0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.23.14863–14875.2005
Specific Recognition and Cleavage of the Plus-Strand Primer by
Reverse Transcriptase†
Angela Atwood-Moore, Kenechi Ejebe, and Henry L. Levin*
Section on Eukaryotic Transposable Elements, Laboratory of Gene Regulation and Development, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892
Received 3 June 2005/Accepted 7 September 2005
Reverse transcriptases (RTs) of retroviruses and long terminal repeat (LTR)-retrotransposons possess DNA polymerase and RNase H activities. During reverse transcription these activities are necessary for the pro-grammed sequence of events that include template switching and primer processing. Integrase then inserts the completed cDNA into the genome of the host cell. The RT of the LTR-retrotransposon Tf1 was subjected to random mutagenesis, and the resulting transposons were screened with genetic assays to test which mutations reduced reverse transcription and which inhibited integration. We identified a cluster of mutations in the RNase H domain of RT that were surprising because they blocked integration without reducing cDNA levels. The results of immunoblots demonstrated that these mutations did not reduce levels of RT or integrase. DNA blots showed that the mutations did not lower the amounts of full-length cDNA. The sequences of the 3ⴕends of the cDNA revealed that mutations within the cluster in RNase H specifically reduced the removal of the polypurine tract (PPT) primer from the ends of the cDNA. These results indicate that primer removal is not a necessary component of reverse transcription. The residues mutated in Tf1 RNase H are conserved in human immunodeficiency virus type 1 and make direct contact with DNA opposite the PPT. Thus, our results identify a conserved element in RT that contacts the PPT and is specifically required for PPT removal.
Reverse transcription of retroviruses requires a sequence of programmed steps that include highly specific switching of templates and precise cleavages of RNA primers. The enzyme responsible for these processes, reverse transcriptase (RT), possesses DNA polymerase activity and an RNase H activity that degrades RNA annealed to DNA. The RNase H activity is critical because it degrades RNA templates, allowing strong-stop intermediates to transfer to new templates. RNase H activity is also responsible for precise cleavages flanking the polypurine tract (PPT) that “select” the plus-strand primer of reverse transcription. After plus-strand priming has occurred, RNase H removes the PPT. In addition, the RNase H also removes the minus-strand primer. The highly precise cleavages that remove the primers are absolutely critical because ulti-mately they define the sequence at the ends of the cDNA. For integrase (IN) to complete insertion, the highly conserved CA dinucleotide must be positioned at the 3⬘ends (32).
Long terminal repeat (LTR) retrotransposons are closely related to retroviruses and use the same multistep process of reverse transcription to copy their mRNA into full-length dou-ble-stranded DNA (32). As a result, retrotransposons are ex-cellent models for the reverse transcription of retroviruses. Previous work revealed that the Tf1 transposon of
Schizosac-charomyces pombeis highly active and that the individual steps
of reverse transcription can be studied inS. pombe(14, 15, 19). The presence of all seven conserved domains in the RT of Tf1
indicates that functional information obtained from the study of Tf1 will be applicable to the RT of retroviruses (5, 34).
A surprising observation came from studying mutations throughout Tf1 that specifically inhibited integration in vivo. Two mutations in RT were found to block integration without reducing the levels of cDNA (1). The mutations may have reduced the ability of RT to recognize specific cDNA struc-tures, and as a result, one of the later steps of reverse tran-scription could have been restricted. Currently, it is not clear whether the specificity of the strand transfers and the precision of the primer cleavages result from a specific recognition of the substrate by RT or are due strictly to structures of the sub-strates, such as the PPT, that resist the actions of RT.
To identify the amino acids of Tf1 RT that may contribute to this specificity, we used a novel combination of genetic assays to screen an extensive library of random mutations. We ob-tained a cluster of mutations that produced normal levels of cDNA which nevertheless could not be integrated. Analysis of 526 cDNA sequences showed the mutations specifically inhib-ited removal of the plus-strand primer (PPT). The mutations correspond to the amino acids of human immunodeficiency virus type 1 (HIV-1) RT that, in a crystal structure, contact DNA opposite the PPT (30). These results provide strong evidence that RT makes contacts with the PPT that are re-quired specifically for PPT removal.
MATERIALS AND METHODS
Media and culture growth.Selective medium contained Edinburgh minimal medium (EMM) (21) and 2 g per liter of dropout mix (29). When indicated, 10M vitamin B1 (thiamine: T-4625, Sigma, St. Louis, MO) was added to repress thenmt1promoter. EMM plates with 5-fluoroorotic acid (5-FOA) con-tained 1 mg/ml 5-fluoroorotic Acid (F5050, United States Biologicals, Swamp-scott, MA). YES FOA/G418 contained 500g/ml (corrected for purity) of geneticin (11811-031, Life Technologies, Rockville, MD) and 1 mg/ml 5-FOA in
* Corresponding author. Mailing address: Section on Eukaryotic Transposable Elements, Laboratory of Gene Regulation and Devel-opment, National Institute of Child Health and Human DevelDevel-opment, National Institutes of Health, Bethesda, Maryland 20892. Phone: (301) 402-4281. Fax: (301) 496-4491. E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi .asm.org/.
14863
on November 8, 2019 by guest
http://jvi.asm.org/
YES rich medium (5 g Difco yeast extract with 30 g glucose and 2 g complete dropout mix per liter).
Yeast strains and plasmid constructions.The yeast strains used in this study and the specific plasmids each strain carried are described in Tables S3 and S4, respectively, in the supplemental material. The plasmids were introduced into strains ofS. pombeusing lithium acetate transformation (21).
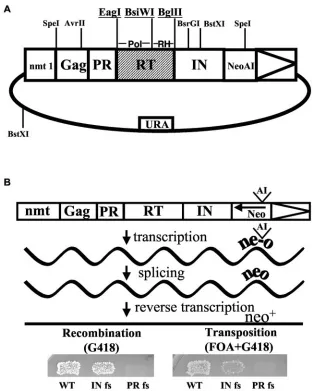
Plasmid pHL1690 expressed Tf1-neoAI with the unique restriction sites EagI, BsiWI, and BglII flanking the polymerase and RNase H sequences of RT (Fig. 1). The restriction sites were created using fusion PCR as previously de-scribed and did not alter the coding sequence of Tf1 (14). The oligonucleotides used are described in Table S5 in the supplemental material. Four quarter products were PCR amplified using pHL449-1 as the template. Quarter products were used as the template to produce half products. Finally, half products were combined as the template to produce the full product, which was ligated into pHL449-1 digested with AvrII and BsrGI. The sequence of the insert was con-firmed in the context of the completed plasmid, pHL1690. An alternative version of pHL1690 possessing an SpeI site in the 5⬘untranslated region immediately upstream of Gag was created by swapping the 4.5-kb Mlu I to AvrII fragment from pHL891-19 (containing the extra SpeI site) into pHL1690 to create pHL2006. The construction of pHL891-19 was described previously (19).
For the complementation experiments, versions of the wild-type Tf1-neoAI and Tf1 with the IN frameshift (INfs) that lackedneowere created in a vector
that contained theLEU2gene ofSaccharomyces cerevisiae. A 2.0-kb BamHI to SphI fragment withLEU2from pHL385 was inserted into the 10.4-kb BamHI to SphI fragment of pHL411-62. This wild-type version of Tf1 withoutneowas pHL691-1. To make a similar plasmid with the INfs the 4.1-kb BstXI fragment from Tf1-INfs (pHL476-3) was first ligated into the BstXI-digested backbone of pHL 411-62 to create a version of INfs that lackedneo, pHL1223-1. Then the 4.5-kb AvrII to BamHI fragment containing the Tf1 ORF from pHL1223-1 was ligated into the AvrII to BamHI backbone of the leu-marked transposon vector pHL691-1. This construct was pHL1224-1.
[image:2.585.135.450.67.458.2]Transposition and homologous recombination assays.Following transforma-tion intoSchizosaccharomyces pombe, the mutant versions of Tf1 were assayed for transposition and recombination as previously described (1, 14). Briefly, strains containing mutant versions of Tf1-neoAI were grown as patches for 2 days on EMM-Ura supplemented with 10 uM thiamine to repress the transcription of Tf1-neoAI from thenmt1promoter. Patches were then replica printed to similar plates lacking thiamine to activate transcription. Following four days growth, induced patches were further replica printed to EMM plates containing 5-FOA. Patches that were 5-FOA resistant were then replica printed to YES plates supplemented with 5-FOA and G418 to measure integration of Tf1-neoAI. Homologous recombination between the cDNA of Tf1 and the Tf1-neoAI se-quence on the expression plasmid was detected by printing induced patches
FIG. 1. Assays for Tf1 activity. (A) The plasmid used to express Tf1-neoAI pHL1690-1 is shown. The restriction sites created for the mutagenesis are underlined. (B) The expression of Tf1-neoAI and the splicing of the artificial intron resulted in cDNA with an activeneo.To detect transposition cells were first replica printed to plates with FOA then to plates with FOA and G418. To measure recombination, cells induced for Tf1-neoAI expression were replica printed directly to plates with G418.
14864 ATWOOD-MOORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
directly to YES-G418 as described (1). Mutants identified with these assays were subjected to secondary and tertiary screens to confirm their phenotypes.
Random PCR mutagenesis.Primers for mutagenic PCR, HL87 and HL88, were designed to anneal upstream of the 5⬘restriction site (EagI) and down-stream of the 3⬘restriction site (BglII). Two independent PCRs (A1 and A2) using condition set A were performed. Condition set A included 4 ng of tem-plate, 500 ng of each primer, 5 units of AmpliTaqDNA polymerase (Applied Biosystems; Foster City, CA), 5 mM MgCl2, 0.2 mM MnCl2, 0.4 mM of an equal
ratio mix of deoxynucleoside triphosphates, and an additional 0.2 mM each of dATP and dCTP. Reaction conditions for set A required an initial denaturation step at 95°C, followed by 30 cycles of 1 min of denaturation at 95°C, 2 min of annealing at 45°C, and 2 min of extension at 72°C. Two additional independent reactions (B1 and B2) were identical to A1 and A2 except that the B reactions contained a 0.2 mM supplemental amount of dGTP and dTTP in place of the extra dATP and dCTP. A third set of reactions (C1, C2, C3, and C4) was likewise identical to the A and B reactions but contained all deoxynucleoside triphos-phates represented equally at 0.2 mM and no MnCl2was included.
Following PCR, all A, B, and C reactions (8 total) were pooled, ethanol precipitated and concentrated. The entire mutagenized DNA pool was digested with EagI and BglII, phenol extracted, gel purified, and ligated directly into pHL1690 digested with EagI and BglII. Over 62,000 bacterial transformants were harvested from transformation plates, pooled into a 500-ml LB-ampicillin cul-ture, and the resulting DNA was transformed into YHL912.
Identification of mutations in RT.Once the plasmids with the mutant trans-posons were isolated from the yeast strains, the EagI to BsiWI and the BsiWI to BglII fragments were subcloned separately into a wild-type version of the
Tf1-neoAI to determine whether the mutations were in the polymerase domain or in RNase H. The recipient vector, pHL2006, contained a unique SpeI site in the 5⬘
untranslated region of Tf1 to distinguish it from the original expression vector, pHL1690-1. A representative clone of each fragment for each mutant was se-quenced to identify the position of the mutation and to verify that no other changes existed.
Protein preparation and immunoblots.Total proteins were extracted from cultures grown under induction conditions according to established protocols (2). Equal amounts of total protein were run on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA). The ECL detection system (Amersham Biosciences) was used in conjunction with the polyclonal antisera for the detec-tion of IN (producdetec-tion bleed 657, 1:10,000) and RT (bleed 12-31-93 of 1571, 1:1,000). The filters were hybridized for 1 hour at room temperature and 16 h at 4°C, respectively (7, 16).
Preparation of DNA for blotting and ligation-mediated PCR.cDNA for DNA-blot analysis was extracted from 100 OD units of cells as previously described (2, 15). One tenth of this purified nucleic acid was restriction digested with BstXI, subjected to agarose gel electrophoresis and transferred to Gene Screen mem-brane (Perkin Elmer Life Sciences, Wellesley, MA) for DNA blot analysis. After transfer, filters were hybridized with a32P labeled, 1.0-kbneofragment derived
from a BamHI digest of pHL765.
DNA for LM PCR, two LTR circle characterization, and primer extension was isolated from virus like particles purified from 500-ml cultures as previously described (14, 16). Following growth at 32°C for 40 h, the harvest, wash, and breakage were as previously described (14, 16). The supernatant from the break-age procedure was subjected to a 3,000 rpm spin (SS34 rotor), and 3.5 ml of that supernatant was loaded onto a 20 to 70% linear sucrose density gradient in buffer B (15 mM NaCl, 10 mM HEPES [pH 7.8], 5 mM EDTA, and 5 mM dithiothre-itol). Gradients were spun at 25,000 rpm (Beckman, SW28 rotor) for 1.5 h, and 1.2-ml fractions were collected. One-seventeenth of each fraction was precipi-tated with tricholoroacetic acid, washed extensively (three to five times) with acetone and immunoblotted to identify the peak fractions of virus-like particles. The peak fractions were pooled and spun at 35,000 rpm (Beckman SW 41 rotor) for 1.5 h. Pellets were resuspended in 0.4 ml 25 mM EDTA, 0.1% SDS and subjected to proteinase K (Roche) treatment (50g/ml) for 1 hour at room temperature. Extensive phenol extraction (30 min vortex at room temperature followed by two additional 5-min vortex extractions) and an ethanol precipitation completed the treatment. Final DNA extracted from particle preps was resus-pended in a 50l volume of Tris-EDTA.
Ligation-mediated PCR.The cloning and characterization of the 3⬘ends of Tf1 cDNAs was performed as previously published (3, 22, 23). Briefly, the oligonucleotide to be ligated is blocked at the 3⬘end with a C3 spacer to drive ligation exclusively of its 5⬘end to the 3⬘ends of the cDNAs. Approximately 50 ng of particle prep purified cDNA was added to 6l of 3 M blocked oligonucleotide. This reaction was heated to 95°C for 3 min. Tubes were trans-ferred immediately to ice for an additional 3-minute incubation. To this cooled
reaction we added 15l of 2X Quick DNA Ligase Reaction buffer (NEB), 3l dimethyl sulfoxide, and 1l of T4 RNA ligase (NEB). Following overnight ligation at 16°C, 1l was used as the template in a PCR also containing 1l of 10M each of two oligonucleotides specific for amplification of the 3⬘end of the minus strand (HL 973 and JB 54.4).
Reactions also contained the following components from the TitaniumTaq
polymerase kit (Clontech/BD Biosciences, Palo Alto, CA): deoxynucleoside triphosphates, reaction buffer,Taqpolymerase, and distilled H2O to a final
volume of 50l. These reactions were performed using the following steps: 95°C denaturation for 2 min, 30 cycles of 15 seconds at 95°C denaturation, 1 min at 66°C annealing, 1 min at 68°C extension, followed by a final 3 min of extension at 68°C. Primary PCR products of the appropriate molecular weights were gel purified and 1l of each gel slice was used in a secondary amplification with identical conditions. Secondarily amplified PCR products were purified from gel slices and eluted in a final volume of 30l. One microliter of purified PCR product was ligated into pCR2.1 TOPO vector (Invitrogen) and transformed into TOP10F (Invitrogen) cells according to the manufacturer’s specifications.
Two-LTR circle characterizations.Three microliters (25 ng) of DNA purified from virus-like particles was used as the template in a PCR containing 2.5l of 20M stocks of two oligonucleotides (HL917 and HL923) specific for amplifi-cation of the junction between two Tf1 LTRs in partially purified two-LTR circles. Two microliters (3 ng) of purified PCR product were ligated to 50 ng pT-Adv vector (Biosciences/Clontech) and transformed into Top10F⬘ (Invitro-gen) cells. Six independent transformants of each PCR product were sequenced.
Primer extension.Approximately 50 ng of the cDNA was mixed with 2.5 pmol of32
P-labeled oligonucleotide (JB54.3 with wild-type cDNA and JB54.4 with cDNA produced by the mutants) in reactions containing 2.5 mM MgCl2, 200 uM
deoxynucleoside triphosphate mix, 5 units AmpliTaq (Perkin Elmer/Cetus, Wellesley, MA) and the manufacturer’s extension buffer. PCR conditions con-sisted of 50 cycles of 1 min of denaturation at 94°C, 2 min of annealing at 45°C and 2 min of extension at 72°C, and concluded with a final 5 min of extension at 72°C. Extended products were run on 6% buffer gradient denaturing polyacryl-amide gels next to sequencing ladders generated with the same oligonucleotides used in the extension reactions. The oligonucleotides used in the sequencing reactions were 5⬘ phosphorylated with unlabeled ATP and the ladders were produced using the Sequenase 2.0 kit (70770, USB). RNase A treatments con-sisted of 30-min incubations at 37°C with 16g/ml RNase A (109142, Roche) followed by phenol-chloroform extraction and ethanol precipitation.
RESULTS
Random mutagenesis of RT identified mutations in RNase H that reduced integration.A previous study of Tf1 identified two mutations in RT that reduced integration without lowering amounts of cDNA (1). To determine what feature of reverse transcription was altered by these mutations we set out to identify other residues in RT that made specific contributions to the cDNA that were required for integration. We used random mutagenesis of the RT sequence and screened for mutations that allowed reverse transcription but not integra-tion. Mutagenized DNA spanning RT was introduced into a copy of Tf1 used to induce transposition in
Schizosaccharomy-ces pombe. For this purpose we generated unique restriction
sites at the beginning of RT, between the polymerase domain and RNase H, and at the end of RT (Fig. 1A). The nucleotide changes used to make the restriction sites did not alter the amino acid sequence or the activity of Tf1.
To identify residues that were important for integration but not for producing cDNA, mutant versions of Tf1 were screened with genetic assays that independently detected re-verse transcription and transposition (Fig. 1B). Both assays, as previously developed (1, 15), were based on strains of
Schizo-saccharomyces pombethat expressed Tf1-neoAI, a version of
Tf1 with neodisrupted by an artificial intron (Fig. 1B). The recombination assay measured the products of Tf1 reverse transcription that, as the result of homologous recombination, generated copies of Tf1 in the plasmid that lacked the artificial
on November 8, 2019 by guest
http://jvi.asm.org/
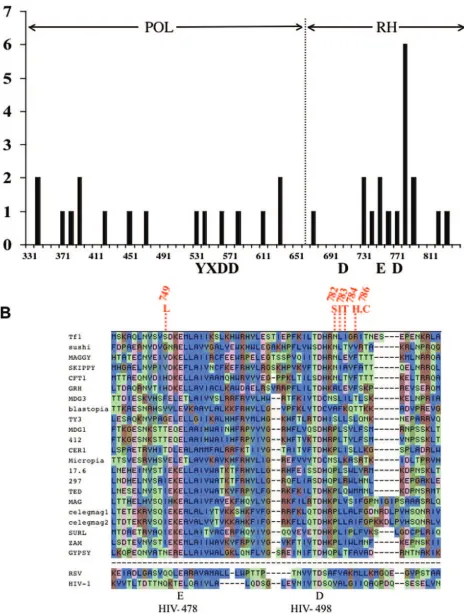
FIG. 2. Position of mutations in RT. (A) The numbers of single amino acid mutations identified with the genetic assays were mapped in bins of 10 amino acids. YXDD and DED indicate the invariant residues of the polymerase and RNase H domains, respectively. (B) The alignment of RNases H as previously reported is shown for a representative set of elements (20). The cluster of mutations in Tf1 RNase H are shown in red. The invariant amino acids labeled below.
14866
on November 8, 2019 by guest
http://jvi.asm.org/
intron. The resulting copies of neo generated resistance to G418 (Fig. 1B). Frameshifts in the coding sequence of Tf1 indicated that resistance to G418 required expression of RT but not IN (Fig. 1B, left panel). Because the synthesis of minus strand strong stop cDNA and its transfer must occur before
neocan be reverse transcribed, resistance to G418 in this assay demonstrated that both the DNA polymerase and RNase H domains were active. Of the 6,250 yeast strains screened with this assay, 3,288 exhibited normal activity and 2,962 had sig-nificantly less activity.
The versions of Tf1 capable of reverse transcription were subsequently screened with a transposition assay to determine which were unable to integrate the products of reverse tran-scription. The assay for transposition is similar to the recom-bination assay and used the same expression plasmid contain-ing Tf1-neoAI. However, after expression of Tf1-neoAI, the plasmid was removed from the strain by growing cells on 5-FOA. This prevented resistance to G418 due to homologous recombination with the plasmid. Under these conditions the bulk of the resistance to G418 was due to integration as indi-cated by the dependence on IN expression (Fig. 1B, right panel). Of the 3,288 copies of Tf1 that had normal activity in the recombination assay, 121 were defective for transposition. As a result, these mutants were candidates for being defective specifically in a late step of reverse transcription.
Of the 121 mutant version of Tf1, 34 were found to have single-amino-acid substitutions that were the sole cause of the transposition defects. The details of the mutagenesis and screening are summarized in Table S1 in the supplemental material.
Mutations formed a tight cluster within a region of RNase H that is highly conserved.Of the 34 mutants with single nucle-otide substitutions, 16 were in the polymerase domain and 18 were in RNase H. The distribution of the mutations was mapped graphically into bins of 10 amino acids (Fig. 2A). Their exact positions were tabulated in Table S2 in the sup-plemental material. Although the mutations in the polymerase portion of RT were evenly distributed, the mutations in the RNase H domain were surprisingly clustered. Within one block of 70 amino acids, there were 15 separate mutations. As a result, 75% of the mutations in RNase H clustered within 33% of the domain. At the most concentrated region of the cluster, five mutations fell within a window of just five amino acids (Fig. 2B). Two different mutations of Arg786 were included in the cluster.
We examined the possibility that the cluster of mutations in RNase H identified a domain with critical function by asking whether the residues were conserved in other RTs. A compre-hensive alignment of RTs was published previously and a por-tion of it containing RNases H from elements related to Tf1 is shown in Fig. 2B (20). The region of RNase H that includes the cluster is well conserved among all the elements shown, includ-ing the retroviruses Rous sarcoma virus and HIV-1. The five mutations that make up the core of the cluster started just three amino acids after Asp779, one of the active-site residues that is invariant (Fig. 2B).
The sharp clustering of the mutations and the general con-servation of these residues suggested these amino acids pos-sessed a critical function. Interestingly, the mutation S749L was one of the alleles isolated from the previous mutagenesis
and screen (1). That study found S749L caused a significant defect in integration and not in the accumulation of cDNA. With the goal of revealing the role of RNase H in late steps of reverse transcription, we focused our experiments on the spe-cific function of the four amino acids at the core of the cluster (N782, L783, I784, and R786) and on S749. The results of the assays used for the screen described above clearly showed the mutations in these five residues caused a reduction in trans-position without lowering the level of cDNA recombination (Fig. 3). In comparison, mutations RH1⫺ and RH2⫺in the catalytic residues of RNase H abolished recombination and transposition (Fig. 3).
Tf1 with the mutations that clustered in RNase H produced normal levels of double-stranded cDNA. The high levels of cDNA recombination generated by the mutants in this study indicated these elements were capable of reverse transcription. DNA blots were used as a direct method for characterizing the products of reverse transcription. DNA from cells expressing the mutant elements was digested with BstXI and the resulting blot was hybridized with aneo-specific probe. Wild-type
Tf1-neoAI produced a single cDNA band of 2.1 kb, the size ex-pected based on the location of the only BstXI site in Tf1 (Fig. 4A). The band of 9.5 kb is theneo-containing fragment of the expression plasmid and the 5.5-kb signal is a single-LTR intermediate of reverse transcription. Both the 2.1-kb and the 5.5-kb species were absent from a version of Tf1 that lacked RT (Fig. 4A, PRfs).
The cDNA produced by the 34 mutants in this study exhib-ited a variety of patterns (Table S2 in the supplemental mate-rial). Some mutations caused a reduction in the 2.1-kb frag-ment of cDNA and a corresponding increase in a smear of species. These smears may have been due to heterogeneous double- or single-stranded species. The mutations that clus-tered in RNase H were particularly surprising in that except for I784T, they produced wild-type amounts of the 2.1-kb frag-ment of cDNA (Fig. 4A). Because the 2.1-kb fragfrag-ment was generated with a restriction enzyme, its wild-type amounts indicated the DNA at the cut site was double-stranded. The position of the BstXI site was just upstream of the polypurine tract (PPT). Thus, in the process of reverse transcription this restriction site was among the last sequences to become dou-ble-stranded. Therefore, the presence of the 2.1-kb species was also evidence that the cDNA products were full-length and double-stranded. The large number of mutants that produced wild-type levels of the 2.1-kb species was surprising because their reduced transposition was the result of mutations in RT, the enzyme responsible for cDNA synthesis.
Mutations in the RNase H cluster did not affect the pro-cessing or stability of RT or IN.One explanation for how the mutations in the cluster could have reduced integration with-out effecting reverse transcript was that they caused defects in the processing or stability of IN. This possibility was plausible because IN is positioned next to RT in the single primary translation product generated by Tf1 (2, 16). In addition, the RT and IN of Tf1 have been shown in two-hybrid experiments to interact with each other (31). Immunoblot analysis using anti-IN antibodies was performed on whole-cell extracts of cultures harvested in log phase. The results for all 34 mutants are summarized in Table S2 in the supplemental material. The vast majority of the mutations caused no change in the
on November 8, 2019 by guest
http://jvi.asm.org/
amounts of IN. The levels of IN were not changed by the mutations that clustered in RNase H (Fig. 4B). As expected, frameshift mutations at the beginning of IN (INfs) or in PR (PRfs) blocked expression of IN.
The Gag and IN proteins of Tf1 are expressed at equal molar ratios in log-phase cultures. However, during the log to stationary phase transition most of the IN is degraded, result-ing in the molar excess of Gag that is required for the produc-tion of virus-like particles (2). All mutants in this study exhib-ited the appropriate degradation of IN (data not shown).
The possibility was also considered that the mutations changed the stability of RT or the proteolytic processing that separated RT from PR and IN. The anti-RT antibodies de-tected the 60-kDa and 72-kDa species of RT (Fig. 4C). The 60-kDa protein was RT and the 72-kDa species was the PR-RT precursor (7). All but one of the mutations in the cluster had no effect on the amounts of the RT proteins (Fig. 4C). L783I produced significantly less 60-kDa RT. Despite this low level of RT, L783I was capable of producing normal amounts of cDNA (Fig. 4A). This mutant did produce wild-type levels of PR-RT, suggesting this protein was sufficient for generating the cDNA. The INfs was another mutant that produced the 72-kDa species without the accumulation of the mature RT. Like L783I, INfs also generated normal levels of full-length cDNA (Fig. 4A).
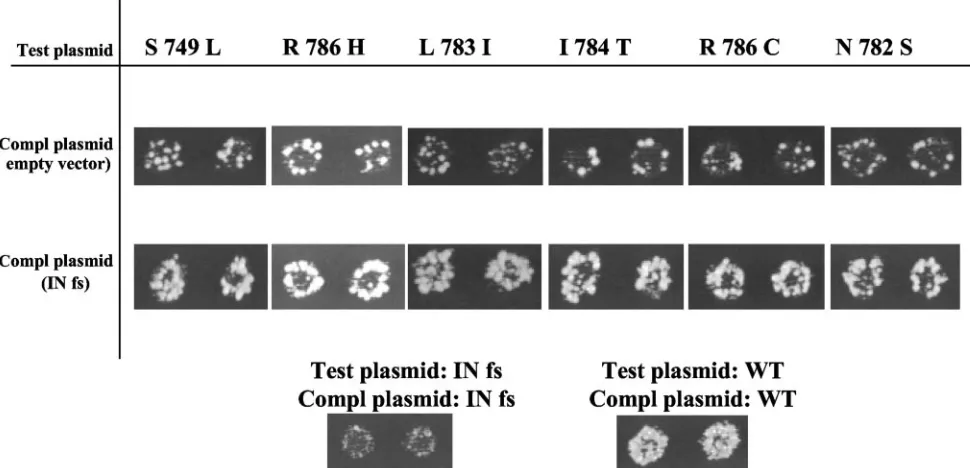
Transposon complementation assays indicated the mutants retained IN activity.The immunoblot data indicated that the cluster of mutations in RNase H did not reduce the accumu-lation of IN. However, the possibility remained that the RNase H mutations reduced IN activity either by altering the confor-mation of IN or by causing small shifts in the protease cleavage sites of IN that could result in an altered protein with a similar
molecular weight to IN. Either of these effects could result in transposons unable to integrate cDNA.
To test whether the mutants retained IN activity, we devel-oped a complementation assay based on coexpressing two transposons. The transposon being tested for IN activity con-tained theneomarker and was assayed in the expression plas-mid (test plasplas-mid) shown in Fig. 1A. The complementing ver-sion of Tf1 was INfs and because its only defect was a lack of IN expression, it had the potential to complement any defect other than a lack of IN activity. The baseline transposition activity of the elements with the RNase H mutations was mea-sured in strains that contained the mutation in the test plasmid and an empty vector in place of the INfs complementing plas-mid (Fig. 5, top row of assay patches). The baseline transpo-sition activity of Tf1 that contained INfs was measured in a strain where both plasmids encoded INfs (Fig. 5, INfs/INfs). If the transposons with the RNase H mutations in the test plas-mid had reduced IN activity, coexpression of the INfs version of Tf1 would be unable to complement the defect. If however, the RNase H mutations only reduced RT activity, the INfs would be expected to rescue the transposition defect. Combin-ing the RNase H mutants with the INfs revealed significant increases in transposition as indicated by their increased resis-tance to G418 (Fig. 5, second row of assay patches). These data indicate that the transposons with mutations that clustered in RNase H retained IN activity.
Cluster of amino acids in the RNase H of Tf1 corresponded to the amino acids of HIV-1 RNase H that can bind DNA opposite the PPT.The mutations that clustered in RNase H caused no detectable defects in the expression of Tf1 proteins, in the production of cDNA, or in the activity of IN. These observations left open the possibility that the mutations
af-FIG. 3. Transposition and recombination activities of Tf1-neoAI with the mutations that clustered in RNase H. mcc indicates the Tf1 with unique restriction sites in RT. RH1⫺is D710N and RH2⫺is D779N.
14868 ATWOOD-MOORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.138.446.69.330.2]fected the ability of RNase H to process the primers of reverse transcription. Were RNase H to miscleave either the plus or minus-strand primers, changes in sequence would occur at the 3⬘ends of the cDNA that would prevent IN from completing strand transfer.
A high-resolution structure of HIV-1 RT bound to an RNA/ DNA hybrid identified a set of amino acids that interact di-rectly with the RNA:DNA hybrid containing the PPT (30). Collectively, these residues were referred to as the RNase H primer grip. Based on their model, the authors suggested these residues could contribute to cleavage specificity. The align-ment in Fig. 2B allowed us to make the observation that four of the six mutations in the Tf1 cluster corresponded to amino acids in the RNase H of HIV-1 that make direct contact with the RNA-DNA hybrid (Fig. 6). The strong correspondence between the concentrated cluster of Tf1 mutations and the residues of HIV-1 that contact the PPT hybrid suggested the
possibility that the mutations in Tf1 inhibited transposition because they altered the ability of RNase H to cleave the PPT.
Cluster of mutations in the RNase H of Tf1 inhibited re-moval of the PPT.The effect of the mutations in RNase H on the recognition of the PPT was examined by sequencing the 3⬘ ends of the Tf1 cDNA. One method used to obtain sequence from the ends of HIV-1 cDNA is to amplify by PCR the LTR-LTR junctions of two-LTR circles. These circles result from the fortuitous action of cellular ligases on full-length cDNAs. The sequences at the LTR-LTR junctions are used as surrogates for the sequences at the ends of the linear DNAs (25). Approximately 60% of the two-LTR circles produced by wild-type HIV-1 contain the consensus sequence found at the ends of mature cDNA (8). However, of the six two-LTR circles generated by wild-type Tf1-neoAI, none contained the consen-sus sequences of the termini.
Three of the isolates had the same 28-nucleotide deletion of the U5 sequence of the downstream LTR. Another had a single nucleotide substitution, C to A, in the U5 end of this LTR. One isolate contained an insertion of a single A between the LTRs, while another had an eight-nucleotide deletion from the U3 of the upstream LTR. This high frequency of alter-ations in the ends of the LTRs indicated that the DNA ligated into circles may have derived from a pool of damaged reverse transcripts.
Ligation-mediated PCR is a direct method for sequencing the ends of linear cDNA (3, 22, 23). We used this technique to sequence the 3⬘ends of the minus strand because they were the substrate for IN and because this end was defined by the PPT. Oligonucleotide Rag208 with a 3⬘block was ligated to the 3⬘ ends of cDNA isolated from virus-like particles. Oligonucleo-tides were then used to PCR the upstream LTR so that the junction with Rag208 could be sequenced.
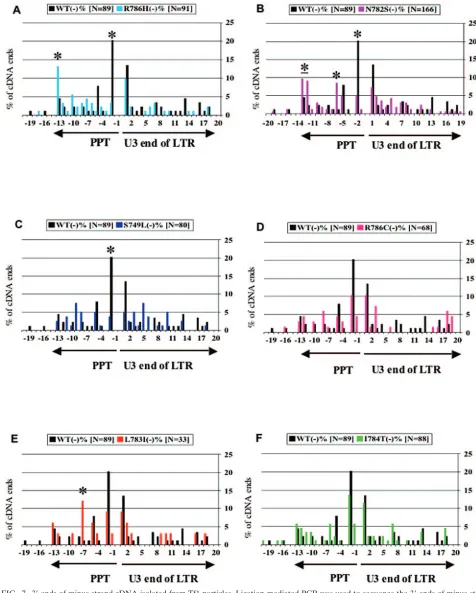
Approximately 100 clones were sequenced for each version of Tf1 tested. The sequences produced by wild-type Tf1-neoAI exhibited a surprising heterogeneity of lengths (Fig. 7A, black). The dominant species represented 20.2% of the cDNA and contained the entire LTR and two Ts likely templated by the PPT. The DNAs that extended beyond the two Ts also con-tained sequences templated by the PPT. Their presence sug-gested that wild-type RNase H inefficiently processed the PPT. Perhaps the most surprising feature of this cDNA was that a full 85% terminated with one to eight untemplated nucleo-tides, with the average being 1.8. The cDNAs with untem-plated 3⬘ ends were included in the histograms of Fig. 7 by mapping their last templated nucleotide on the X axis. Inter-estingly, the sequence of the untemplated nucleotides was largely random. This revealed that the multiple clones of each ligated species were derived from independent cDNAs. While retroviruses and LTR-retrotransposons are known to have cDNA with nontemplated additions, 85% is the largest per-centage observed (8, 22).
The mutation R786H caused specific changes in the cDNA profile. The most significant was that the major species pro-duced by wild-type Tf1-neoAI, the cDNA with two Ts beyond the LTR (Fig. 7A, position ⫺2), was reduced by 6.1-fold. R786H also caused a corresponding 3.0-fold increase in cDNA that ended with the intact PPT. It was particularly significant that the magnitude of the increases in cDNA species with the PPT matched the amount of the decrease observed in the
FIG. 4. Levels of protein and cDNA produced by the mutant ver-sions of Tf1-neoAI. (A) The cDNA produced by Tf1-neoAI with the cluster of mutations in RNase H was measured on a blot. The cDNA was digested with BstXI and probed withneo. The 2.1-kb band repre-sents theneocontaining fragment of Tf1 cDNA and the 9.5-kb band resulted from the expression plasmid. (B) The levels of IN and RT produced by the mutant versions of Tf1 were measured on immuno-blots. Antibodies specific for IN (left panel) detected the mature 56-kDa species. Antibodies specific for RT (right panel) detected the mature 60-kDa species and the 72-kDa PR-RT protein.
on November 8, 2019 by guest
http://jvi.asm.org/
major cDNA species. The cDNA ending at⫺2 represented 20.2% of the total cDNA ends produced by wild-type RT. The 6.1-fold drop was a loss of 16.9% of the total ends. The R786H mutation also caused a substantial increase in species that ended with some or all of the PPT sequence (positions⫺3 to
⫺13). The resulting increase constituted 18.2% of the total material, a number very close to the 16.9% that was lost from the cDNA ending at⫺2. These specific changes in the cDNA profile indicated that the R786H mutation inhibited removal of PPT sequences from the 5⬘end of the plus strand and as a result, the PPT sequences were templated into the 3⬘ end of the minus strand. Alternatively, the increase in the PPT se-quences could have resulted from a defect in the cleavages of the PPT RNA that select the primer.
R786 of Tf1 corresponded to I505 of HIV-1, one of the residues that makes a direct contact with the PPT hybrid in the crystal structure (30). N782 corresponded to Y501 of HIV-1, another residue that makes direct contact with the PPT hybrid. Similar to R786H, N782S also caused a dramatic reduction in the dominant species ending at position⫺2 (Fig. 7B). Here, the species ending at position⫺2 dropped by 4.2-fold, a quan-tity that represented 15.4% of the total cDNA ends. Corre-sponding increases of 2.8- and 7.5-fold were observed in cDNA ending at positions ⫺12 to ⫺13 and ⫺6, respectively. The mutation caused an increase in species ending with PPT se-quence that constituted 17.9% of the cDNA, a number similar to the 15.4% that was lost from the cDNA ending at⫺2. Thus, the magnitude of the increases in the cDNA profiles caused by N782S largely matched the decreases observed.
S749 of Tf1 RNase H corresponded to Q475 of HIV-1, a third amino acid of the primer grip that binds the RNA:DNA hybrid. The S749L mutation also caused a substantial
(5.4-fold) reduction in the cDNA ending at⫺2 that corresponded to 16.5% of the total cDNA (Fig. 7C). However in this case, no corresponding increase was detected within the window of species observed with the technique we used. The mutation R786C exhibited a more modest reduction in the species end-ing at ⫺2 that nevertheless constituted 9.9% of the total cDNA. However, no corresponding increases in other species were statistically significant (Fig. 7D). L783I was a mutation that corresponded to residue A502 of HIV-I RNase H, an amino acid adjacent to one that binds the PPT hybrid. Al-though the number of cDNA clones isolated from L783I was just 33, there was a reduction in the species ending at position
⫺2 that constituted 11.1% of the total cDNA (Fig. 7E). There was also a corresponding increase in cDNA with PPT se-quence. In this case there was an 10.8-fold increase in the species ending at⫺7 that constituted 11.0% of the total cDNA. These data are in line with those of R786H and N782S, indi-cating a defect in PPT processing. The mutation at I784T did not change the cDNA profile significantly (Fig. 7F). I784T was the only mutation that caused a reduction in the amount of cDNA on the blot, suggesting that this substitution caused a defect earlier in the process of reverse transcription.
Alterations in either the selection or the removal of the PPT could result in cDNA that contains PPT sequence at the 3⬘end of the minus strand. A defect in the selection of the PPT would introduce PPT sequence as DNA at the 5⬘ end of the plus-strand cDNA. A reduction in PPT removal would only add RNA not DNA sequence to the 5⬘ end of the plus-strand cDNA. To determine which of these two types of defects were caused by the mutations in RNase H, we analyzed the 5⬘ends of the plus-strand cDNA by primer extension. The cDNA isolated from virus-like particles was annealed to a
minus-FIG. 5. Complementation analysis of Tf1-neoAI with the mutations that clustered in RNase H. Transposition activities of strains containing two versions of Tf1 were measured using the assay described in Fig. 1B. The test plasmids contained the Tf1 versions with the mutations in RNase H expressed as Tf1-neoAI. The second plasmid expressed Tf1 INfs that lackedneoAI.
14870 ATWOOD-MOORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.46.531.74.308.2]strand primer that was then extended withTaqpolymerase in 50 cycles of polymerization. The cDNA produced by wild-type Tf1 generated an extension product that corresponded to the first nucleotide of the LTR (Fig. 8, arrow in left panel). This indicates that the selection of the PPT was precise. The cDNA from the two mutations that caused the highest levels of PPT at the 3⬘ end of the minus strand, R786H and N782S, also resulted in extension products that mapped to the first nucle-otide of the LTR (Fig. 8, arrow in right panel). Pretreatment of the cDNAs with RNase A did not change the size of the bands, indicating that the extension products were templated by DNA only. This technique was not a quantitative method for com-paring the amounts of cDNA produced and consequently, the intensities of the extension products did vary substantially.
The mutants did not produce bands corresponding to cDNA with the PPT sequence added to the beginning of the LTR (Fig. 8, asterisk). This demonstrates that the PPT sequence at the 3⬘end of the plus strand was not the result of defects in the selection of the PPT. Although a reduction in the ability of RNase H to remove the PPT could also generate an extension product corresponding to the PPT added to the beginning of the LTR, the absence of such products was likely due to the hydrolysis of RNA that occurs during the repeated cycles of heating to 95°C (Fig. 8, asterisks). Taken together, our data indicate that the mutations in RNase H caused the PPT se-quence to accumulate at the 3⬘ end of the minus strand by
inhibiting the removal of the PPT RNA from the 5⬘end of the plus strand.
The presence of the PPT sequence on the 3⬘ end of the minus strand generated by the mutants indicates that the amino acids altered in RNase H mediate the removal of the primer from the 5⬘end of the plus strand. We also asked whether the same mutations affected primer removal from the 5⬘ end of the minus strand. The mutations exhibited profiles that were very similar to that of wild-type Tf1-neoAI indicating that the altered amino acids did not contribute to processing of the primer of the minus strand (data not shown). These data will be presented elsewhere.
DISCUSSION
The genetic assays that measure cDNA recombination sep-arately from transposition were used previously to screen a library of Tf1 elements for mutations that were capable of reverse transcription but not integration (1). Although the entire transposon was mutagenized, the majority of the ele-ments identified had mutations in IN. This observation cou-pled with other analyses of the mutants demonstrated that the genetic assays were able to identify transposons that were spe-cifically defective for integration. These results suggested that the same assays used with mutagenesis of just RT could iden-tify a specific class of mutations that inhibited specialized
func-FIG. 6. Cluster of mutations in the RNase H of Tf1 correspond to amino acids of HIV-1 RT that contact the PPT hybrid. The molecular model shown was derived from the crystal structure of HIV-1 RT bound to an RNA-DNA hybrid containing the PPT (30). The five amino acids in HIV-1 RT that correspond to Tf1 amino acids altered in the cluster of mutations are labeled using both Tf1 and HIV-1 numbers.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:9.585.117.470.68.375.2]FIG. 7. 3⬘ends of minus-strand cDNA isolated from Tf1 particles. Ligation-mediated PCR was used to sequence the 3⬘ends of minus-strand cDNA produced by wild-type Tf1-neoAI and the Tf1-neoAI elements with the cluster of mutations in RNase H. The number of clones that terminated at each position within a 40 bp window was shown in a histogram. The clones were categorized by the last nucleotide derived from Tf1 sequence. The histograms compare the ends of wild-type (black) to those of the mutants. (A) R786H (blue); (B) N782S (pink); (C) S749L (purple); (D) R786C (red); (E) L783I (orange); (F) I784T (green). The asterisk indicates cDNAs from the mutants that differ significantly from that of wild-type withPvaluesⱕ0.05 as tested by chi square analyses.
14872 ATWOOD-MOORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
tions of RT. The sharp clustering of the current mutations in RNase H was another indication that the genetic assays could identify amino acids with a specific function not required for cDNA synthesis.
Tf1 with the mutations that clustered in RNase H produced cDNA that was indistinguishable in size and amount from that of wild-type Tf1. The use of a restriction digest and the loca-tion of the cut site in the cDNA was designed to provide a sensitive assay for intermediates of reverse transcription. The wild-type amounts of the BstXI fragment indicated that the mutants produced normal levels of full-length double stranded cDNA. The one exception was I784T, a mutation in the cluster that showed reduced cDNA. One possible explanation for why this mutant exhibited high levels of homologous recombination is that it produced single-stranded intermediates of heteroge-neous length that did not resolve on the blot.
Immunoblots indicated that the mutations in the cluster did not change IN levels and except for one mutation, they did not change amounts of RT either. Interestingly, L783I caused a significant reduction in the 60-kDa species of RT without af-fecting the 72-kDa PR-RT species. Although it is not known which form of RT is responsible for reverse transcription in vivo, the dramatic reduction in the 60-kDa species suggests that the 72-kDa RT has sufficient activity to produce wild-type levels of cDNA (Fig. 4A). This possibility is supported
by a study of a closely related transposon, Tf2, that found the PR-RT species was likely responsible for reverse tran-scription (7).
The cDNA produced by the mutants was characterized fur-ther to identify defects that could inhibit integration. We ex-amined the sequences at the 3⬘ ends of the cDNA because small changes could remove or reposition the CA dinucleotide that, for integration to occur, must be at the 3⬘terminus. Our initial analysis of cDNA produced by wild-type Tf1-neoAI re-vealed that the sequences at the 3⬘end of the minus strands were surprisingly heterogeneous. The dominant species con-stituted just 20% of the cDNA. In comparison, analyses of cDNA produced by HIV-1, Moloney murine leukemia virus, and the LTR-retrotransposon Ty1 indicate that 50 to 60% of the cDNA terminates precisely at the consensus site (3, 8, 22). In addition to the broad distribution of the Tf1 cDNA, another unusual feature was that 85% of the cDNAs terminated with 3⬘ untemplated additions. This is a significantly higher level than that of Ty1, where 25% of the minus-strand cDNA had non-templated additions (22). The nonnon-templated additions present at the 3⬘ends of cDNAs are thought to result from RT because in purified form it has terminal transferase activity (24, 26).
The high proportion of untemplated additions at the end of cDNA produced by wild-type Tf1-neoAI would in principle inhibit integration by placing the CA dinucleotide at an inter-nal position. Although the INs of some retroviruses and LTR-retrotransposons have processing activities that remove nucle-otides past the CA, the position of the minus-strand primer suggests that the IN of Tf1 should lack such an activity (17). Nevertheless, recent work with recombinant protein demon-strated that Tf1IN does possess a processing activity capable of removing nontemplated additions (6a). Thus, the products with termini that include an internal CA have the potential to be integrated.
The most prevalent species of cDNA produced by wild-type Tf1-neoAI terminated beyond the CA with two T’s that likely were templated by the last two A’s of the PPT. The resulting species of cDNA would have a two nucleotide extension that would have to be removed by the processing activity of IN to participate in integration. That this species is the dominant product of wild-type Tf1-neoAI is consistent the idea that it is the principal substrate for integration in vivo. Other evidence that this species ending at the⫺2 position was the major donor for integration in vivo came from analyses of cDNA produced by Tf1 with the mutations that clustered in RNase H. Of the six mutations in RNase H that were studied, five caused substan-tial reductions in the dominant species of cDNA ending at position⫺2. For R786H, R786C, and L783I, the major prod-uct was the only species to be significantly reduced. The cor-relation of reduced integration with the specific drop in the major cDNA indicates that, in vivo, this species ending at position⫺2 is likely the dominant substrate for integration.
[image:11.585.50.278.67.326.2]The changes in the sequence profiles at the 3⬘end of minus-strand cDNA revealed important information about how the mutations in RNase H altered the cDNA. Of the five muta-tions that caused significant reducmuta-tions in the major species of cDNA, N782S, L783I, and R786H, exhibited corresponding increases in cDNAs with PPT sequence. In each case, the amount of the cDNA lost from the population of the major species matched quantitatively the increase in species that
FIG. 8. Analysis of the 5⬘ end of plus-strand cDNA by primer extension. The cDNA produced by wild-type Tf1 and the mutants R786H and N782S was isolated from virus-like particles and analyzed by primer extension. The cDNAs were either not treated (⫺), or were pretreated with RNase A (⫹). The extension products were mapped using sequence ladders generated with the same primers used in the extension reactions. The triangles indicate the products that corre-sponded to the first nucleotide of the LTR. The asterisks mark the positions where products would map if PPT sequence were present at the 5⬘end of the cDNA.
on November 8, 2019 by guest
http://jvi.asm.org/
ended with the PPT sequence. This strong correlation indicates that the mutations we studied caused a severe defect in the ability of RNase H to either recognize or remove the PPT. Such a defect would cause the residual PPT sequence at the 5⬘ end of the plus strand to be templated into the 3⬘end of the minus strand. The evidence that the mutations altered the processing of the PPT is supported by the observation that all six of the mutations we identified corresponded to residues associated with the RNase H primer grip, the domain of HIV-1 RNase H that in a crystal structure binds the DNA annealed to the PPT (30). Of the three mutations that caused quantitative shifts of cDNA from the major class to species with PPT sequence, R786H and N782S correspond to amino acids of HIV-1 that interact directly with nucleotides opposite the PPT. The third mutation resulting in a quantitative shift of cDNA, L783I maps one amino acid from a contact with the nucleic acid at a residue that forms the base of a critical alpha helix (Fig. 6). Thus, L783I is also likely to disrupt specific interac-tions with nucleic acid.
Two of the mutants, S749L and R786C, showed reductions in the major cDNA species without a corresponding increase in cDNA ending with PPT sequence. Neither of these mutants exhibited corresponding increases in cDNA anywhere in the 40 nucleotide windows (Fig. 7C and 7D). Thus, S749L and R786C may have increased levels of cDNA that terminate prematurely in positions that were close to or downstream of the primers we used for ligation mediated PCR. Evidence supporting this pos-sibility for S749L was that approximately 25% of the cDNA species we sequenced terminated hundreds of nucleotides be-fore the 3⬘of the minus strand. The only reason we were able to clone and sequence these species was that tandem ligations of Rag208 allowed the product to migrate within the window of the agarose gels that we analyzed. Such tandem ligations were not detected or expected with cDNA from other mutants be-cause Rag208 was 3⬘ blocked. Apparently this block was in-complete in the ligation with cDNA from the S749L mutant.
Despite the complexities of the S749L and R786C substitu-tions, the other three mutations that reduced the amounts of the⫺2 species, N782S, L783I, and R786H, caused increases in the cDNA that ended with PPT sequences. Along with the crystal structure of HIV-1 RT, these results suggest the three amino acids in Tf1 RNase H recognize the PPT due to direct contacts they make with the nucleic acid. Damaging this ability did not cause any apparent defects in the other functions of RNase H required for reverse transcription. The function dam-aged by these mutations was likely the specific removal of the PPT RNA from the 5⬘end of the plus strand. This is supported by the preponderance of full-length PPT associated with the cDNA of R786H and N782S. However, there was a more complex possibility that the mutations move the positions of the RNase H-mediated cleavages that “select” the PPT such that the plus-strand primer was 5⬘of the PPT and as a result the PPT sequence in the plus strand would be copied into DNA. In DNA form, the PPT could not then be removed by RNase H. This scenario was ruled out by the primer extension experiment that showed there was no PPT DNA at the 5⬘end of the plus strand.
The PPT sequence at the 3⬘end of the minus-strand cDNA that resulted from the RNase H mutants was likely the reason transposition was reduced since the critical CA was no longer
at the 3⬘terminus. Even though the IN of Tf1 has processing activity, the processing activities of retroviral INs are unable to remove double-stranded sequences longer than 4 base pairs from the CA (4, 9, 13, 33). The processing activity of Tf1 IN has a similar inability to process long double-stranded se-quences distal to the CA (6a).
One of the most interesting results from these experiments was that reverse transcription could proceed through the strong stop transfers in the absence of primer degradation. This indicates that primer removal is not a necessary compo-nent of reverse transcription. A suggestion that this is true for another element is that the retrotransposon Ty1 proceeds through plus-strand transfer without first copying the tRNA primer into cDNA (12).
Our study provides strong evidence that the RNase H primer grip of Tf1 makes contacts with DNA that are specifi-cally required for the removal of the PPT. Other reports have suggested that the primer grip may have a role in recognition of the PPT. The crystal structure of HIV-1 RT in complex with the PPT RNA-DNA hybrid clearly documents direct interac-tions between the RNase H primer grip and the DNA of the RNA-DNA hybrid (30). This provided invaluable information about the amino acids that make contacts and suggests they may participate in interactions with the PPT in vivo. However, it is not clear from the structure what function these interac-tions may have in vivo and whether these interacinterac-tions are specific for PPT sequence.
More physiological support for a role of the RNase H primer grip in PPT recognition came from mutations in the C-helix of Moloney murine leukemia virus RT. This helix is adjacent to the RNase H primer grip and is absent in other retroviruses. Targeted alanine scanning mutations cause sig-nificant reductions in overall RNase H cleavage activity. How-ever, some increases occur in the two-LTR circles with PPT sequence between the LTRs (18). In the case of HIV-1, mu-tations I505A, Y501A, and N474A⫹Q475A were created in a vector system that supports single round replication (8). While I505A has little effect on virus titer, Y501A and N474A⫹Q475A reduced the titer significantly. These mutants were found to have 10-fold less initiation of DNA synthesis, a defect that is independent of the PPT. Analyses of two-LTR circles revealed that the majority of the cDNA produced by Y501A and N474A⫹Q475A had deletions in the U3 and U5 regions of the LTRs. Nevertheless, an increase in DNAs with PPT sequence between the LTRs was observed. The lack of specificity of the mutations in HIV-1 and Moloney murine leukemia virus compared to the mutations we isolated may be because the substitutions used were arbitrary and likely dis-rupted other important functions of RT.
The function of the RNase H primer grip of HIV-1 was further examined using recombinant RT with alanine muta-tions (28). Although the mutamuta-tions in the amino acids of the primer grip did not reduce DNA polymerase activity of RT, they did cause sharp reductions in polymerase independent RNase H cleavage of nonspecific RNA-DNA hybrids. How-ever, the extent of these defects were more extensive when the mutant enzymes were challenged to cleave the PPT in the context of RNA or to remove the PPT from the 5⬘ end of a plus-strand RNA-DNA chimera. These data suggested the RNase H primer grip contributes specificity to the cleavage
14874 ATWOOD-MOORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
reactions. This specificity is not as pronounced as was observed with the RNase H of Tf1, perhaps again because the substitu-tions with alanine may have had secondary effects.
There is considerable evidence that the structure of the PPT plays a key role in resisting the RNase H mediated cleavages (27). High resolution analyses of RNA-DNA hybrids with the sequence of the HIV-1 PPT identified structural anomalies (6, 10, 11). The position of the anomalies corresponds to the cleavage sites, suggesting that it may be the PPT itself that positions the cleavages. However, the mutations we isolated indicate that Tf1 RT also plays a key role in directing the PPT cleavages. Taken together, our data are the first to indicate that RT contains amino acids that function solely to recognize the PPT. If the corresponding residues in HIV-1 RT provide the same specificity, then they become an important target for antiviral strategies. Inhibitors of PPT processing in combina-tion with compounds that affect other activities of RT would significantly reduce the number of drug-resistant forms of HIV-1 RT.
ACKNOWLEDGMENTS
We thank Meenal Gore for technical assistance.
This research was supported by the Intramural Research Program of the NIH from the National Institute of Child Health and Human Development and the NIH Intramural AIDS Targeted Antiviral Program.
REFERENCES
1.Atwood, A., J. Choi, and H. L. Levin.1998. The application of a homologous recombination assay revealed amino acid residues in an LTR-retrotranspo-son that were critical for integration. J. Virol.72:1324–1333.
2.Atwood, A., J. H. Lin, and H. L. Levin.1996. The retrotransposon Tf1 assembles virus-like particles that contain excess Gag relative to integrase because of a regulated degradation process. Mol. Cell. Biol.16:338–346. 3.Bacharach, E., J. Gonsky, D. Lim, and S. P. Goff.2000. Deletion of a short,
untranslated region adjacent to the polypurine tract in moloney murine leukemia virus leads to formation of aberrant 5⬘plus-strand DNA ends in vivo. J. Virol.74:4755–4764.
4.Craigie, R., T. Fujiwara, and F. Bushman.1990. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell62:829–837.
5.Eickbush, T. H. (ed.).1994. The evolutionary biology of viruses. Raven Press, New York, N.Y.
6.Fedoroff, O. Y., Y. Ge, and B. R. Reid.1997. Solution structure of r(gag-gacug):d(CAGTCCTC) hybrid: Implications for the initiation of HIV-1 (⫹ )-strand synthesis. J. Mol. Biol.269:225–239.
6a.Hizi, A., and H. L. Levin.J. Biol. Chem., in press.
7.Hoff, E. F., H. L. Levin, and J. D. Boeke.1998. Schizosaccharomyces pombe retrotransposon Tf2 mobilizes primarily through homologous cDNA recom-bination. Mol. Cell. Biol.18:6839–6852.
8.Julias, J. G., M. J. McWilliams, S. G. Sarafianos, E. Arnold, and S. H. Hughes.2002. Mutations in the RNase H domain of HIV-1 reverse tran-scriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc. Natl. Acad. Sci. USA99:9515–9520.
9.Katz, R. A., G. Merkel, J. Kulkosky, J. Leis, and A. M. Skalka.1990. The avian retroviral IN protein is both necessary and sufficient for integrative recombination in vitro. Cell63:87–95.
10.Kopka, M. L., L. Lavelle, G. W. Han, H. L. Ng, and R. E. Dickerson.2003. An unusual sugar conformation in the structure of an RNA/DNA decamer of the polypurine tract may affect recognition by RNase H. J. Mol. Biol.
334:653–665.
11.Kvaratskhelia, M., S. R. Budihas, and S. F. J. Le Grice.2002. Pre-existing distortions in nucleic acid structure aid polypurine tract selection by HIV-1 reverse transcriptase. J. Biol. Chem.277:16689–16696.
12.Lauermann, V., and J. D. Boeke.1994. The primer transfer-rna sequence is not inherited during Ty1 retrotransposition. Proc. Natl. Acad. Sci. USA
91:9847–9851.
13.Leavitt, A. D., R. B. Rose, and H. E. Varmus.1992. Both substrate and target oligonucleotide sequences affect in vitro integration mediated by human immunodeficiency virus type 1 integrase protein produced inSaccharomyces cerevisiae. J. Virol.66:2359–2368.
14.Levin, H. L.1995. A novel mechanism of self-primed reverse transcription defines a new family of retroelements. Mol. Cell. Biol.15:3310–3317. 15.Levin, H. L.1996. An unusual mechanism of self-primed reverse
transcrip-tion requires the RNase H domain of reverse transcriptase to cleave an RNA duplex. Mol. Cell. Biol.16:5645–5654.
16.Levin, H. L., D. C. Weaver, and J. D. Boeke.1993. Novel gene expression mechanism in a fission yeast retroelement: Tf1 proteins are derived from a single primary translation product. EMBO J.12:4885–4895.
17.Levin, H. L., D. C. Weaver, and J. D. Boeke.1990. Two related families of retrotransposons fromSchizosaccharomyces pombe. Mol. Cell. Biol.10:6791– 6798.
18.Lim, D., M. Orlova, and S. P. Goff.2002. Mutations of the RNase HC helix of the Moloney murine leukemia virus reverse transcriptase reveal defects in polypurine tract recognition. J. Virol.76:8360–8373.
19.Lin, J. H., and H. L. Levin.1997. A complex structure in the mRNA of Tf1 is recognized and cleaved to generate the primer of reverse transcription. Genes Dev.11:270–285.
20.Malik, H. S., and T. H. Eickbush.1999. Modular evolution of the integrase domain in the Ty3/Gypsy class of LTR retrotransposons. J. Virol.73:5186– 5190.
21.Moreno, S., A. Klar, and P. Nurse.1991. Molecular genetic analysis of fission yeastSchizosaccharomyces pombe. Methods Enzymol.194:795–823. 22.Mules, E. H., O. Uzun, and A. Gabriel.1998. In vivo Ty1 reverse
transcrip-tion can generate replicatranscrip-tion intermediates with untidy ends. J. Virol.72:
6490–6503.
23.Mules, E. H., O. Uzun, and A. Gabriel.1998. Replication errors during in vivo Ty1 transposition are linked to heterogeneous RNase H cleavage sites. Mol. Cell. Biol.18:1094–1104.
24.Patel, P. H., and B. D. Preston.1994. Marked Infidelity of Human-Immu-nodeficiency-Virus Type-1 Reverse-Transcriptase at Rna and DNA-Tem-plate Ends. Proc. Natl. Acad. Sci. USA91:549–553.
25.Pauza, C. D., P. Trivedi, T. S. McKechnie, D. D. Richman, and F. M. Graziano.1994. 2-Ltr circular viral-DNA as a marker for human-immuno-deficiency-virus type-1 infection in-vivo. Virology205:470–478.
26.Peliska, J. A., and S. J. Benkovic.1992. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science258:1112–1118. 27.Rausch, J. W., and S. F. J. Le Grice.2004. Binding, bending and bonding’: polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. International J. Biochem. Cell. Biol.36:1752–1766. 28.Rausch, J. W., D. Lener, J. T. Miller, J. G. Julias, S. H. Hughes, and S. F. Le Grice.2002. Altering the RNase H primer grip of human immunodefi-ciency virus reverse transcriptase modifies cleavage specificity. Biochemistry
41:4856–4865.
29.Rose, M. D., F. Winston, and P. Hieter.1990. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
30.Sarafianos, S. G., K. Das, C. Tantillo, A. D. Clark, J. Ding, J. M. Whitcomb, P. L. Boyer, S. H. Hughes, and E. Arnold.2001. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J.20:1449–1461.
31.Steele, S. J., and H. L. Levin.1998. A map of interactions between the proteins of a retrotransposon. J. Virol.72:9318–9322.
32.Telesnitsky, A., and S. P. Goff.1997. Reverse transcription and the genera-tion of retroviral DNA. Cold Spring Harbor Press, Plainview, N.Y. 33.Vink, C., D. C. Vangent, Y. Elgersma, and R. H. A. Plasterk.1991. Human
immunodeficiency virus integrase protein requires a subterminal position of its viral-DNA recognition sequence for efficient cleavage. J. Virol.65:4636– 4644.
34.Xiong, Y., and T. H. Eickbush.1990. Origin and evolution of retroelements based on their reverse transcriptase sequences. EMBO J.9:3353–3362.