Proteins at the
trans
-Golgi Network To Promote Virus Replication
Kathryne E. Taylor, Karen L. Mossman
Department of Pathology and Molecular Medicine, McMaster Immunology Research Centre, Institute for Infectious Disease Research, McMaster University, Hamilton, Ontario, Canada
ABSTRACT
It has recently been proposed that the herpes simplex virus (HSV) protein ICP0 has cytoplasmic roles in blocking antiviral
sig-naling and in promoting viral replication in addition to its well-known proteasome-dependent functions in the nucleus.
How-ever, the mechanisms through which it produces these effects remain unclear. While investigating this further, we identified a
novel cytoplasmic interaction between ICP0 and the poorly characterized cellular protein WDR11. During an HSV infection,
WDR11 undergoes a dramatic change in localization at late times in the viral replication cycle, moving from defined perinuclear
structures to a dispersed cytoplasmic distribution. While this relocation was not observed during infection with viruses other
than HSV-1 and correlated with efficient HSV-1 replication, the redistribution was found to occur independently of ICP0
ex-pression, instead requiring viral late gene expression. We demonstrate for the first time that WDR11 is localized to the
trans
-Golgi network (TGN), where it interacts specifically with some, but not all, HSV virion components, in addition to ICP0.
Knock-down of WDR11 in cultured human cells resulted in a modest but consistent decrease in yields of both wild-type and ICP0-null
viruses, in the supernatant and cell-associated fractions, without affecting viral gene expression. Although further study is
re-quired, we propose that WDR11 participates in viral assembly and/or secondary envelopment.
IMPORTANCE
While the TGN has been proposed to be the major site of HSV-1 secondary envelopment, this process is incompletely
under-stood, and in particular, the role of cellular TGN components in this pathway is unknown. Additionally, little is known about the
cellular functions of WDR11, although the disruption of this protein has been implicated in multiple human diseases. Therefore,
our finding that WDR11 is a TGN-resident protein that interacts with specific viral proteins to enhance viral yields improves
both our understanding of basic cellular biology as well as how this protein is co-opted by HSV.
W
ith worldwide seroprevalence rates reaching 60 to 90% (
1
),
herpes simplex virus 1 (HSV-1) is a tremendously successful
human pathogen. HSV-1 particles consist of a double-stranded
DNA genome encased by an icosahedral capsid, surrounded by a
proteinaceous layer known as the tegument, which is in turn
en-closed by an envelope of host-derived lipids (
2
). During the lytic
replication cycle, viral glycoprotein-mediated fusion of the
enve-lope with the host cell plasma membrane releases the capsid and
tegument proteins into the cytosol (
3
). HSV capsids are then
transported to the nucleus along microtubules via molecular
mo-tor proteins (
4
). The release of the genome into the nucleus is
followed by viral gene expression in a sequential manner,
begin-ning with the immediate-early (IE) genes and then progressing to
the early (E) and late (L) classes, resulting in viral genomic DNA
replication and packaging into capsids.
Although subsequent steps have been controversial, it is now
generally accepted that capsids escape from the nucleus by first
budding from the inner nuclear membrane, acquiring a primary
envelope, and then fusing with the outer nuclear membrane,
re-leasing the unenveloped capsids into the cytoplasm (
5
,
6
). To
be-come mature particles, these capsids must then undergo
second-ary envelopment. Although the exact mechanism and subcellular
location at which this event takes place have been highly debated
(
7–12
), current evidence appears to support the
trans
-Golgi
net-work (TGN) as the major site of secondary envelopment (
8
,
9
,
13–17
). This network of tubules connected with the
trans
-face of
the Golgi apparatus functions in the secretory pathway as a sorting
station, directing cargo into distinct carriers which transport them
to their final cellular destinations (reviewed in references
18
and
19
). Although the exact details of secondary envelopment remain
unclear, interactions between the tegument proteins and
glyco-proteins that gather at the TGN and the capsid and
capsid-associ-ated tegument proteins are thought to help drive this process (
5
,
20
). The resulting enveloped viral particles within transport
vesi-cles travel to and fuse with the plasma membrane, releasing
ma-ture viruses, via the classical pathway of cargo transport from the
TGN to the cell surface (
17
).
Infected cell protein 0 (ICP0) is a multifunctional IE HSV-1
protein with roles in enhancing viral gene expression during lytic
replication, promoting reactivation from latency, and opposing
both the intrinsic and the interferon-mediated antiviral response
(reviewed in reference
21
). Traditionally, the majority of ICP0
functions have been thought to occur in the nucleus—where the
Received6 July 2015 Accepted12 July 2015
Accepted manuscript posted online15 July 2015
CitationTaylor KE, Mossman KL. 2015. Cellular protein WDR11 interacts with specific herpes simplex virus proteins at thetrans-Golgi network to promote virus replication. J Virol 89:9841–9852.doi:10.1128/JVI.01705-15.
Editor:R. M. Sandri-Goldin
Address correspondence to Karen L. Mossman, [email protected].
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01705-15
on November 7, 2019 by guest
http://jvi.asm.org/
protein localizes early in infection (
22
)—through its ability to
target proteins for degradation via the proteasome using the E3
ubiquitin ligase activity of its RING finger domain (
23–25
).
How-ever, ICP0 is found largely in the cytoplasm as the infection
pro-gresses (
26–30
), and evidence is mounting that it also has
impor-tant activities in this location (
31–33
). Interestingly, a RING
finger-independent cytoplasmic function for ICP0 in promoting
viral replication has recently been suggested (
33
), implying that
this protein may have activities that are distinct from its
func-tion in proteasome-mediated protein degradafunc-tion. However, the
mechanism through which ICP0 produces this effect remains
un-clear.
Although widely expressed in human tissues (
34
) and highly
conserved in vertebrates (
35
), the cellular protein WD repeat
do-main 11 (WDR11) is poorly understood. Disruption of the
WDR11 gene has been found in both human glioma cells (
34
) and
breast cancer cells (
36
), leading to the suggestion that WDR11
may act as a tumor suppressor, and mutations in WDR11 have
also been reported in patients with idiopathic hypogonadotropic
hypogonadism (IHH), Kallmann syndrome (KS), and combined
pituitary hormone deficiency (CPHD) (
37
), conditions
character-ized by low sex steroids and delayed puberty (
35
,
38
).
Addition-ally, depletion of WDR11 was found to sensitize cells to the
AB-type toxin ricin (
39
). Although a potential function in autophagy
for WDR11 has been proposed to explain some of these results
(
39
,
40
), the molecular mechanisms through which WDR11 is
involved in these diverse activities remain unknown.
To further characterize the mechanism of action of ICP0 in the
cytosol, we recently performed quantitative mass spectrometry to
identify proteins that coimmunoprecipitated with ICP0 from
cy-toplasmic extracts (unpublished data). One of the proteins
iden-tified in this screen was WDR11. Here, we characterize the
ICP0-WDR11 interaction in more detail. We found that ICP0-WDR11 has a
tight perinuclear distribution in uninfected human fibroblasts,
but undergoes a profound redistribution at late times
postinfec-tion in an HSV-1-specific manner. Interestingly, although ICP0
was found to interact strongly with WDR11, this protein is not
degraded during an HSV-1 infection, and the relocation of
WDR11 was found to occur independently of ICP0. To begin to
explain this finding, we show for the first time that WDR11 is
localized to the TGN and interacts specifically with some, but not
all, HSV-1 virion components. Additionally, depletion of WDR11
was found to cause a modest but reproducible decrease in the
yields of both cell-associated and secreted virus for wild-type
HSV-1 as well as an ICP0-null mutant, without affecting viral gene
expression, suggesting a possible role for this protein in viral
as-sembly in the cytoplasm or secondary envelopment.
MATERIALS AND METHODS
Cells, viruses, reagents, and plasmids.Human embryonic lung (HEL) fibroblasts and U2OS osteosarcoma cells were purchased from the Amer-ican Type Culture Collection (ATCC) and grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mML-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. Polyinosinic/poly(C) (poly I·C; GE Healthcare) was directly added to cul-ture medium at a concentration of 100g/ml. Wild-type HSV-1 strain 17 syn (41) was propagated on Vero cells, while the ICP0-null strain dl1403 (42) was grown on U2OS in the presence of 3 mM hexamethylene bisac-etamide (HMBA). Both viruses were purified on a 36% sucrose cushion. Human cytomegalovirus (HCMV; strain AD169; kindly provided by T. Compton) was propagated on HEL fibroblasts; Sendai virus (SeV; strain
Cantell) was purchased from Charles River Laboratories; and vesicular stomatitis virus expressing green fluorescent protein (VSV-GFP; kindly provided by B. Lichty) was propagated on Vero cells. All viral infections were performed for 1 h in serum-free medium at 37°C, at the following multiplicities of infection (MOIs): 10 PFU/cell for 17 syn and dl1403, 0.5 PFU/cell for HCMV, 80 hemagglutinating units (HAU)/106cells for SeV,
and 0.1 PFU/cell for VSV-GFP. Transfections were performed with 15g of pCI-D8 (57) or the empty vector and 48l Lipofectamine 2000 (Invit-rogen) per 10-cm dish of U2OS cells, according to the manufacturer’s instructions, 24 h before harvest.
Immunoprecipitation.For immunoprecipitation (IP) experiments, cytoplasmic extracts were generated as previously described (33). Five micrograms of anti-ICP0 (Virusys) or 0.8g anti-WDR11 (ab93871; Ab-cam) antibody was incubated with 20l of Dynabeads protein G (Life Technologies) for 1 h at 4°C. Beads were washed three times with immu-noprecipitation buffer (50 mM Tris [pH 8.0], 150 mM NaCl, and 1% NP-40) and incubated with 500g (HEL) or 250g (U2OS) of cytoplas-mic extract for 16 h at 4°C. Beads were washed 5 times and eluted either by boiling in sample buffer containing SDS and-mercaptoethanol (for WDR11 IPs) or via two consecutive 2-min incubations with 20l of elution buffer (100 mM glycine, pH 2.0) and neutralization with 4l 2 M Tris, pH 8.0 (for ICP0 IPs).
Western blotting.For Western blotting studies, radioimmunopre-cipitation assay (RIPA) extracts and cytoplasmic extracts were generated as previously described (33), and extract protein levels were quantified via Bradford assay (Bio-Rad Laboratories). Twenty-five-microgram aliquots of the indicated extracts were separated via electrophoresis on 10% dena-turing polyacrylamide gels, transferred onto polyvinylidene difluoride membranes (Millipore), and blocked in 5% skim milk. Blots were probed with 1:1,000 dilutions of the following primary antibodies: anti-ICP0, ICP4, ICP27, gC, and gB (Virusys Corporation), actin and ICP8 (Santa Cruz Biotechnology), WDR11, and anti-VP16 (LP1; ab110226; Abcam). Secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1:5,000, and the signals were visualized via chemiluminescence.
Immunofluorescence.For the immunofluorescence assays, HEL or U2OS cells were seeded onto coverslips and infected at 50% confluence. At the indicated times, cells were fixed with 10% formalin (Sigma), per-meabilized with 0.1% Triton X-100, and blocked with 3% goat serum, 3% bovine serum albumin, and 0.02% Tween 20. Cells were incubated with 1:250 anti-ICP0, 1:250 anti-WDR11, or 1:100 anti-TGN46 (AHP500; Se-rotec), and then 1:250 anti-rabbit Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen) or 1:250 anti-sheep Alexa Fluor 488 (Life Technologies) sec-ondary antibodies. Nuclei were stained with 1:10,000 Hoechst 33258 dye. All images were captured with a Leica DM-IRE2 inverted microscope and analyzed using Openlab software (Improvision).
siRNA transfections.For small interfering RNA (siRNA) transfec-tions, the WDR11 ON-TARGETplus SMARTpool (Dharmacon) (se-quences: 5=-GCCAAGAAAGCUCUAAAUA-3=, 5=-GGAUGUAGCAGC AGGAGUA-3=, 5=-GGUAUUGAAUGGACAAGUU-3=, and 5=-GCAGU CGUAUUCAGAGAUA-3=) and the ON-TARGETplus nontargeting pool (Dharmacon) were resuspended in sterile nuclease-free water to make 5 M stocks. Next, 25 nM siRNA was combined with 3l Dharmafect 1 (Dharmacon) in serum-free DMEM per well of a 12-well plate, according to the manufacturer’s instructions, and added to quarter-seeded (HEL) or half-seeded (U2OS) cells. Medium was replaced after 6 h. Cells were left to grow for 3 days, then challenged with 17 syn or dl1403 for 24 h in 1 ml of medium. The medium was then removed and spun at 1,500 rpm in a GH-3.8 rotor (Beckman Coulter) for 10 min to remove any cells, and the supernatant was frozen. Meanwhile, the cells were scraped into 1 ml se-rum-free medium and freeze-thawed three times.
Plaque assays.Serial dilutions of the appropriate samples were used to infect U2OS cells in the presence of HMBA and 2% human serum, and after 3 days cells were fixed with methanol and stained with Giemsa (Sigma), and plaques were counted.
on November 7, 2019 by guest
http://jvi.asm.org/
Statistical analysis.Statistical analysis was performed using Graph-Pad Prism. Where necessary, values were first adjusted via logarithmic transformation to equalize variances.
RESULTS
ICP0 interacts with the cellular protein WDR11.
In a screen for
cytoplasmic proteins that bind to ICP0, we recently identified the
poorly characterized cellular protein WDR11. To further
investi-gate this potential interaction, we first performed reciprocal
co-immunoprecipitations (co-IPs) in cells infected with wild-type
HSV-1 strain 17 syn (
Fig. 1A
). Accordingly, endogenous WDR11
was confirmed to be present after IP with an anti-ICP0 antibody,
and ICP0 was found after IP with an anti-WDR11 antibody.
Iden-tical results were also observed when cells were infected with D8/
FXE (data not shown), a virus encoding an ICP0 mutant lacking
both the RING finger of ICP0 as well as the nuclear localization
signal, which causes the protein to localize to the cytoplasm (
33
).
We next tested whether ICP0 induces the proteasome-mediated
degradation of WDR11, given that the RING finger is not required
for this interaction, by determining the levels of WDR11 after
infection (
Fig. 1B
), and we found no loss of WDR11, even at late
times postinfection with either 17 syn, which expresses high levels
of ICP0 at this time, or the ICP0-null HSV-1 strain dl1403.
WDR11 becomes relocalized at late times postinfection in an
HSV-specific but ICP0-independent manner.
We next examined
the subcellular localization of endogenous WDR11. In primary
human HEL fibroblasts (
Fig. 2A
), WDR11 is found in the
cyto-plasm in a characteristic pattern adjacent to the nucleus in
mock-treated cells. However, beginning approximately 8 h after
infec-FIG 1ICP0 interacts with WDR11 but does not cause its degradation. (A) HEL cells were infected with the indicated viruses at 10 PFU/cell for 8 h, and then samples were harvested via cytoplasmic extract and immunoprecipitated with the indicated antibodies. Eluents were then analyzed via Western blotting for ICP0 and WDR11. (B) HEL cells were infected with the indicated viruses at 10 PFU/cell for 10 h and then harvested in RIPA extract and analyzed for WDR11, ICP0, and actin levels via Western blotting.
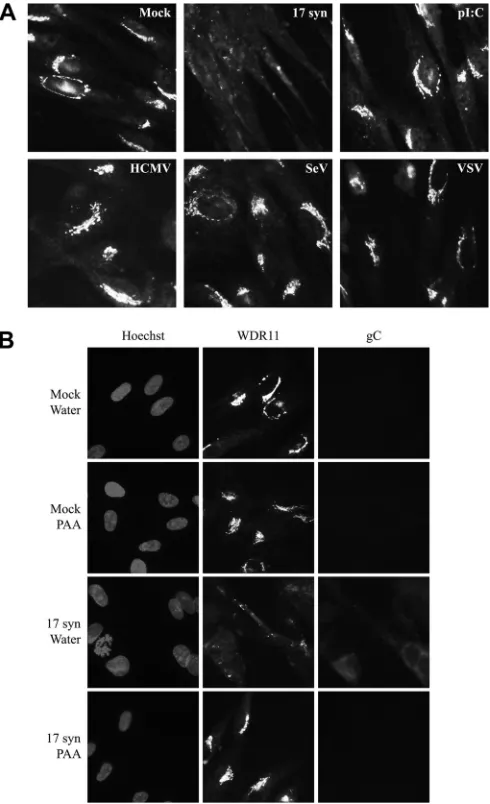
FIG 2WDR11 has a distinct perinuclear distribution that becomes dispersed at late times after HSV-1 infection in an ICP0-independent manner. HEL cells (A) or U2OS cells (B) were mock treated or infected with the indicated viruses at 10 PFU/cell for the times shown and then fixed and analyzed for WDR11 localization via immunofluorescence. Nuclei were identified using Hoechst stain.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.63.267.66.148.2] [image:3.585.111.477.330.690.2]tion with 17 syn (data not shown) and becoming pronounced by
12 h postinfection (hpi), WDR11 undergoes a dramatic
redistri-bution, losing the tight perinuclear localization and becoming
dis-persed throughout the cytoplasm. In contrast, even at the very late
time point of 24 hpi in HEL cells, no redistribution of WDR11 was
observed with dl1403, suggesting that this effect may be
depen-dent on ICP0. However, because ICP0-null viruses show a
repli-cation defect in human fibroblasts at the MOI we used here, it was
not clear whether the lack of WDR11 relocation was directly due
to the absence of ICP0 or was rather a by-product of the decreased
replication of dl1403. Therefore, the experiment was repeated in
U2OS cells (
Fig. 2B
), which complement the growth of ICP0-null
viruses (
43
). Although these highly permissive cells were
begin-ning to show morphological changes as a result of the
HSV-in-duced cytopathic effect at late times postinfection, the relocation
of WDR11 was clearly detectable with dl1043 as well as 17 syn by
12 hpi, demonstrating that this effect correlates with the efficiency
of viral replication and not specifically with the expression of
ICP0.
Given that WDR11 relocation was not dependent on ICP0
ex-pression, we next examined whether this change in localization
was a general consequence of viral infection or was due to
activa-tion of the antiviral response (
Fig. 3A
). HEL cells were therefore
infected with HCMV, VSV-GFP, or SeV at MOIs adjusted to
pro-duce approximately equal levels of cytopathic effect at 16 hpi, or
the cells were treated with the synthetic double-stranded RNA
analogue poly I·C. At that time, WDR11 redistribution was
ob-served only after infection with 17 syn and remained intact with all
other treatments. While this time point is late in the replication
cycles of SeV, VSV, and HSV (
44–48
), it occurs relatively early for
HCMV (
49
). Therefore, we also monitored WDR11 localization
over 4 days after infection with HCMV (data not shown), and
although we never observed a dispersal of WDR11 comparable to
that seen with HSV-1, we did find a subtle but consistent
relocal-ization of WDR11, in a pattern similar to the previously reported
reorganization of TGN markers into HCMV assembly sites (
50
).
To begin to determine how HSV-1 produces this effect on
WDR11, we next investigated whether the dispersal could be
pre-vented by blocking viral DNA synthesis using phosphonoacetate
(PAA) (
Fig. 3B
). As expected, this resulted in the inhibition of viral
late gene expression, as monitored by observing gC production,
and also prevented the redistribution of WDR11, implicating one
or more late gene products in this process.
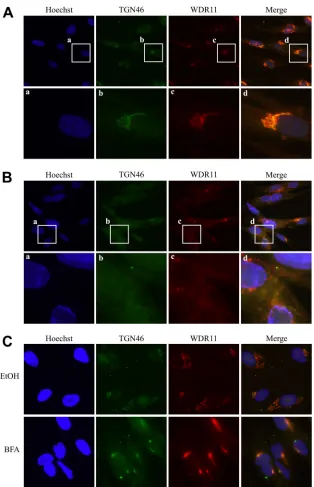
WDR11 colocalizes with the TGN marker TGN46.
This
HSV-1 late gene-specific dispersal of WDR11 in the cytoplasm at
late times postinfection is similar to the disruption of both the
Golgi apparatus and the TGN that has been reported during HSV
infections (
8
,
15
,
51–54
). Indeed, markers for both structures have
been shown to lose their tight perinuclear organization and
be-come scattered throughout the cytoplasm at approximately 8 to 12
h after infection (
15
,
52
). Therefore, we wondered if WDR11
might in fact be a TGN component, becoming redistributed upon
the dispersal of the TGN during HSV infection. To investigate
this, we examined the localization of WDR11 relative to the TGN
marker TGN46 via immunofluorescence. In uninfected cells (
Fig.
4A
), the two proteins showed strong colocalization, suggesting
that WDR11 does in fact reside in the TGN. During 17 syn
infec-tion (
Fig. 4B
), the relocation of WDR11 mirrored the dispersal of
TGN46, although the two proteins did not maintain their
colocal-ization upon breakdown of the TGN. This demonstrated, for the
first time, that WDR11 is a TGN component. To further confirm
this observation, we next treated the cells with brefeldin A (BFA),
which causes the collapse of TGN membranes (
55
), and we found
that this treatment also caused the relocalization of WDR11 into
distinctive perinuclear spots that continued to colocalize with
TGN46 (
Fig. 4C
), verifying that WDR11 resides in the TGN. We
also observed strong colocalization of WDR11 and gE at early
times postinfection, when gE has been previously shown to
local-ize to the TGN (
54
), but not at later times, when gE moves to
cell-cell junctions (data not shown).
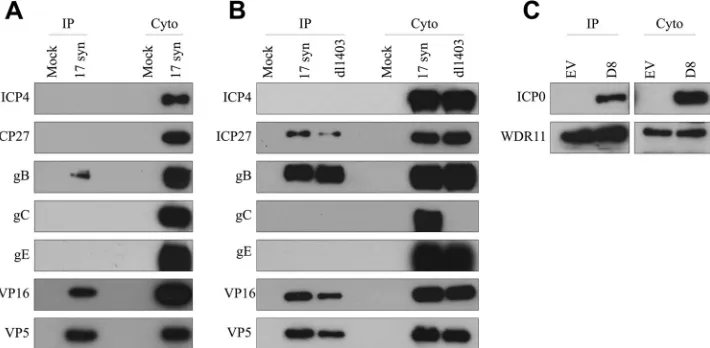
WDR11 interacts with specific additional HSV-1 proteins.
Although WDR11 is neither relocated nor degraded by ICP0, the
strong interaction between these two proteins suggests that
FIG 3WDR11 relocalization is specific to HSV infection and requires viral late gene expression. (A) HEL cells were infected with 17 syn (10 PFU/cell), HCMV (0.5 PFU/cell), SeV (80 HAU/106cells), or VSV (0.1 PFU/cell) or
treated with 100g/ml pI·C for 16 h. Infected cells were then fixed and ana-lyzed for WDR11 localization via immunofluorescence. (B) HEL cells were mock treated or infected with 17 syn at 10 PFU/cell for 10 h in the presence or absence of 400g/ml PAA and then analyzed for WDR11 localization and gC expression via immunofluorescence. Nuclei were identified using Hoechst stain.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.299.544.67.469.2]WDR11 may have a role in HSV-1 replication. To begin to
exam-ine this further, we investigated whether additional viral proteins
interacted with WDR11 via co-IP in HEL cells (
Fig. 5A
).
Intrigu-ingly, WDR11 was found to bind to gB, VP16, and VP5 in addition
to ICP0, but not to ICP4, ICP27, gC, or gE, suggesting that it
interacts specifically with some, but not all, HSV-1 proteins.
How-ever, it was possible that the proteins not detected after WDR11 IP
are simply expressed at lower levels than the interacting proteins.
To rule out this possibility, we repeated this experiment in U2OS
cells (
Fig. 5B
), which allow very high levels of expression of viral
proteins. As these cells permit efficient replication of dl1403, we
also determined whether ICP0 was required for the interaction of
the various viral proteins with WDR11. A similar pattern was
ob-served after WDR11 IP with U2OS cells compared to HEL cells,
with the exception of ICP27, which was now observed to interact
with WDR11. However, despite abundant expression, neither
gC, gE, nor ICP4 was found in the eluents, confirming our
observations that WDR11 interacts only with specific viral
pro-teins. Interestingly, there was no change in the proteins found
to interact with WDR11 in the presence or absence of ICP0. It
is important to note that no gC expression was detected after
infection with dl1403, consistent with the recent observation
that this virus contains an unintentional secondary mutation
disrupting the gC gene (
56
).
FIG 4WDR11 colocalizes with the TGN marker TGN46. HEL cells were mock treated (A), infected with 17 syn at 10 PFU/cell for 10 h (B), or treated with 10 g/ml BFA or the vehicle control for 30 min (C) and then fixed and analyzed for TGN46 and WDR11 localization via immunofluorescence.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.136.450.62.549.2]Since ICP0 expression is not necessary for ICP27, gB, VP16, or
VP5 to bind to WDR11, we were interested in determining
whether ICP0 itself requires another viral protein to bridge its
interaction with WDR11. To investigate this, we performed the
WDR11 IPs in U2OS cells transfected with pCI-D8 (
57
), which
encodes a mutant form of ICP0 lacking its nuclear localization
signal, as exogenous wild-type ICP0 expressed in the absence of
other viral proteins is restricted to the nucleus (
26
,
58
), where it
may not have access to WDR11. Interestingly, we found that ICP0
is capable of interacting with WDR11 in the absence of all other
viral proteins, demonstrating that while ICP0 is sufficient to bind
to WDR11, it is not necessary for the binding of the other viral
proteins, suggesting that multiple independent interactions occur
between WDR11 and HSV-1 virion components.
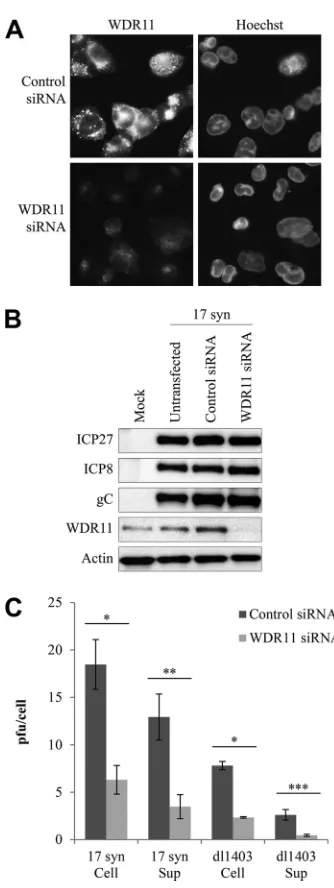
WDR11 depletion reduces viral yields without decreasing
vi-ral gene expression.
To further investigate the potential role of
WDR11 in HSV-1 replication, we used siRNA to deplete WDR11,
using U2OS cells to permit a comparison of the effect of WDR11
knockdown on the growth of both 17 syn and dl1403. Cells
de-pleted of WDR11 remained healthy, showing no changes in
mor-phology or growth rate (data not shown). While siRNA treatment
reduced WDR11 levels in U2OS cells, depletion was not complete,
which was particularly evident in infected cells (
Fig. 6A
). We next
examined the expression of IE (ICP27), E (ICP8), and L (gC)
proteins after infection with 17 syn in control cells and WDR11
siRNA-treated cells (
Fig. 6C
). No detectable change in the levels of
any of these proteins was observed after depletion of WDR11,
suggesting that any possible function of this protein in the viral
replication cycle is downstream of virus entry and gene
expres-sion. We also did not observe any change in the localization of
ICP0, ICP4, gB, gC, gE, VP5, or VP16 via immunofluorescence in
cells treated with WDR11 siRNA compared to the control-treated
cells after 10 h of infection with 17 syn (data not shown),
suggest-ing that WDR11 is not involved in directsuggest-ing the intracellular
tar-geting of viral proteins.
To investigate this further, cells treated with control or WDR11
siRNA were subsequently infected with either 17 syn or dl1403,
and cell-associated or secreted virus was separately quantified
af-ter 24 h (
Fig. 6B
). Interestingly, reducing WDR11 levels caused a
modest but reproducible decrease in both supernatant and
cell-associated virus, equally for 17 syn and dl1403. No such decrease
in titer was observed for VSV (data not shown), demonstrating
that the loss of WDR11 specifically impacts HSV replication and
does not result from nonspecific effects on cell health or density.
Therefore, WDR11 appears to promote HSV replication in an
ICP0-independent manner. Since the equal decrease in both
se-creted and cell-associated virus appears to rule out a function for
WDR11 in egress itself, these data, taken together, suggest a
po-tential role for WDR11 in viral assembly.
DISCUSSION
Currently, the function of the cellular protein WDR11 remains
unclear. A clue to its possible role is that it contains a series of
loosely conserved motifs known as WD repeat domains, making it
a member of the WD repeat family of proteins. These repeats fold
to form a propeller-like arrangement, with each WD repeat
result-ing in a blade in a circular structure (
59–61
). Despite their shared
sequence motif and structure, WD repeat proteins have a great
deal of functional diversity, and the WD repeats themselves do not
exhibit any catalytic activity (
62
). Instead, the common function
of members of the WD repeat family seems to be the coordination
of multiprotein complexes, with the propeller structure providing
a stable scaffold for several simultaneous protein-protein
interac-tions (
62
,
63
). Indeed, WDR11 has been suggested to bind to
mul-tiple proteins, including STAT3 (
64
), EMX1, Tagln2, Ndrg4,
Nrxn3, and Hey1 (
35
), C17orf75 (
39
), and UBXD7 (
65
), although
the significance of these interactions requires further study.
In this work, we identified WDR11 as a novel binding partner
for ICP0. At first glance, this interaction is particularly intriguing,
because it does not result in the loss of the WDR11 protein.
Tra-ditionally, it was thought that proteins binding to ICP0 are
tar-geted for proteasome-mediated degradation (
66–79
). However,
several binding partners have been identified that are not directed
to the proteasome by ICP0 (
27
,
80–88
). This is consistent with our
recent observation that ICP0 has two distinct cytoplasmic
func-tions, neither of which involve protein degradation; first, it blocks
FIG 5Interaction of specific additional HSV proteins with WDR11. (A and B) HEL cells (A) or U2OS cells (B) were mock treated or infected with the indicated viruses at 10 PFU/cell for 10 h. (C) U2OS cells were transfected with either the empty vector (EV) or a plasmid carrying a mutant ICP0 expressed in the cytoplasm (D8) for 24 h. In all three cases, cytoplasmic extracts were then harvested, immunoprecipitations were performed with an anti-WDR11 antibody, and eluents were analyzed via Western blotting with the indicated antibodies.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.115.470.68.242.2]antiviral signaling via a mechanism that requires the RING finger
but not the proteasome, and second, that it promotes viral
repli-cation in a RING-independent manner (
33
). The exact
mecha-nisms behind the nondegradative effects of ICP0 remain to be
fully characterized.
Although ICP0 interacts strongly with WDR11, its relocation
during HSV infection is not dependent on ICP0. Instead, WDR11
relocalization correlates with efficient viral replication. This
find-ing is analogous to what has been observed with cyclin D3 and
IFI16, ICP0-binding proteins that were initially reported to be
stabilized or degraded, respectively, in an ICP0-dependent
man-ner (
78
,
80
). However, when conditions were adjusted to equalize
replication of the ICP0-null and wild-type viruses, the alleged
“ICP0-mediated” effects were observed to occur in the absence of
ICP0 (
89
,
90
). This highlights the importance of considering viral
replication levels during ICP0-null infection.
Here, we have shown for the first time that WDR11 is a
TGN-resident protein and that its redistribution is a result of the
virus-induced fragmentation of the Golgi apparatus and TGN (
8
,
15
,
51–54
). Previous descriptions of WDR11 localization have been
variable. Endogenous WDR11 in neuroblasts was reported to be
cytoplasmic, although the precise subcellular distribution was not
determined (
35
), while exogenous, GFP-tagged WDR11 was
shown to have either a diffuse cytoplasmic distribution in U2OS
cells (
35
) or a punctate perinuclear localization in HeLa cells
which partially colocalized with the autophagosome marker LC3
(
39
). LC3 can associate with the TGN membrane during
au-tophagy (
91
), consistent with our observations. It is currently
un-known whether the dispersal of the Golgi apparatus and TGN is a
specific effect of HSV late proteins to promote egress (
52
) or
sim-ply a by-product of envelopment (
92
), as neither inhibiting nor
augmenting the fragmentation of the Golgi apparatus impacts
HSV replication in cell culture (
51
). Similarly, we observed that
late gene expression is required for WDR11 dispersal, although
whether this is a direct or indirect effect is unclear. Regardless, our
observed relocalization of WDR11 with similar kinetics to the
dis-persal of TGN46 further substantiated the idea that WDR11 is a
TGN component, as did our observation that disruption of the
TGN with BFA also caused the relocation of WDR11.
Collectively, our data suggest that HSV recruits WDR11 for
optimal assembly or secondary envelopment, in an
ICP0-inde-pendent manner. Although the effect of WDR11 depletion of virus
production was modest, the knockdown of WDR11 was not
com-plete. Moreover, observations in cultured cells do not always
re-flect the biological outcome in an animal. For example, although
HSV-1 lacking the virion host shutoff protein vhs replicates to
wild-type levels in cultured cells, it is highly attenuated in mice
(
93
). Similarly, depletion of STAT1 does not affect HSV-1
repli-cation in culture (
94
), but it renders mice highly susceptible to
infection
in vivo
(
95–100
). Furthermore, high levels of WDR11 are
found in the brains of embryonic and adult mice (
35
), and
disrup-tions to WDR11 are associated with IHH/KS/CPHD (
35
,
38
),
con-ditions that result from abnormal migration of specific neurons
during embryogenesis (reviewed in reference
101
). Additionally,
WDR11 interacts with dysbindin, a neuronal protein enriched in
synapses (
102
), as well as EMX1 (
35
), a protein with important
functions in the developing nervous system (reviewed in reference
103
). Therefore, it is possible that the function of WDR11 may be
more crucial for viral replication
in vivo
in neurons as opposed to
cells of the periphery. Indeed, the pathway to assembly and egress
in neurons is thought to require specialized adaptations, as a result
of the long distances between the cell body and the axon termini
over which HSV virions must be transported (reviewed in
refer-ence
104
). Unfortunately, the current lack of a WDR11 knockout
mouse, potentially due to embryonic lethality (
35
), precludes
di-rectly addressing the
in vivo
relevance.
FIG 6WDR11 depletion reduces yields of both cell-associated and secreted HSV-1 without decreasing viral gene expression. U2OS cells were treated with WDR11 or nontargeting control siRNA for 72 h. (A) Cells on coverslips were infected with 17 syn at 10 PFU/cell for 10 h and then fixed and analyzed for WDR11 levels via immunofluorescence. Nuclei were identified using Hoechst stain. (B) Cells were infected with 17 syn at 10 PFU/cell for 10 h, and then RIPA extracts were harvested and analyzed for the expression of the indicated pro-teins via Western blotting. (C) Cells were infected with the indicated viruses at 10 PFU/cell for 24 h. Cells and supernatant medium were then harvested separately, the cell-associated fraction was freeze-thawed three times, and then titers in both fractions were determined on U2OS cells in the presence of HMBA. The data are averages of 3 independent replicates⫾standard errors of the means. Statistical analysis was performed using one-way analysis of vari-ance and Bonferroni’s multiple-comparison posttest. *,P⬍0.05; **,P⬍0.01; ***,P⬍0.001.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.77.244.59.503.2]Perhaps the most intriguing of our observations is that WDR11
not only interacts with ICP0 but also with specific viral proteins,
including gB, VP16, and VP5, but not ICP4, gC, or gE. It is
well-known that virion components form an intricate meshwork, with
tegument proteins binding to one another as well as to the
cyto-plasmic tails of envelope glycoproteins on one side and the capsid
proteins on the other (reviewed in reference
6
). The specificity of
WDR11 binding suggests that it selectively targets particular viral
proteins and that our results cannot simply be explained by the
complex protein-protein interactions among virion components.
The basis for this selectivity is currently unclear. For example,
both ICP0 (
105
) and ICP4 (
106
) are tegument components, and
indeed, these two proteins have been found to interact directly
(
107
), but only ICP0 binds WDR11. Similarly, both gC and gB are
major virion glycoproteins (
108
), and yet only gB interacts with
WDR11. Like gC, minor virion component gE does not bind
WDR11, despite its early localization to the TGN (
54
,
109
). VP16
is a major tegument component (
108
), and interestingly, it has
been shown to associate with gB but not gC (
110
). The interaction
of WDR11 with major capsid protein VP5 is intriguing, as it
sug-gests that WDR11 not only binds specific tegument and envelope
proteins but also the capsid itself. Since VP5 is a component of the
outer shell of the capsid and has been shown to be accessible for
interactions with tegument components (
111–115
), it may also be
available for binding to WDR11. ICP27 has been reported to be
packaged in virions grown on BHK cells (
116
), and not those
grown on Vero cells (
105
,
117
), suggesting that the incorporation
of this protein may be cell type dependent, consistent with our
observations that ICP27 interacts with WDR11 in U2OS but not
HEL cells. Our results also fit with reports that gC and gB are
independently incorporated into the virion (
118
), and likewise
that ICP4 is packaged independently of ICP0 (
119
). However,
further study is clearly necessary to determine the basis for the
ability of WDR11 to discriminate between specific viral proteins.
Given that WDR11 both localizes to the TGN, a major site of
secondary envelopment (
8
,
9
,
13–17
), and specifically interacts
with several HSV virion components, and further that it is a
mem-ber of a protein family involved in coordinating multiple
simulta-neous protein-protein interactions, we propose that this protein
plays a role in HSV morphogenesis. Although WDR11 has not
been found to be among the known cellular proteins incorporated
into mature HSV virions (
120
), our hypothesis is supported by
our observations that WDR11 depletion decreases viral yields
without affecting viral gene expression, while the fact that both
secreted and cell-associated viruses were reduced after WDR11
knockdown suggests that this protein could be functioning in
re-envelopment as opposed to egress. Although the exact mechanism
is not yet clear, secondary envelopment is thought to be driven by
a complex series of protein-protein interactions between
glyco-proteins associated with the TGN membrane, tegument
compo-nents, and capsid proteins (
10
,
121–144
)—a process that could be
coordinated through the actions of WDR11. However,
transmis-sion electron microscopy (TEM) did not reveal any striking
dif-ferences in virion morphogenesis after 17 syn infection of cells
treated with either control or WDR11 siRNA (data not shown).
Although this could indicate that WDR11 is not involved in viral
assembly, it is more likely that TEM, while useful for identifying
gross defects in secondary envelopment or egress, is not
appropri-ate for detecting the subtle differences expected given the modest
decrease in the viral titer observed in the cells depleted of WDR11.
The role of cellular proteins in the reenvelopment process
re-mains poorly characterized. The ESCRT (endosomal sorting
com-plex required for transport) machinery, which has a multitude of
roles in processes involving membrane curvature and fission
(re-viewed in reference
145
), has been implicated in HSV
envelop-ment, with Vps24/CHMP3 (
10
) and Vps4 (
10
,
11
) suggested to
perform the physical budding step. Moreover, inhibiting the
ESCRT-III complex has also been reported to block secondary
envelopment (
146
). In addition, several Rab GTPases have been
implicated in HSV reenvelopment. Depletion of Rab1 and Rab43
results in decreased viral assembly, although this may be partially
explained by indirect effects, such as decreased processing of
gly-coproteins and their impaired transport to the TGN in the absence
of Rab1 and extensive disruption to the TGN structure upon
de-pletion of Rab43 (
147
). Similar to what we observed with WDR11,
Rab27a, which is involved in exocytosis and membrane trafficking
(
148
), has been found to colocalize with HSV proteins at the TGN
and increase viral yields and has therefore been suggested to play a
role in HSV morphogenesis and/or egress (
149
).
Altogether, this work identifies WDR11 as a TGN resident
pro-tein that interacts specifically with certain HSV-1 virion
compo-nents to augment viral yields, leading us to propose that this
pro-tein plays a role in viral assembly and/or secondary envelopment.
Although further study is necessary to confirm this hypothesis,
this work provides new insights into both the function of a poorly
characterized cellular protein as well as the incompletely
under-stood mechanism of HSV-1 secondary envelopment.
ACKNOWLEDGMENTS
This work is supported by Canadian Institutes of Health Research (CIHR) grant MOP-57669. K.T. was supported by a Natural Sciences and Engi-neering Research Council (NSERC) Alexander Graham Bell Canada grad-uate doctoral scholarship.
We thank P. Ezzati and K. Coombs for technical and intellectual con-tributions.
REFERENCES
1.Smith JS, Robinson NJ.2002. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis186(Suppl 1):S3–S28.http://dx.doi.org/10.1086/343739.
2.Mettenleiter TC, Muller F, Granzow H, Klupp BG.2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol15:170 –178.http://dx.doi.org/10.1111/cmi.12044.
3.Campadelli-Fiume G, Menotti L, Avitabile E, Gianni T.2012. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr Opin Virol2:28 –36.http://dx.doi.org/10.1016/j.coviro.2011.12.001. 4.Liashkovich I, Hafezi W, Kuhn JM, Oberleithner H, Shahin V.2011.
Nuclear delivery mechanism of herpes simplex virus type 1 genome. J Mol Recognit24:414 – 421.http://dx.doi.org/10.1002/jmr.1120. 5.Mettenleiter TC, Klupp BG, Granzow H.2006. Herpesvirus assembly:
a tale of two membranes. Curr Opin Microbiol9:423– 429.http://dx.doi .org/10.1016/j.mib.2006.06.013.
6.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol76:
1537–1547.http://dx.doi.org/10.1128/JVI.76.4.1537-1547.2002. 7.Siminoff P, Menefee MG.1966. Normal and
5-bromodeoxyuridine-inhibited development of herpes simplex virus. An electron microscope study. Exp Cell Res44:241–255.
8.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol79:8847– 8860.http: //dx.doi.org/10.1128/JVI.79.14.8847-8860.2005.
9.Harley CA, Dasgupta A, Wilson DW.2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J Virol75:
1236 –1251.http://dx.doi.org/10.1128/JVI.75.3.1236-1251.2001.
on November 7, 2019 by guest
http://jvi.asm.org/
10. Calistri A, Sette P, Salata C, Cancellotti E, Forghieri C, Comin A, Gottlinger H, Campadelli-Fiume G, Palu G, Parolin C.2007. Intracel-lular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J Vi-rol81:11468 –11478.http://dx.doi.org/10.1128/JVI.01364-07. 11. Crump CM, Yates C, Minson T.2007. Herpes simplex virus type 1
cytoplasmic envelopment requires functional Vps4. J Virol81:7380 – 7387.http://dx.doi.org/10.1128/JVI.00222-07.
12. Nozawa N, Yamauchi Y, Ohtsuka K, Kawaguchi Y, Nishiyama Y.2004. Formation of aggresome-like structures in herpes simplex virus type 2-infected cells and a potential role in virus assembly. Exp Cell Res299:
486 – 497.http://dx.doi.org/10.1016/j.yexcr.2004.06.010.
13. Komuro M, Tajima M, Kato K.1989. Transformation of Golgi mem-brane into the envelope of herpes simplex virus in rat anterior pituitary cells. Eur J Cell Biol50:398 – 406.
14. Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, Metten-leiter TC. 2001. Egress of alphaherpesviruses: comparative ultra-structural study. J Virol75:3675–3684.http://dx.doi.org/10.1128/JVI .75.8.3675-3684.2001.
15. Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y.2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluores-cent herpes simplex virus 1. J Virol82:5198 –5211.http://dx.doi.org/10 .1128/JVI.02681-07.
16. van Genderen IL, Brandimarti R, Torrisi MR, Campadelli G, van Meer G.1994. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology200:831– 836.http: //dx.doi.org/10.1006/viro.1994.1252.
17. Remillard-Labrosse G, Lippe R.2009. Meeting of conventional and unconventional pathways at the TGN. Commun Integr Biol2:434 – 436. http://dx.doi.org/10.4161/cib.2.5.9217.
18. Anitei M, Hoflack B.2011. Exit from the trans-Golgi network: from molecules to mechanisms. Curr Opin Cell Biol23:443– 451.http://dx.doi .org/10.1016/j.ceb.2011.03.013.
19. Gu F, Crump CM, Thomas G.2001. Trans-Golgi network sorting. Cell Mol Life Sci58:1067–1084.http://dx.doi.org/10.1007/PL00000922. 20. Henaff D, Radtke K, Lippe R.2012. Herpesviruses exploit several host
compartments for envelopment. Traffic13:1443–1449.http://dx.doi.org /10.1111/j.1600-0854.2012.01399.x.
21. Boutell C, Everett RD.2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol94:465– 481.http://dx.doi.org /10.1099/vir.0.048900-0.
22. Mullen MA, Ciufo DM, Hayward GS.1994. Mapping of intracellular localization domains and evidence for colocalization interactions be-tween the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J Virol68:3250 –3266.
23. Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol76:841– 850.http://dx.doi.org/10 .1128/JVI.76.2.841-850.2002.
24. Everett RD.2000. ICP0 induces the accumulation of colocalizing con-jugated ubiquitin. J Virol74:9994 –10005.http://dx.doi.org/10.1128/JVI .74.21.9994-10005.2000.
25. Everett RD, Orr A, Preston CM.1998. A viral activator of gene expres-sion functions via the ubiquitin-proteasome pathway. EMBO J17:7161– 7169.http://dx.doi.org/10.1093/emboj/17.24.7161.
26. Lopez P, Van Sant C, Roizman B.2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J Virol75:3832–3840.http://dx.doi.org/10.1128/JVI.75.8.3832 -3840.2001.
27. Kawaguchi Y, Bruni R, Roizman B.1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1␦: ICP0 affects translational machinery. J Virol71:1019 –1024.
28. Maul GG, Everett RD.1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rear-ranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol75:1223–1233.http://dx.doi.org/10.1099/0022-1317 -75-6-1223.
29. Everett RD.1988. Analysis of the functional domains of herpes simplex virus type 1 immediate-early polypeptide Vmw110. J Mol Biol202:87– 96.http://dx.doi.org/10.1016/0022-2836(88)90521-9.
30. Everett RD, Maul GG.1994. HSV-1 IE protein Vmw110 causes redis-tribution of PML. EMBO J13:5062–5069.
31. Liu M, Schmidt EE, Halford WP.2010. ICP0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS One5:e10975. http://dx.doi.org/10.1371/journal.pone.0010975.
32. Paladino P, Collins SE, Mossman KL.2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One5:e10428.http://dx.doi .org/10.1371/journal.pone.0010428.
33. Taylor KE, Chew MV, Ashkar AA, Mossman KL.2014. Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING fin-ger are required to block interferon-stimulated gene production but not to promote viral replication. J Virol88:8091– 8101.http://dx.doi.org/10 .1128/JVI.00944-14.
34. Chernova OB, Hunyadi A, Malaj E, Pan H, Crooks C, Roe B, Cowell JK.2001. A novel member of the WD-repeat gene family, WDR11, maps to the 10q26 region and is disrupted by a chromosome translocation in human glioblastoma cells. Oncogene20:5378 –5392.http://dx.doi.org /10.1038/sj.onc.1204694.
35. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, Ha KS, Itokawa Y, Meliciani I, Wenzel W, Lee D, Rosenberger G, Ozata M, Bick DP, Sherins RJ, Nagase T, Tekin M, Kim SH, Kim CH, Ropers HH, Gusella JF, Kalscheuer V, Choi CY, Layman LC.
2010. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet87:465– 479.http://dx.doi .org/10.1016/j.ajhg.2010.08.018.
36. Jonsson G, Staaf J, Olsson E, Heidenblad M, Vallon-Christersson J, Osoegawa K, de Jong P, Oredsson S, Ringner M, Hoglund M, Borg A.2007. High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer46:543–558.http://dx.doi.org/10.1002 /gcc.20438.
37. Izumi Y, Suzuki E, Kanzaki S, Yatsuga S, Kinjo S, Igarashi M, Maruy-ama T, Sano S, Horikawa R, Sato N, Nakabayashi K, Hata K, Umezawa A, Ogata T, Yoshimura Y, Fukami M.2014. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril102:1130 –1136.e3. http://dx.doi.org/10.1016/j.fertnstert.2014.06.017.
38. Quaynor SD, Kim HG, Cappello EM, Williams T, Chorich LP, Bick DP, Sherins RJ, Layman LC.2011. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kall-mann syndrome. Fertil Steril 96:1424 –1430.e6. http://dx.doi.org/10 .1016/j.fertnstert.2011.09.046.
39. Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, Weibezahn J, Horlbeck MA, Chen S, Mann M, Hyman AA, Leproust EM, McManus MT, Weissman JS. 2013. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell152:909 –922.http://dx.doi.org/10.1016/j.cell.2013.01.030. 40. Behrends C, Sowa ME, Gygi SP, Harper JW.2010. Network
organiza-tion of the human autophagy system. Nature466:68 –76.http://dx.doi .org/10.1038/nature09204.
41. Brown SM, Ritchie DA, Subak-Sharpe JH.1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mu-tants, their arrangement into complementation groups and recombina-tion analysis leading to a linkage map. J Gen Virol18:329 –346. 42. Stow ND, Stow EC.1986. Isolation and characterization of a herpes
simplex virus type 1 mutant containing a deletion within the gene encod-ing the immediate early polypeptide Vmw110. J Gen Virol67:2571– 2585.http://dx.doi.org/10.1099/0022-1317-67-12-2571.
43. Yao F, Schaffer PA.1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol69:6249 – 6258.
44. Kato A, Kiyotani K, Sakai Y, Yoshida T, Nagai Y.1997. The paramyxo-virus, Sendai paramyxo-virus, V protein encodes a luxury function required for viral pathogenesis. EMBO J16:578 –587.http://dx.doi.org/10.1093/emboj/16 .3.578.
45. Koyama AH, Irie H, Kato A, Nagai Y, Adachi A. 2003. Virus multiplication and induction of apoptosis by Sendai virus: role of the C proteins. Microbes Infect 5:373–378. http://dx.doi.org/10.1016 /S1286-4579(03)00043-1.
46. McClain ME, Hackett AJ.1958. A comparative study of the growth of vesicular stomatitis virus in five tissue culture systems. J Immunol80:
356 –361.
47. Cheung P, Banfield BW, Tufaro F.1991. Brefeldin A arrests the
on November 7, 2019 by guest
http://jvi.asm.org/
ration and egress of herpes simplex virus particles during infection. J Virol65:1893–1904.
48. Smith JD, De Harven E.1973. Herpes simplex virus and human cyto-megalovirus replication in WI-38 cells. I. Sequence of viral replication. J Virol12:919 –930.
49. Zerbini M, Musiani M, La Placa M.1986. Stimulating effect of heat shock on the early stage of human cytomegalovirus replication cycle. Virus Res6:211–216.http://dx.doi.org/10.1016/0168-1702(86)90070-5. 50. Cepeda V, Esteban M, Fraile-Ramos A.2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol12:386 – 404.http://dx.doi.org /10.1111/j.1462-5822.2009.01405.x.
51. Avitabile E, Di Gaeta S, Torrisi MR, Ward PL, Roizman B, Cam-padelli-Fiume G.1995. Redistribution of microtubules and Golgi appa-ratus in herpes simplex virus-infected cells and their role in viral exocy-tosis. J Virol69:7472–7482.
52. Campadelli G, Brandimarti R, Di Lazzaro C, Ward PL, Roizman B, Torrisi MR.1993. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc Natl Acad Sci U S A90:2798 –2802.http://dx.doi.org/10.1073/pnas.90.7.2798. 53. Cheng SB, Ferland P, Webster P, Bearer EL.2011. Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS One
6:e17966.http://dx.doi.org/10.1371/journal.pone.0017966.
54. McMillan TN, Johnson DC.2001. Cytoplasmic domain of herpes sim-plex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J Virol75:
1928 –1940.http://dx.doi.org/10.1128/JVI.75.4.1928-1940.2001. 55. Reaves B, Banting G.1992. Perturbation of the morphology of the
trans-Golgi network following Brefeldin A treatment: redistribution of a TGN-specific integral membrane protein, TGN38. J Cell Biol116:85–94. http://dx.doi.org/10.1083/jcb.116.1.85.
56. Cunha CW, Taylor KE, Pritchard SM, Delboy MG, Sari TK, Aguilar HC, Mossman KL, Nicola AV.2015. Widely used herpes simplex virus 1 ICP0 deletion mutant strain dl1403 and its derivative viruses do not express glycoprotein C due to a secondary mutation in the gC gene. PLoS One10:e0131129.http://dx.doi.org/10.1371/journal.pone.0311129. 57. Everett RD, Meredith M, Orr A.1999. The ability of herpes simplex
virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expres-sion and stimulation of virus replication. J Virol73:417– 426. 58. Everett RD, Parsy ML, Orr A.2009. Analysis of the functions of herpes
simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J Virol83:4963– 4977.http://dx.doi.org/10.1128/JVI.02593-08.
59. Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR.1995. The structure of the G protein heterotrimer Gi␣11␥2. Cell 83:1047–1058.http://dx.doi.org/10.1016/0092-8674(95)90220-1. 60. Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB.1996. Crystal
structure of a G-protein beta gamma dimer at 2.1 Å resolution. Nature
379:369 –374.http://dx.doi.org/10.1038/379369a0.
61. Neer EJ, Smith TF.1996. G protein heterodimers: new structures propel new questions. Cell 84:175–178. http://dx.doi.org/10.1016 /S0092-8674(00)80969-1.
62. Smith TF, Gaitatzes C, Saxena K, Neer EJ.1999. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci24:181– 185.http://dx.doi.org/10.1016/S0968-0004(99)01384-5.
63. Li D, Roberts R.2001. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci58:2085–2097.http://dx.doi.org/10.1007/PL00000838. 64. Blumert C, Kalkhof S, Brocke-Heidrich K, Kohajda T, von Bergen M,
Horn F.2013. Analysis of the STAT3 interactome using in-situ biotiny-lation and SILAC. J Proteomics94:370 –386.http://dx.doi.org/10.1016/j .jprot.2013.08.021.
65. Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ.2008. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1␣ turnover. Cell 134:804 – 816.http://dx.doi.org/10.1016/j .cell.2008.06.048.
66. Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J.1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol72:6581– 6591.
67. Chelbi-Alix MK, de The H.1999. Herpes virus induced
proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene18:935–941.http://dx.doi.org/10.1038/sj .onc.1202366.
68. Lomonte P, Sullivan KF, Everett RD.2001. Degradation of nucleo-some-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J Biol Chem276:5829 –5835. http://dx.doi.org/10.1074/jbc.M008547200.
69. Everett RD, Earnshaw WC, Findlay J, Lomonte P. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell divi-sion by herpes simplex virus immediate-early protein Vmw110. EMBO J
18:1526 –1538.http://dx.doi.org/10.1093/emboj/18.6.1526.
70. Parkinson J, Lees-Miller SP, Everett RD.1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-proteasome-dependent pro-tein kinase. J Virol73:650 – 657.
71. Boutell C, Canning M, Orr A, Everett RD.2005. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J Virol79:12342–12354. http://dx.doi.org/10.1128/JVI.79.19.12342-12354.2005.
72. Boutell C, Everett RD.2003. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and ubiquitinates p53. J Biol Chem278:36596 –36602.http://dx.doi.org/10.1074/jbc.M300776200. 73. Diao L, Zhang B, Fan J, Gao X, Sun S, Yang K, Xin D, Jin N, Geng Y,
Wang C.2005. Herpes virus proteins ICP0 and BICP0 can activate NF-B by catalyzing IB␣ubiquitination. Cell Signal17:217–229.http: //dx.doi.org/10.1016/j.cellsig.2004.07.003.
74. Kummer M, Turza NM, Muhl-Zurbes P, Lechmann M, Boutell C, Coffin RS, Everett RD, Steinkasserer A, Prechtel AT.2007. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J Virol81:
6326 – 6338.http://dx.doi.org/10.1128/JVI.02327-06.
75. van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, Kurt-Jones EA.2010. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-B signaling. J Virol84:10802–10811. http://dx.doi.org/10.1128/JVI.00063-10.
76. Fukuyo Y, Horikoshi N, Ishov AM, Silverstein SJ, Nakajima T.2011. The herpes simplex virus immediate-early ubiquitin ligase ICP0 induces degradation of the ICP0 repressor protein E2FBP1. J Virol85:3356 – 3366.http://dx.doi.org/10.1128/JVI.02105-10.
77. Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD.2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J29:943–955.http://dx.doi.org/10.1038 /emboj.2009.400.
78. Orzalli MH, DeLuca NA, Knipe DM.2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A109:E3008 –E3017.http: //dx.doi.org/10.1073/pnas.1211302109.
79. Lin AE, Greco TM, Dohner K, Sodeik B, Cristea IM.2013. A proteomic perspective of inbuilt viral protein regulation: pUL46 viral tegument protein is targeted for degradation by the viral ICP0 protein during HSV-1 infection. Mol Cell Proteomics12:3237–3252.http://dx.doi.org /10.1074/mcp.M113.030866.
80. Van Sant C, Kawaguchi Y, Roizman B. 1999. A single amino acid substitution in the cyclin D binding domain of the infected cell protein no. 0 abrogates the neuroinvasiveness of herpes simplex virus without affecting its ability to replicate. Proc Natl Acad Sci U S A96:8184 – 8189. http://dx.doi.org/10.1073/pnas.96.14.8184.
81. Kawaguchi Y, Van Sant C, Roizman B.1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol71:7328 –7336.
82. Kawaguchi Y, Tanaka M, Yokoymama A, Matsuda G, Kato K, Kagawa H, Hirai K, Roizman B.2001. Herpes simplex virus 1 alpha regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc Natl Acad Sci U S A98:1877–1882.http://dx.doi.org/10 .1073/pnas.041592598.
83. Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL.
2004. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol78:6744 – 6757.http://dx.doi .org/10.1128/JVI.78.13.6744-6757.2004.
84. Liang Y, Kurakin A, Roizman B.2005. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the
on November 7, 2019 by guest
http://jvi.asm.org/
degradation of EGF receptor from cell surfaces. Proc Natl Acad Sci U S A
102:5838 –5843.http://dx.doi.org/10.1073/pnas.0501253102.
85. Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A102:7571–7576.http://dx.doi.org/10.1073/pnas.0502658102. 86. Gu H, Roizman B.2009. The two functions of herpes simplex virus 1
ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J Virol83:181–187.http: //dx.doi.org/10.1128/JVI.01940-08.
87. Liang Y, Roizman B.2006. State and role of SRC family kinases in replication of herpes simplex virus 1. J Virol80:3349 –3359.http://dx.doi .org/10.1128/JVI.80.7.3349-3359.2006.
88. Nagel CH, Albrecht N, Milovic-Holm K, Mariyanna L, Keyser B, Abel B, Weseloh B, Hofmann TG, Eibl MM, Hauber J.2011. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for protea-somal degradation. J Virol85:7644 –7657.http://dx.doi.org/10.1128/JVI .02207-10.
89. Everett RD.2004. Herpes simplex virus type 1 regulatory protein ICP0 does not protect cyclins D1 and D3 from degradation during infection. J Virol78:9599 –9604.http://dx.doi.org/10.1128/JVI.78.18 .9599-9604.2004.
90. Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, Everett RD.2013. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol87:13422–13432.http://dx.doi.org/10.1128/JVI .02474-13.
91. Guo Y, Chang C, Huang R, Liu B, Bao L, Liu W.2012. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci125:1706 –1715.http://dx.doi.org/10.1242/jcs.093203.
92. Wisner TW, Johnson DC.2004. Redistribution of cellular and herpes simplex virus proteins from the trans-Golgi network to cell junctions without enveloped capsids. J Virol78:11519 –11535.http://dx.doi.org/10 .1128/JVI.78.21.11519-11535.2004.
93. Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW.1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med189:663– 672.http://dx.doi.org /10.1084/jem.189.4.663.
94. Everett RD, Young DF, Randall RE, Orr A.2008. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J Virol82:8871– 8881. http://dx.doi.org/10.1128/JVI.00613-08.
95. Halford WP, Weisend C, Grace J, Soboleski M, Carr DJ, Balliet JW, Imai Y, Margolis TP, Gebhardt BM.2006. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol J 3:44. http://dx.doi.org/10.1186 /1743-422X-3-44.
96. Katzenell S, Chen Y, Parker ZM, Leib DA. 2014. The differential interferon responses of two strains of Stat1-deficient mice do not alter susceptibility to HSV-1 and VSV in vivo. Virology450 – 451:350 –354. 97. Pasieka TJ, Cilloniz C, Carter VS, Rosato P, Katze MG, Leib DA.2011.
Functional genomics reveals an essential and specific role for Stat1 in protection of the central nervous system following herpes simplex virus corneal infection. J Virol85:12972–12981.http://dx.doi.org/10.1128/JVI .06032-11.
98. Pasieka TJ, Collins L, O’Connor MA, Chen Y, Parker ZM, Berwin BL, Piwnica-Worms DR, Leib DA.2011. Bioluminescent imaging reveals divergent viral pathogenesis in two strains of Stat1-deficient mice, and in ␣␥interferon receptor-deficient mice. PLoS One6:e24018.http://dx .doi.org/10.1371/journal.pone.0024018.
99. Pasieka TJ, Cilloniz C, Lu B, Teal TH, Proll SC, Katze MG, Leib DA.
2009. Host responses to wild-type and attenuated herpes simplex virus infection in the absence of Stat1. J Virol83:2075–2087.http://dx.doi.org /10.1128/JVI.02007-08.
100. Pasieka TJ, Lu B, Leib DA.2008. Enhanced pathogenesis of an attenu-ated herpes simplex virus for mice lacking Stat1. J Virol82:6052– 6055. http://dx.doi.org/10.1128/JVI.00297-08.
101. Valdes-Socin H, Rubio Almanza M, Tome Fernandez-Ladreda M, Debray FG, Bours V, Beckers A.2014. Reproduction, smell, and neu-rodevelopmental disorders: genetic defects in different hypogonado-tropic hypogonadal syndromes. Front Endocrinol (Lausanne)5:109. http://dx.doi.org/10.3389/fendo.2014.00109.
102. Han MH, Hu Z, Chen CY, Chen Y, Gucek M, Li Z, Markey SP.2014.
Dysbindin-associated proteome in the P2 synaptosome fraction of mouse brain. J Proteome Res13:4567– 4580.http://dx.doi.org/10.1021 /pr500656z.
103. Sen S, Reichert H, Vijay Raghavan K.2013. Conserved roles of ems/ Emx and otd/Otx genes in olfactory and visual system development in Drosophila and mouse. Open Biol3:120177.http://dx.doi.org/10.1098 /rsob.120177.
104. Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL.
2008. Transport and egress of herpes simplex virus in neurons. Rev Med Virol18:35–51.http://dx.doi.org/10.1002/rmv.560.
105. Yao F, Courtney RJ.1992. Association of ICP0 but not ICP27 with purified virions of herpes simplex virus type 1. J Virol66:2709 –2716. 106. Yao F, Courtney RJ.1989. A major transcriptional regulatory protein
(ICP4) of herpes simplex virus type 1 is associated with purified virions. J Virol63:3338 –3344.
107. Yao F, Schaffer PA.1994. Physical interaction between the herpes sim-plex virus type 1 immediate-early regulatory proteins ICP0 and ICP4. J Virol68:8158 – 8168.
108. Heine JW, Honess RW, Cassai E, Roizman B.1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J Virol14:640 – 651.
109. Para MF, Baucke RB, Spear PG.1982. Glycoprotein gE of herpes sim-plex virus type 1: effects of anti-gE on virion infectivity and on virus-induced fc-binding receptors. J Virol41:129 –136.
110. Zhu Q, Courtney RJ.1994. Chemical cross-linking of virion envelope and tegument proteins of herpes simplex virus type 1. Virology204:590 – 599.http://dx.doi.org/10.1006/viro.1994.1573.
111. McNabb DS, Courtney RJ.1992. Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology190:221–232. http://dx.doi.org/10.1016/0042-6822(92)91208-C.
112. Ko DH, Cunningham AL, Diefenbach RJ.2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J Virol84:1397–1405.http://dx.doi.org/10.1128/JVI.01721-09. 113. Bowman BR, Baker ML, Rixon FJ, Chiu W, Quiocho FA. 2003. Structure of the herpesvirus major capsid protein. EMBO J22:757–765. http://dx.doi.org/10.1093/emboj/cdg086.
114. Cardone G, Newcomb WW, Cheng N, Wingfield PT, Trus BL, Brown JC, Steven AC.2012. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J Virol86:
4058 – 4064.http://dx.doi.org/10.1128/JVI.00012-12.
115. Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W.1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol73:3210 –3218.
116. Maringer K, Elliott G.2010. Recruitment of herpes simplex virus type 1 immediate-early protein ICP0 to the virus particle. J Virol84:4682– 4696. http://dx.doi.org/10.1128/JVI.00126-10.
117. Sedlackova L, Rice SA.2008. Herpes simplex virus type 1 immediate-early protein ICP27 is required for efficient incorporation of ICP0 and ICP4 into virions. J Virol 82:268 –277.http://dx.doi.org/10.1128/JVI .01588-07.
118. Rodger G, Boname J, Bell S, Minson T.2001. Assembly and organiza-tion of glycoproteins B, C, D, and H in herpes simplex virus type 1 particles lacking individual glycoproteins: no evidence for the formation of a complex of these molecules. J Virol75:710 –716.http://dx.doi.org/10 .1128/JVI.75.2.710-716.2001.
119. Delboy MG, Siekavizza-Robles CR, Nicola AV.2010. Herpes simplex virus tegument ICP0 is capsid associated, and its E3 ubiquitin ligase domain is important for incorporation into virions. J Virol84:1637– 1640.http://dx.doi.org/10.1128/JVI.02041-09.
120. Loret S, Guay G, Lippe R.2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol82:8605– 8618. http://dx.doi.org/10.1128/JVI.00904-08.
121. Mettenleiter TC, Klupp BG, Granzow H.2009. Herpesvirus assembly: an update. Virus Res143:222–234.http://dx.doi.org/10.1016/j.virusres .2009.03.018.
122. Mossman KL, Sherburne R, Lavery C, Duncan J, Smiley JR.2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J Virol74:6287– 6299. http://dx.doi.org/10.1128/JVI.74.14.6287-6299.2000.
123. Baines JD, Roizman B.1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J Virol66:5168 –5174.
on November 7, 2019 by guest
http://jvi.asm.org/
124. Leege T, Fuchs W, Granzow H, Kopp M, Klupp BG, Mettenleiter TC.
2009. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J Virol83:896 –907. http://dx.doi.org/10.1128/JVI.01842-08.
125. Starkey JL, Han J, Chadha P, Marsh JA, Wills JW.2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument