
1LRT in bovine TG at 7 days p.i. was spliced
differ-ently than at 15 or 60 days p.i., (ii) poly(A)
2LRT detected in
FIG. 6. Summary of spliced poly(A)1or poly(A)2LRT. (A) Schematic of
[image:5.612.309.546.72.400.2]BHV-1 genome, positions of repeats, and positions of map units (mu). Locations of immediate-early transcripts with respect to LRT are shown (82). Introns of immediate-early transcripts are presented as dashed lines. Exons are listed as e1 and e2. The e1 of IE2.9 and that of IE4.2 are identical but not protein coding. The E2.6 transcript does not contain e1, and transcription initiates from a novel promoter at the 59terminus of e2. Consequently, the proteins encoded by IE2.9 and E2.6 are identical. (B) Alternatively spliced variants of LRT. The numbers on the left indicate that RNA was prepared from TG of calves infected for 1, 7, 15, or 60 days. Tr, spliced LRT which was synthesized in COS-7 cells after transfection with pcDNA1/LRT; In, spliced LRT which was synthesized in MDBK cells infected with BHV-1 for 24 h. Numbers above the lines indicate the 59and 39boundaries of the intron. The schematic in panel A is not drawn to scale with respect to panel B.
FIG. 5. Determination of DNA sequences of the LRT splice junctions. The splice junction site was amplified with primers as described in Fig. 1. The sequence presented on the right is the sense strand. Double asterisks mark the splice sites of LRT and the sequence which was spliced out (41). The 59and 39 splice sites are marked with asterisks. The nucleotides that differ from the published LR gene sequence (41) are underlined. The nucleotides in parentheses (T at position 1368 in panel A and C and T at positions 1173 and 1368, respec-tively, in panel B) were detected in this study but not in the previous study (41). (A) 1 day p.i.; (B) 7 days p.i.; (C) 15 days p.i.
on November 9, 2019 by guest
http://jvi.asm.org/
bovine TG at 1 day p.i. was not the same as LRT detected at
7, 15, or 60 days p.i., (iii) poly(A)
1LRT in transfected or
infected cells which migrated with viral genomic DNA was not
spliced, (iv) poly(A)
2LRT was spliced in productively infected
MDBK cells, and (v) poly(A)
2LRT was apparently spliced at
two positions in transiently transfected COS-7 cells. Although
DNA sequence analysis of splice junction sites suggested that
samples from different times p.i. contained one spliced
prod-uct, the procedures used for amplifying and cloning splice
junction sites would yield the major spliced product. It is also
possible that at other times p.i., different spliced versions of
LRT exist or certain splice variants were not stably cloned. The
finding that nonconsensus splice sites were utilized suggests
that splicing was regulated by a combination of cell-specific
and viral or virus-induced factors.
Several LRT introns are smaller than the 80-nt minimum
intron size which has been proposed for eukaryotes (81). For
example, we detected a 35-nt intron at 60 days p.i. in TG, a
44-nt intron at 1 day p.i. in TG, and a 44-nt intron in transiently
transfected COS-7 cells. The ciliate Paramecium tetraurelia has
introns which are 20 to 33 nt long, and these introns have
consensus eukaryotic splice signals, G(T/A)G (21, 60). Most
fungi or insects, including Drosophila, have introns which range
in size from 31 to 70 nt (51, 61, 73; reviewed in references 31
and 48)). More importantly, the polyomavirus small tumor
antigen transcript contains a 48-nt-long intron which is excised
by a novel mechanism (27), demonstrating that small introns
can be excised in mammalian systems. We hypothesize that
cis-acting sequences within LRT regulate alternative splicing
and mediate excision of short introns. The three in-frame stop
codons at the C terminus of ORF 2 (reference 41 and Fig. 7)
may be important for alternative splicing because it is known
that multiple in-frame stop codons influence cell-specific
splic-ing (2). Although splicsplic-ing of LRT has unusual features (intron
length and nonconsensus splicing signals, for example), there is
precedence for unusual introns in a variety of organisms.
[image:6.612.51.544.81.217.2]Splicing is regulated by a complex array of trans-acting
fac-tors, some of which are cell or tissue specific (reviewed in
references 12 and 42). Although 59
splice signals are usually
recognized by small ribonucleoprotein complexes (snRNPs)
which contain the U1 small nuclear RNA (reviewed in
refer-ences 5 and 8), introns containing nonconsensus splice sites are
frequently spliced by less abundant snRNPs (30, 75, reviewed
in reference 69). Serine/arginine (SR) proteins are also
impor-tant for selection of 59
and 39
splice sites (reviewed in
refer-ences 13, 25, 45, 49, and 78). Adenovirus (33, 38), bovine
papillomavirus (84), and HSV-1 (46, 63) alter the distribution
or activity of SR proteins. Neuron-specific or brain-specific
alternative splicing of specific mRNAs has frequently been
observed (1, 6, 7, 44, 72, 77). A neuron-specific splicing
regu-lator (KSRP) is crucial for neuron-specific splicing of c-src (43;
reviewed in reference 29). Finally, alternative splicing of
HSV-1 LAT occurs in neural cells (44) or murine TG (3),
FIG. 7. Organization of ORFs in the LR gene or in alternatively spliced LRT [image:6.612.65.271.368.624.2]isoforms. The organization of ORFs in the LR gene was described originally by Kutish et al. (41). The reading frame in B (open box) that follows LR ORF 2 (stippled box) does not contain a methionine at its amino terminus. The reading frame in C (black box) does not contain a methionine at its amino terminus. Asterisks indicate the positions of in-frame stop codons. The hatched box de-notes ORF 1. Numbers at the top indicate nucleotide positions, and those on the left indicate that RNA was prepared from TG of calves infected for 1, 7, 15, or 60 days. Tr, LRT synthesized in COS-7 cells transfected with pcDNA1/LRT; In, LRT synthesized in MDBK cells infected with BHV-1 for 24 h. The sequence of each LRT variant was analyzed and translated by using the IBI MacVector sequence analysis software (Kodak).
TABLE 1. Summary of the 5
9
and 3
9
splice junctions observed in poly(A)
1and poly(A)
2LR RNAs
aRNAb Sequence
59splice site 39splice site
Poly(A)
1LRT
7
CTG:GCCTTC
GTAACAGCGGGGCTC:GGC
15
GCC:CGGGCG
GCCGAGGCCGGCCCC:GGG
60
GGT:GCCGCC
CCGCCGCGGCGGCAG:TTA
Poly(A)
2LRT
1
GGT:GCCGCC
CGGCGGCAGTTACTG:CCG
Tr
GGC:CGGGGT
GTGCTGGTAGCCGCC:GCG
GGT:GCCGCC
CGGCGGCAGTTACTG:CCG
In
TCT:CGGGGC
GTTACTGCCGCCGCC:GCG
mRNA consensus
(
2
3)A/CAG:GTA/GAGT(
1
5)
(
2
15)T/C11NC/TAG:NNN(
1
3)
aThe underlined sequences represent the splice signals. The sequences matched to the consensus splice sites are indicated by italicized letters.
bNumbers indicate the LR RNA obtained from TG of infected cattle at the indicated days p.i. Tr and In indicate LR RNA from transfected and infected cells.
on November 9, 2019 by guest
http://jvi.asm.org/
suggesting that neuron-specific splicing has functional
signifi-cance.
Latency has conveniently been divided into three distinct
steps: (i) establishment, (ii) maintenance, and (iii) reactivation.
The finding that LRT is alternatively spliced during
establish-ment (1 to 15 days p.i.) relative to maintenance (60 days p.i.)
suggests that LR gene products have specialized functions
which are necessary for the various stages of latency. During
establishment of latency, it is reasonable to hypothesize that a
viral function represses viral gene expression and enhances
neuronal survival. BHV-1 gene expression in TG, early or
immediate-early, is detected as early as 2 days p.i. and peaks at
7 days p.i. (66). Spliced LRT was detected at 1 day p.i. in TG,
suggesting that it accumulates prior to productive viral gene
expression and thus participates in establishment of latency.
HSV-1 LAT promotes establishment of latency (64, 76) in
mice by repressing productive viral gene expression (16, 26),
adding support to the hypothesis that the LR gene plays a role
in establishment. During maintenance of latency, promoting
neuronal survival would still be important but repression of
viral gene expression does not appear to be as important. A
viral function which promotes viral gene expression or DNA
replication but prevents neuronal death would be
advanta-geous during reactivation from latency. A number of studies
have concluded that HSV-1 LAT mutants do not reactivate
from latency efficiently in vivo (reviewed in references 57 and
58), but the mechanism by which LAT functions in this
capac-ity is unknown. Although it is unlikely that LRT regulates every
aspect of latency, we hypothesize that alternative splicing of
LRT yields novel proteins with specialized functions and that
these protein isoforms are important for certain steps of
la-tency. Cloning and characterizing the various LRT cDNAs
should allow a better understanding of how LR gene products
regulate latency.
ACKNOWLEDGMENTS
We are grateful to Luis Schang and Maria Teresa Winkler for
assisting with the animal studies and preparing RNA from TG. We
appreciate suggestions made by Harikrishna Nakshatri (IU Medical
Center) and Stephen Mount (University of Maryland) for advice on
nonconsensus splice sites and on intron lengths. Finally, we thank
Ruben Donis for critically reading the manuscript.
This work was supported by grants 9402117, 9502236, and 9702394
from the USDA.
REFERENCES
1. Amara, S. G., V. Jonas, and M. G. Rosenfeld. 1982. Alternative RNA pro-cessing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature 298:240–244.
2. Aoufouchi, S., J. Yelamos, and C. Milstein. 1996. Nonsense mutations inhibit RNA splicing in a cell-free system: recognition of mutant codon is indepen-dent of protein synthesis and tissue specific. Cell 85:415–422.
3. Arthur, J. L., R. Everett, I. Brierley, and S. Efstathiou. 1998. Disruption of the 59and 39splice sites flanking the major latency-associated transcripts of herpes simplex virus type 1: evidence for alternate splicing in lytic and latent infections. J. Gen. Virol. 79:107–116.
4. Bennett, J. L., and D. H. Gilden. 1996. The molecular genetics of herpes simplex virus latency and pathogenesis: a puzzle with many pieces still miss-ing. J. Neuro-Virol. 2:225–229.
5. Berget, S. M. 1995. Exon recognition in vertebrate splicing. J. Biol. Chem.
270:2411–2414.
6. Black, D. L. 1991. Does steric interference between splice sites block the splicing of a short c-src neuron-specific exon in non-neuronal cells? Genes Dev. 5:389–402.
7. Black, D. L. 1992. Activation of c-src neuron-specific splicing by an unusual RNA element in vivo and in vitro. Cell 69:795–807.
8. Black, D. L. 1995. Finding splice sites within a wilderness of RNA. RNA
1:763–771.
9. Block, T. M., and J. M. Hill. 1997. The latency associated transcript (LAT) of herpes simplex virus: still no end in sight. J. Neuro-Virol. 3:313–321. 10. Bratanich, A. C., and C. Jones. 1992. Localization of cis-acting sequences in
the latency-related promoter of bovine herpesvirus 1 which are regulated by neuronal cell type factors and immediate-early genes. J. Virol. 66:6099–6106. 11. Bratanich, A. C., N. Hanson, and C. Jones. 1992. The latency-related gene of bovine herpesvirus 1 inhibits the activity of immediate-early transcription unit 1. Virology 191:988–991.
12. Chabot, B. 1996. Directing alternative splicing, cast and scenarios. Trends Genet. 12:472–478.
13. Chandler, S. D., A. Mayeda, J. M. Yeakley, A. R. Krainer, and X.-D. Fu. 1997. RNA splicing specificity determined by the coordinated action of RNA recognition motifs in SR proteins. Proc. Natl. Acad. Sci. USA 94:3596–3601. 14. Chelly, J., and A. Kahn. 1994. RT-PCR and mRNA quantitation, p. 97–109.
In K. B. Mullis et al. (ed.), The polymerase chain reaction. Birkhauser,
Boston, Mass.
15. Chelly, J., G. Hamard, A. Koulakoff, J. C. Kaplan, A. Kahn, and Y.
Berward-Netter.1990. Dystrophin gene transcribed from different promoters in
neu-ronal and glial cells. Nature 344:64–65.
16. Chen, S.-H., M. F. Kramer, P. A. Schaffer, and D. M. Coen. 1997. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 71: 5878–5884.
17. Delhon, G. A., and C. Jones. 1997. Identification of DNA sequences in the latency related promoter of bovine herpes virus type 1 which are bound by neuronal specific factors. Virus Res. 51:93–103.
18. Devi-Rao, G. B., S. A. Goodart, L. S. Hecht, R. Rochford, M. K. Rice, and
E. K. Wagner.1991. Relationship between polyadenylated and
nonpolyade-nylated herpes simplex virus type 1 latency-associated transcripts. J. Virol.
65:2179–2190.
19. Devireddy, L. R., K. U. Kumar, M. M. Pater, and A. Pater. 1996. Evidence for a mechanism of demyelination by human JC virus: negative transcrip-tional regulation of RNA and protein levels from myelin basic protein gene by large tumor antigen in human glioblastoma cells. J. Med. Virol. 49:205– 211.
20. Dilworth, D. D., and J. R. McCarrey. 1992. Single-step elimination of con-taminating DNA prior to reverse transcriptase-PCR. PCR Methods Appl.
1:279–282.
21. Dupuis, P. 1992. The beta-tubulin genes of Paramecium are interrupted by two 27 bp introns. EMBO J. 11:3713–3719.
22. Farrell, M. J., A. T. Dobson, and L. T. Feldman. 1991. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA
88:790–794.
23. Feener, C. A., M. Koenig, and L. M. Kunkel. 1989. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Na-ture 338:509–511.
24. Fraser, N. W., T. M. Block, and J. G. Spivack. 1992. The latency-associated transcripts of herpes simplex virus: RNA in search of function. Virology
191:1–8.
25. Fu, X.-D. 1995. The superfamily of arginine/serine-rich splicing factors. RNA
1:663–680.
26. Garber, D. A., P. A. Schaffer, and D. M. Knipe. 1997. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 71:5885– 5893.
27. Ge, H., J. Noble, J. Colgan, and J. L. Manley. 1990. Polyoma virus small tumor antigen pre-mRNA splicing requires cooperation between two 39
splice sites. Proc. Natl. Acad. Sci. USA 87:3338–3342.
28. Goldenberg, D., N. Mador, M. J. Ball, A. Panet, and I. Steiner. 1997. The abundant latency-associated transcripts of herpes simplex virus type 1 are bound to polyribosomes in cultured neuronal cells and during latent infec-tion in mouse trigeminal ganglia. J. Virol. 71:2897–2904.
29. Grabowski, P. J. 1998. Splicing regulation in neurons: tinkering with cell-specific control. Cell 92:709–712.
30. Hall, S. L., and R. A. Padgett. 1996. Requirement for U12 snRNA for in vivo splicing of a minor class of eukaryotic nuclear pre-mRNA introns. Science
271:1716–1718.
31. Hawkins, J. D. 1988. A survey on intron and exon lengths. Nucleic Acids Res.
16:9893–9908.
32. Hayashi, K. 1994. Manipulation of DNA by PCR, p. 3–13. In K. B. Mullis et al. (ed.), The polymerase chain reaction. Birkhauser, Boston, Mass. 33. Himmelshpach, M., Y. Cavaloc, K. Chebli, J. Stevenin, and R. Gattoni. 1995.
Titration of serine/arginine (SR) splicing factors during adenoviral infection modulates E1A pre-mRNA splicing. RNA 1:794–806.
34. Ho, D. Y. 1992. Herpes simplex virus latency: molecular aspects. Prog. Med. Virol. 39:76–115.
35. Hossain, A., L. M. Schang, and C. Jones. 1995. Identification of gene prod-ucts encoded by the latency-related gene of bovine herpesvirus 1. J. Virol.
69:5345–5352.
36. Jackson, I. J. 1991. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res. 19:3795–3798.
37. Jones, C., G. A. Dehlon, A. C. Bratanich, G. Kutish, and D. L. Rock. 1990. Analysis of the transcription promoter which regulates the latency-related transcript of bovine herpesvirus 1. J. Virol. 64:1164–1170.
38. Kanopka, A., O. Muhlemann, and G. Akusjarvi. 1996. Inhibition by SR
on November 9, 2019 by guest
http://jvi.asm.org/
proteins of splicing of a regulated adenovirus pre-mRNA. Nature 381:535– 538.
39. Kozasa, T., H. Itoh, T. Tsukamoto, and Y. Kaziro. 1988. Isolation and characterization of the human Gsagene. Proc. Natl. Acad. Sci. USA 85: 2081–2085.
40. Krummenacher, C., J. M. Zabolotny, and N. W. Fraser. 1997. Selection of a nonconsensus branch point is influenced by an RNA stem-loop structure and is important to confer stability to the herpes simplex virus 2-kilobase latency-associated transcript. J. Virol. 71:5849–5860.
41. Kutish, G., T. Mainprize, and D. L. Rock. 1990. Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. J. Virol. 64:5730–5737.
42. Latchman, D. S. 1990. Cell-type-specific splicing factors and the regulation of alternative RNA splicing. New Biol. 2:297–303.
43. Lin, Z., S. Haus, J. Edgerton, and D. Lipscombe. 1997. Identification of functionally distinct isoforms of the N-type Ca21channel in rat sympathetic
ganglia and brain. Neuron 18:153–166.
44. Mador, N., A. Panet, D. Latchman, and I. Steiner. 1995. Expression and splicing of the latency-associated transcripts of herpes simplex virus type 1 in neuronal and non-neuronal cell lines. J. Biochem. 117:1288–1297. 45. Manley, J. L., and R. Tacke. 1996. SR proteins and splicing control. Genes
Dev. 10:1569–1574.
46. Martin, T. E., S. C. Barghusen, G. P. Leaser, and P. G. Spear. 1987. Redis-tribution of nuclear ribonucleoprotein antigens during herpes simplex virus infection. J. Cell Biol. 105:2069–2082.
47. Mount, S. M. 1982. A catalogue of splice junction sequences. Nucleic Acids Res. 10:459–472.
48. Mount, S. M., C. Burks, G. Hertz, G. D. Stormo, O. White, and C. Fields. 1992. Splicing signals in Drosophila: intron size, information content, and consensus sequences. Nucleic Acids Res. 20:4255–4262.
49. Mount, S. M. 1997. Genetic depletion reveals an essential role for an SR protein splicing factor in vertebrate cells. Bioessays 19:189–192.
50. Nicosia, M., J. M. Zabolotny, R. P. Lirette, and N. W. Fraser. 1994. The HSV-1 2 kb latency-associated transcript is found in the cytoplasm comi-grating with ribosomal subunits during productive infection. Virology 204: 717–728.
51. O’Hare, K., C. Murphy, R. Levis, and G. M. Rubin. 1984. DNA sequence of the white locus of Drosophila melanogaster. J. Mol. Biol. 15:437–455. 52. Puga, A., and A. L. Notkins. 1987. Continued expression of a poly(A)1
transcript of herpes simplex virus type 1 in trigeminal ganglia of latently infected mice. J. Virol. 61:1700–1703.
53. Quan, F., and M. A. Forte. 1990. Two forms of Drosophila melanogaster Gs are produced by alternate splicing involving an unusual splice site. Mol. Cell. Biol. 10:910–917.
54. Reyes, A. A., S. J. Small, and R. Akeson. 1991. At least 27 alternatively spliced forms of the neural cell adhesion molecule mRNA are expressed during rat heart development. Mol. Cell. Biol. 11:1654–1661.
55. Rock, D. L., W. A. Hagesmoser, F. A. Osorio, and D. E. Reed. 1986. Detec-tion of bovine herpesvirus type 1 RNA in trigeminal ganglia of latently infected rabbits by in situ hybridization. J. Gen. Virol. 67:2515–2520. 56. Rock, D. L., S. L. Beam, and J. E. Mayfield. 1987. Mapping bovine
herpes-virus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits by in situ hybridization. J. Virol. 61:3827–3831.
57. Rock, D. L. 1993. The molecular basis of latent infection by alphaherpesvi-ruses. Semin. Virol. 4:157–165.
58. Rock, D. L. 1994. Latent infection with bovine herpesvirus type 1. Semin. Virol. 5:233–240.
59. Rodahl, E., and L. Haarr. 1997. Analysis of the 2-kilobase latency-associated transcript expressed in PC12 cells productively infected with herpes simplex virus type 1: evidence for a stable nonlinear structure. J. Virol. 71:1703–1707. 60. Russell, C. B., D. Fraga, and R. D. Hinrichsen. 1994. Extremely short 20-33 nucleotide introns are the standard length in Paramecium tetraurelia. Nucleic Acids Res. 22:1221–1225.
61. Salkoff, L., A. Butler, N. Scavarda, and A. Wei. 1987. Nucleotide sequence of the putative sodium channel gene from Drosophila: the four homologous domains. Nucleic Acids Res. 15:8569–8572.
62. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a
laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
63. Sandri-Goldin, R. M., M. K. Hibbard, and M. A. Hardwicke. 1995. The C-terminal repressor region of herpes simplex virus type 1 ICP27 is required for the redistribution of small nuclear ribonucleoprotein particles and splic-ing factor SC35; however, these alterations are not sufficient to inhibit host cell splicing. J. Virol. 69:6063–6076.
64. Sawtell, N. M., and R. L. Thompson. 1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol. 66:2157–2169. 65. Schang, L. M., A. Hossain, and C. Jones. 1996. The latency-related gene of
bovine herpesvirus 1 encodes a product which inhibits cell cycle progression. J. Virol. 70:3807–3814.
66. Schang, L. M., and C. Jones. 1997. Analysis of bovine herpesvirus 1 tran-scripts during a primary infection of trigeminal ganglia of cattle. J. Virol.
71:6786–6795.
67. Sidrauski, C., J. S. Cox, and P. Walter. 1996. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87:405–413. 68. Smith, K., C. Long, B. Bowman, and M. Manos. 1990. Using cosolvents to
enhance PCR amplification. Amplifications 5:16–17.
69. Smith, C. M., and J. A. Steitz. 1997. Sno storm in the nucleolus. New roles for myriad small RNPs. Cell 89:669–672.
70. Spivack, J. G., and N. W. Fraser. 1987. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol. 61:3841–3847. 71. Spivack, J. G., G. M. Woods, and N. W. Fraser. 1991. Identification of a
novel latency-specific splice donor signal within the herpes simplex virus type 1 2.0-kilobase latency-associated transcript (LAT): translation inhibition of LAT open reading frames by the intron within the 2.0-kilobase LAT. J. Virol.
65:6800–6810.
72. Stamm, S., D. Casper, J. Dinsmore, C. A. Kaufmann, J. Brosius, and D. M.
Helfman.1992. Clathrin light chain B: gene structure and neuron-specific
splicing. Nucleic Acids Res. 20:5097–5103.
73. Talerico, M., and S. M. Berget. 1994. Intron definition in splicing of small
Drosophila introns. Mol. Cell. Biol. 14:3434–3445.
74. Tanaka, S., H. Minagawa, Y. Toh, Y. Liu, and R. Mori. 1994. Analysis by RNA-PCR of latency and reactivation of herpes simplex virus in multiple neuronal tissues. J. Gen. Virol. 75:2691–2698.
75. Tarn, W.-Y., and J. A. Steitz. 1996. A novel spliceosome containing U11, U12, and U5 snRNPs excises a minor class (AT-AC) intron in vitro. Cell
84:801–811.
76. Thompson, R. L., and N. M. Sawtell. 1997. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J. Virol. 71:5432–5440.
77. Ullrich, B., Y. A. Ushkaryov, and T. C. Sudhof. 1995. Cartography of neur-exins: more than 1000 isoforms generated by alternative splicing and ex-pressed in distinct subsets of neurons. Neuron 14:497–507.
78. Valcarcel, J., and M. R. Green. 1996. The SR protein family, pleiotropic functions in pre-mRNA splicing. Trends Biochem. Sci. 21:296–301. 79. Wagner, E. K., W. M. Flanagan, G. B. Devi-Rao, Y.-F. Zhang, J. M. Hill,
K. P. Anderson, and J. G. Stevens.1988. The herpes simplex virus
latency-associated transcript is spliced during the latent phase of infection. J. Virol.
62:4577–4585.
80. Wagner, E. K., and D. C. Bloom. 1997. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 10:419–443.
81. Weiringa, B., E. Hofer, and C. Weissmann. 1984. A minimal intron length but no specific internal sequence is required for splicing the large rabbit beta-globulin intron. Cell 37:915–925.
82. Wirth, U. V., B. Vogt, and M. Schwyzer. 1991. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J. Virol. 65:195–205.
83. Zabolotny, J. M., C. Krummenacher, and N. W. Fraser. 1997. The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J. Virol. 71:4199–4208.
84. Zheng, Z.-M., P. He, and C. C. Baker. 1996. Selection of the bovine papil-lomavirus type 1 nucleotide 3225 39splice site is regulated through an exonic splicing enhancer and its juxtaposed exonic splicing suppressor. J. Virol.
70:4691–4699.
on November 9, 2019 by guest
http://jvi.asm.org/