0022-538X/06/$08.00⫹0 doi:10.1128/JVI.00841-06
Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Host Cell DNA Repair Pathways in Adeno-Associated Viral
Genome Processing
Vivian W. Choi,
1,2† Douglas M. McCarty,
2‡ and R. Jude Samulski
1,2*
Department of Pharmacology1and Gene Therapy Center,2University of North Carolina at Chapel Hill,
Chapel Hill, North Carolina 27599
Received 24 April 2006/Accepted 31 July 2006
Recent studies have shown that wild-type and recombinant adeno-associated virus (AAV and rAAV) genomes persist in human tissue predominantly as double-stranded (ds) circular episomes derived from input linear single-stranded virion DNA. Using self-complementary recombinant AAV (scAAV) vectors, we generated intermediates that directly transition to ds circular episomes. The scAAV genome ends are palindromic hairpin-structured terminal repeats, resembling a double-stranded break repair intermediate. Utilizing this substrate, we found cellular DNA recombination and repair factors to be essential for generating circular episomal products. To identify the specific cellular proteins involved, the scAAV circularization-dependent vector was used as a reporter in 19 mammalian DNA repair-deficient cell lines. The results show that RecQ helicase family members (BLM and WRN), Mre11 and NBS1 of the Mre11-Rad50-Nbs1 (MRN) complex, and ATM are required for efficient scAAV genome circularization. We further demonstrated that the scAAV genome requires ATM and DNA-PKCS, but not NBS1, to efficiently convert to a circular form in nondividing cells in
vivo using transgenic mice. These studies identify specific pathways involved for further elucidating viral and cellular mechanisms of DNA maintenance important to the viral life cycle and vector utilizations.
Adeno-associated virus (AAV) and AAV-derived vectors (recombinant AAV [rAAV]) deliver a single-stranded DNA genome, which must be converted into double-stranded DNA (dsDNA) by the host cell. This process is normally carried out by utilizing the hairpinned inverted terminal repeats (TRs) in a self-priming replication mechanism (3). After this conver-sion, the vector genome undergoes additional changes medi-ated by host cell recombination factors acting on the AAV TR ends (30). Most of the vector DNA circularizes, but when the dose is high enough to deliver multiple genomes to the nu-cleus, they can join end to end to form concatemers, which also can circularize (11–13). A small number of vector genomes are integrated into the host chromosome, putatively through sim-ilar recombination reactions between the TRs and chromo-somal double-strand breaks (DSBs) (30, 32, 34, 45). Though relatively little is known about the cellular factors that partic-ipate in these reactions, these recombinations ultimately gov-ern the stability of rAAV DNA and the potential for adverse interactions with genomic DNA sequences. Persistence of the vector genome in the host cell is a critical parameter for suc-cessful use of rAAV for gene delivery. In preclinical and clin-ical trials, rAAV genomes have been documented to persist in monkeys and humans for over 6 and 3.7 years, respectively (41). More recently, the molecular fate of both wild-type (wt) AAV in humans and rAAV in animals has been demonstrated to be persistence as episomal circles (5, 47, 48). This
observa-tion strongly suggests that wt and vector AAV genomes are utilizing similar host recombination pathways. While the cor-relation between long-term persistence of rAAV genomes and conversion to circular and concatemeric forms has been noted in numerous studies, it is not clear that the linear rAAV ge-nome is inherently subject to loss of gene expression through nucleolytic degradation or by other mechanisms. Rather, the linear form of the genome represents a transient episomal phase in normal cells due to the recombinogenic activity of the free DNA ends. It is therefore important to understand how mammalian DNA repair and recombination systems partici-pate in conversion of linear rAAV genomes to more stable structures.
The repair of DSBs generally involves the recognition of broken DNA ends by signaling molecules followed by repair by several proteins, each performing a specific biochemical reac-tion. DNA breaks with little or no homology can be repaired by the “cut-and-paste” mechanism of nonhomologous end joining (NHEJ), which is carried out by Ku70/80 heterodimers, DNA-dependent protein kinase (DNA-PKCS), ligase IV, XRCC4,
Artemis, Warner protein (WRN), and others. When homolo-gous sequence is available (e.g., sister chromatids), DNA breaks can be repaired with higher accuracy by homologous recombination (HR), carried out by the Mre11/Rad50/NBS1 (MRN) complex, XRCC2 and XRCC3, Rad51 and its paral-ogs, DNA polymerases and ligases, and others.
Several previous studies have demonstrated that DSB repair proteins associate with the rAAV genome and affect its fate in different ways. First, the catalytic subunit of DNA-PKCSaffects
the efficiency of rAAV genome circularization. In mice lacking DNA-PKCS(SCID mice), rAAV vector DNA recovered from
muscle contains a significant fraction of linear molecules, which are not seen in normal mice (14, 50). In contrast, rAAV DNA extracted from liver tissue in SCID mice is circularized,
* Corresponding author. Mailing address: 7119 Thurston Bowles CB#7352, University of North Carolina at Chapel Hill, NC 27599. Phone: (919) 962-3285. Fax: (919) 966-0907. E-mail: [email protected]. † Present address: CBR Institute for Biomedical Research, Chil-dren’s Hospital, Department of Genetics, Harvard Medical School, Boston, MA 02115.
‡ Present address: Center for Gene Therapy, Columbus Children’s Research Institute, The Ohio State University, Columbus, OH 43205.
10346
on November 8, 2019 by guest
http://jvi.asm.org/
though the chromosomal integration frequency of vector DNA is higher in these animals than in the livers of normal mice (51). The involvement of DNA-PKCS implicates the NHEJ
repair pathway when AAV TRs are utilized and further sug-gests that vector DNA template circularization and integration may be competing recombination end products. When AAV vectors were previously characterized for nontargeted integra-tion in dividing cells in vitro, NHEJ appeared to be the likely pathway (32, 43). Host proteins are not only involved in AAV vector persistence and wt AAV integration but also associated with other steps of viral replication. DNA DSB repair proteins Ku80 (involved in NHEJ) and Rad52 (involved in HR) are recruited to AAV nuclear replication compartments during productive infection (24, 40) and have been shown to interact with the rAAV genome, enhancing or regulating transduction (39, 68). These observations, and AAV dependency on host factors and helper virus for successful replication, reflect the virus classification asDependovirus.
The circularization of rAAV genomes in normal cells can be inhibited by coinfection with adenovirus (Ad), and this activity has been mapped to the Ad E4 region (11, 13). Two of the Ad E4 gene products, ORF3 and ORF6, augment Ad growth by preventing the end-to-end joining of the replicating genomes (60). The E4 ORF6 and the Ad E1b55K proteins form a complex that promotes the degradation of two important DNA repair-associated proteins, Mre11 and p53. The Ad E4 ORF3 protein can act alone to sequester Mre11, again inhibiting the end-to-end joining of Ad genomes. These results would sug-gest that either Mre11 or p53, or both, participates in recom-bination between AAV TRs, leading to circularization of the genome.
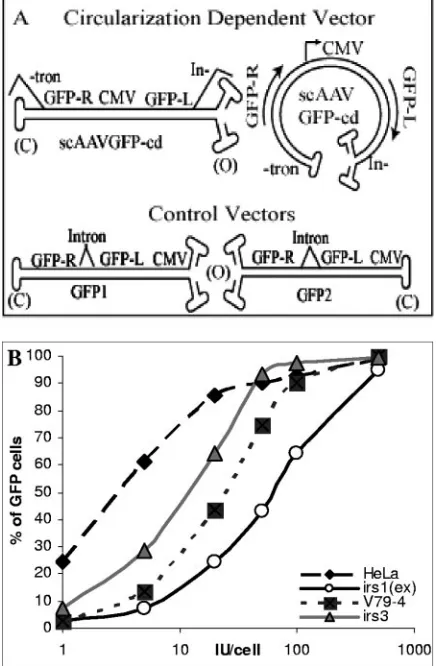
In order to systemically test the involvement of specific DNA DSB repair pathway proteins in the mechanism of AAV TR recombination, we utilized a previously developed reporter system using self-complementary AAV (scAAV) vectors (7). These vectors were designed to express green fluorescent pro-tein (GFP) only after specific recombination events such as intramolecular recombination to form circles (Fig. 1A) or spe-cific intermolecular interactions to form concatemers (7). Us-ing this system, we previously demonstrated that recombina-tion between closed hairpin ends was favored over that between open ends and that closed hairpin end recombination was more sensitive to inhibition of specific cellular factors including topoisomerase I and DNA polymerase␣/␦/ε(7).
In this study we identify cellular proteins required for scAAV TR recombination, in order to better understand which DNA repair pathways are important for this process. We utilize our previously described reporter gene rescue system to provide a sensitive genetic readout of recombination efficiency in specific DNA repair-deficient cell lines and in mutant mice. Our results support the premise that the AAV TR is recog-nized as a DNA DSB in mammalian cells, and many host DSB repair proteins, such as ATM, Mre11, NBS1, BLM, WRN, and DNA-PKCS, contribute to AAV TR recombination. In
addi-tion, using these substrates, we demonstrate for the first time correlation of AAV circle formation and transgene expression in vivo in nondividing tissue and extend this analysis to mutant mice deficient in critical host DNA repair proteins.
MATERIALS AND METHODS
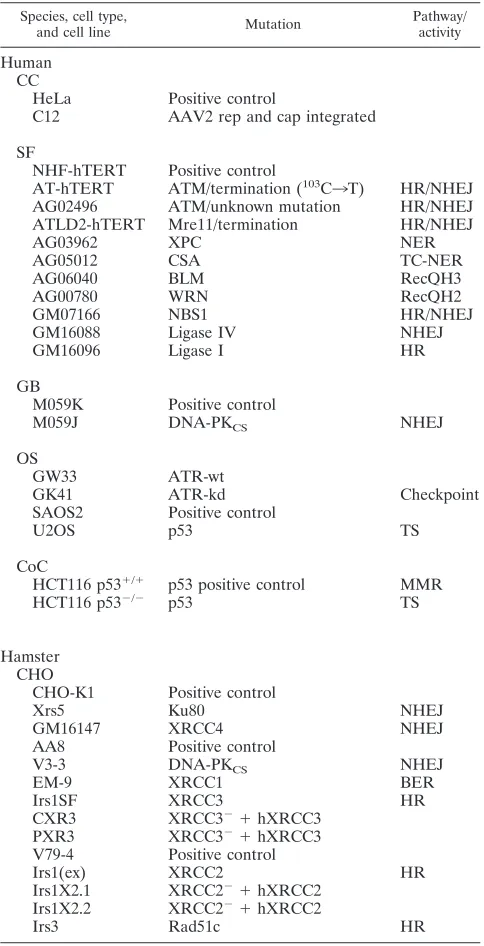
Cell lines and maintenance of cells.Cell lines used in this study were obtained from the ATCC (Manassas, VA) and the Coriell Institute (Camden, NJ) and were generously provided by other investigators (19, 23, 37, 38, 53–57). All cell
lines used in this study (Table 1) were cultured at 37°C in a 5% CO2humidified
incubator in medium and with modifications as described in Table 2. Expression of ATR-wt (GW33 cells) and ATR-kinase dead (kd) (GK41 cells) can be induced
by treating cells with 1g/ml doxycycline for 48 h prior to viral infection. Drug
treatment was continued during the course of viral infection. All cell lines used in this study can be categorized according to their cell types and origins, which are kidney fibroblast, cervical carcinoma, foreskin fibroblast, lung fibroblast, skin fibroblast, glioblastoma, osteosarcoma, colon cancer, and Chinese hamster ovary (CHO) cells (Table 1).
Viral vector construction and vector purification.The construction of viral
vectors GFP1 and GFP2 and the circularization-dependent vector
scAAVGFP-cd was described previously (7) (Fig. 1A). Viruses were generated using the triple transfection method with AAV serotype 2 capsid (16, 63) and purified by discontinuous iodixanol gradient separation and heparin chromatog-raphy (52). The viral vectors were characterized by dot blot hybridization and infectious center assays in C12 cells (8, 69).
FIG. 1. (A) Reporter system to study scAAV genome circulariza-tion. The schematic depicts the gene structures in the positive-control vectors, GFP2 and GFP1, and the circularization-dependent experi-mental vector, scAAVGFP-cd, shown in linear (left) and circular (right) conformations. CMV, cytomegalovirus. (B) Normalization of infectious dose in different recombination-deficient cell lines. A rep-resentative dose-response study was performed to determine the op-timal viral dose to be used for an scAAV circularization experiment on different cell types of recombination-deficient cell lines (Table 2). Cell lines Irs1(ex), V79-4, and Irs3 are derived from CHO cells. All of the cell lines used in this study were normalized in this way.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.585.312.530.73.406.2]Infection and quantification by flow cytometry.Circularization assays were performed by infecting cells in 6-well or 12-well plates at a low multiplicity of infection with equal infectious units of scAAVGFP-cd or the vectors containing the intact GFP cassette in either orientation (GFP1 and GFP2). The dosage was adjusted for equivalent transduction in different cell lines by prior titration with the GFP2 vector. Medium with virus was replaced with fresh medium 3 to 4 h postinfection.
Cells were harvested 24 h postinfection, washed with ice-cold phosphate-buffered saline, and fixed with 1% formaldehyde in phosphate-phosphate-buffered saline on ice. The GFP expression in transduced cells was analyzed by fluorescence-activated cell sorting using a FACScan1 (Becton-Dickinson) cytometer. Forward and side scatter setting parameters were set according to the size of the cell types,
and the setting for fluorochrome detection was adjusted so that the fluorescence intensity of uninfected negative-control cells fell within the first decade of the 4-decade log scale.
Animal procedures.Three mouse lines were used in this study. ATM trans-genic mice (background strains 129 and C57BL/6J) were a generous gift from Terry Van Dyke (University of North Carolina at Chapel Hill) (2, 15, 65).
NBS⌬B transgenic mice (background strains C57BL/6 and 129Sv) were a
gen-erous gift from John Petrini (Memorial Sloan-Kettering Cancer Center) (61). SCID mice (background strain C57BL/6J) were obtained from Jackson Labora-tory and housed in a clean cubicle due to their immunodeficiency (4). ATM and
NBS⌬B mice were bred in-house by crossing heterozygous males and females
because both sexes of ATM homozygous mice and female homozygous mice of
NBS⌬B are nonfertile. The genotypes of the animals were determined by PCR
using primers specific to the gene of interest with exon knockouts as described in previous studies (Table 3) (2, 61). Wild-type littermates were used as positive controls.
Mice were injected with vector under anesthesia at 6 to 10 weeks of age. An incision was made medial to the injection site to avoid scar formation above the
area expressing GFP. After the muscle was exposed, equal infectious units (10l
total volumes) of GFP1 and scAAVGFP-cd vector were injected manually into the right and left gastrocnemius, respectively, of each animal by using a 30-gauge needle over the course of 2 min to minimize vector leakage and dissemination.
Animal imaging procedure for GFP detection and data analysis.A Macro-molecular Illumination System (Lightools) was used to monitor the kinetics of GFP expression in the muscles of the mice over time. At each time point, mice
were anesthetized with Avertin (15l to 17l/g body weight) and the fur on top
of the gastrocnemius was chemically depilated. Images acquired were analyzed by Image J software. Briefly, the total amount of GFP light emission was calcu-lated as the area of visible signal multiplied by the average measured fluores-cence intensity. The efficiency of vector circularization was determined as a ratio of GFP expression calculated by dividing the expression level of scAAVGFP-cd vector (left leg) with the expression level of GFP1 (right leg) for each image. Individual images were taken for each mouse at each time point. The ratios calculated for mice with the same genotype were averaged and presented as means together with standard errors.
RESULTS
[image:3.585.43.285.80.553.2]To determine which DSB repair proteins participate in AAV TR recombination, we utilized the previously described circu-larization-dependent self-complementary adeno-associated vi-ral vector (scAAVGFP-cd) as a reporter system for studying recombination events (Fig. 1A) (7). Human skin fibroblast and CHO cell lines deficient in specific genes required for DSB repair previously established and characterized by other inves-tigators were obtained for this study (19, 23, 37, 38, 53–57). Since AAV is known to transduce cell lines of different origins with varying efficiency (6), we normalized the amount of virus added to each cell type for infection efficiency such that all cell lines were evaluated at the same multiplicity of infection. Us-ing AAV serotype 2 capsid, we performed titration experi-ments on each cell line with control vectors GFP1 and GFP2 as previously described (7). These vector DNA templates have contiguous cytomegalovirus-GFP expression cassettes in two orientations with respect to the closed-end TR and will express GFP regardless of the circularization status of the genome (Fig. 1A). Therefore, the number of GFP-expressing cells rep-resents the number of functional genomes delivered to the nucleus. A dose-response experiment using the positive-con-trol vectors (GFP1 and GFP2) was performed on each cell line (Table 1; data not shown), and a representative titration ex-periment is presented in Fig. 1B. As predicted from the liter-ature, even cell lines of similar origin [CHO Irs1(ex), V79-4, and Irs3] have different susceptibilities to AAV2 infection (Fig. 1B). Equivalent dose-response curves were established for all cell lines (data not shown) to ensure that a similar percentage
TABLE 1. Summary of cell lines used in the current studya
Species, cell type,
and cell line Mutation
Pathway/ activity
Human CC
HeLa Positive control
C12 AAV2 rep and cap integrated
SF
NHF-hTERT Positive control AT-hTERT ATM/termination (103C
3T) HR/NHEJ AG02496 ATM/unknown mutation HR/NHEJ ATLD2-hTERT Mre11/termination HR/NHEJ
AG03962 XPC NER
AG05012 CSA TC-NER
AG06040 BLM RecQH3
AG00780 WRN RecQH2
GM07166 NBS1 HR/NHEJ
GM16088 Ligase IV NHEJ
GM16096 Ligase I HR
GB
M059K Positive control
M059J DNA-PKCS NHEJ
OS
GW33 ATR-wt
GK41 ATR-kd Checkpoint
SAOS2 Positive control
U2OS p53 TS
CoC
HCT116 p53⫹/⫹ p53 positive control MMR
HCT116 p53⫺/⫺ p53 TS
Hamster CHO
CHO-K1 Positive control
Xrs5 Ku80 NHEJ
GM16147 XRCC4 NHEJ
AA8 Positive control
V3-3 DNA-PKCS NHEJ
EM-9 XRCC1 BER
Irs1SF XRCC3 HR
CXR3 XRCC3⫺⫹hXRCC3
PXR3 XRCC3⫺⫹hXRCC3
V79-4 Positive control
Irs1(ex) XRCC2 HR
Irs1X2.1 XRCC2⫺⫹hXRCC2 Irs1X2.2 XRCC2⫺⫹hXRCC2
Irs3 Rad51c HR
a
Abbreviations: BER, base excision repair; CC, cervical carcinoma; CoC, colon cancer; GB, glioblastoma; MMR, mismatch repair; NER, nucleotide ex-cision repair; OS, osteosarcoma; RecQH, RecQ helicase; SF, skin fibroblast; TC-NER, transcription-coupled nucleotide excision repair; TS, tumor suppres-sor; kd, kinase dead.
on November 8, 2019 by guest
http://jvi.asm.org/
of cells was infected when scAAVGFP-cd reagents were tested. In addition, in order to measure only expression due to circularization of monomeric genomes and not from concate-merized genomes, we used a multiplicity of infection that achieves only 10 to 15% infection of cells. Previously under these conditions we established that it is highly unlikely for there to be more than one genome per infected cell nucleus (7). Because all of the vectors used in this study were scAAV, which folds into a double-stranded DNA conformation without the requirement for cellular DNA synthesis, the majority of vector DNA released into the nucleus should be immediately available as functional substrates for recombination. The ob-served differences in transduction efficiencies with control vec-tor in different cell lines reflected expected variations at mul-tiple steps including attachment to the cell, internalization, and trafficking to the nucleus. There were no observed differences in transduction between the two control vectors in any one cell line. Because the control and experimental vectors differ only in the arrangement of primary sequence, and not DNA con-formation, they are expected to be identically manipulated
according to the available recombination factors specific to that cell line. In all cell line-based experiments, the number of GFP-expressing cells infected with scAAVGFP-cd was com-pared to that of cells infected with control vector GFP1 or GFP2 in order to calculate the percent circularization.
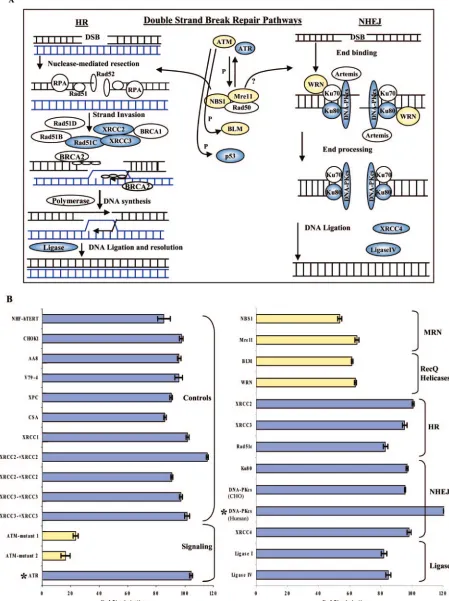
Requirement for specific DSB repair proteins in scAAV ge-nome circularization.Double-strand break repair is a multistep, multipathway mechanism involving a large number of proteins performing specific sequential enzymatic reactions. The two main DSB repair pathways are HR and NHEJ (Fig. 2A) (22, 25, 59), both of which are active to various degrees in dividing cell cul-tures.
[image:4.585.41.541.79.309.2]Figure 2B shows percent vector DNA circularization in each recombination-deficient cell line compared to the analogous wild-type cell line (NHF-hTERT for human fibroblasts and CHO-K1, A8, and V79-4 for CHO cells) and/or the parent recombination-deficient cell line complemented with the miss-ing factor (XRCC2⫺⫹XRCC2 and XRCC3⫺⫹XRCC3, cell lines that are stably transfected with cDNA of the correspond-ing wild-type misscorrespond-ing protein). We also tested cells that were
TABLE 2. Summary of medium formulations used in the current study
Formulation Cell line(s) Componentsa
1 AG05012, AG02496, AG03962, AG06040, GM07166, GM00037
Eagle’s MEM with Earle’s salts (Gibco 11095), 1⫻Pen-Strep, 15% uninactivated FBS (Sigma), and 1⫻nonessential amino acids (Gibco 11140)
2 AG000780H, GM16096, GM16088 Eagle’s MEM with Earle’s salts (Gibco 11095), 1⫻Pen-Strep, 10% uninactivated FBS (Sigma), and 1⫻nonessential amino acids (Gibco 11140)
3 M059K, M059J DMEM–F-12 (ATCC 30-2006), 1⫻Pen-Strep, and 10%
uninactivated FBS (Sigma)
4 GM16147 Ham’s F-12 (Gibco 11765), 1⫻Pen-Strep, 5% uninactivated
FBS (Sigma), and 1 mML-glutamine (Gibco 25030)
5 CHO-K1 Ham’s F-12 (Gibco 11765), 1⫻Pen-Strep, 10% uninactivated
FBS (Sigma), and 1 mML-glutamine (Gibco 25030) 6 Xrs5, AA8, V3-3, Irs1SF, Irs1SFwt5, Irs1SFwt6, EM-9 ␣-MEM (Gibco 12561), 1⫻Pen-Strep, and 10% uninactivated
FBS (Sigma)
7 HeLa, SAOS2 DMEM (Sigma D6429), 1⫻Pen-Strep, and 10% uninactivated
FBS (Sigma)
8 GW33, GK41 ␣-MEM (Gibco 12561), 1⫻Pen-Strep, 10% uninactivated
FBS (Sigma), 200g/ml G418 (Gibco 10131), and 200g/ml hygromycin B (Roche 843 555)
9 C12 DMEM (Sigma D6429), 1⫻Pen-Strep, 500g/ml G418
(Gibco 10131), and 10% uninactivated FBS (Sigma) 10 U2OS, HCT116 p53⫹/⫹, HCT116 p53⫺/⫺ McCoy’s 5A modified, 1⫻Pen-Strep, and 10% uninactivated
FBS (Sigma)
a
[image:4.585.41.543.602.724.2]Abbreviations: MEM, minimal essential medium; Pen, penicillin; Strep, streptomycin; FBS, fetal bovine serum; DMEM, Dulbecco modified Eagle medium.
TABLE 3. PCR primers and conditions for genotyping transgenic animals
Strain Primer names and sequences (melting temps) Step no. and cycling conditions
ATM IMR0013, 5⬘CTT GGG TGG AGA GGC TAT TC (59°C); IMR0014, 5⬘AGG TGA GAT GAC AGG AGA TC (54°C); IMR0640, 5⬘ GCT GCC ATA CTT GAT CCA TG (59°C); IMR0641, 5⬘TCC GAA TTT GCA GGA GTT G (60°C)
1, 94°C for 3 min; 2, 94°C for 20 s; 3, 64°C for 30 s (⫺0.5°C/cycle); 4, 72°C for 35 s (repeat steps 2 to 4 12 times); 5, 94°C for 20 s; 6, 58°C for 30 s; 7, 72°C for 35 s (repeat steps 5 to 7 25 times); 8, 72°C for 2 min; 9, 4°C
NBS⌬B mp95.a7, TCC AGT TGT TTG CGG TAA GTC CTC (64.6°C); mp95.s3, GAT AGA GTT ACC TTT GGG GTG TTT (61.2°C); p95.N, TTT GCC AAG TTC TAA TTC CAT CAG (59.4°C)
1, 94°C for 3 min; 2, 94°C for 1 min; 3, 60°C for 1 min; 4, 72°C for 1 min (repeat steps 2 to 4 34 times); 5, 72°C for 5 min; 6, 4°C
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 2. scAAV circularization experiment using recombination-deficient human and Chinese hamster ovary cells. (A) DNA double-stranded break repair pathways. The major known components of HR and NHEJ are depicted. (B) Each cell line was infected with 5 to 50 IU/cell of scAAVGFP-cd vector and the two control vectors, GFP1 and GFP2 (the values of which were averaged), in triplicate or quadruplicate in multiple experiments. The percent vector circularization was calculated from the percentage of GFP-positive cells infected with scAAVGFP-cd vector divided by the average of the percentages of GFP-positive cells infected with the two control vectors. Asterisk, percent vector circulation in mutant cells compared to that in the analygous wild-type cells, as described in Table 1 and in the text.
on November 8, 2019 by guest
http://jvi.asm.org/
wild type for DSB repair (human fibroblast XPC, CSA, and CHO cell XRCC1 lines) but with deficiencies in nuclear exci-sion repair or base exciexci-sion repair. An average of 87% and 96% of the scAAV genomes circularized in wt human fibro-blast and CHO control cell lines, respectively (Fig. 2). Repair-deficient cell lines were considered negative if the efficiency of circularization was lower than 2 standard deviations below the average of the control cell lines (Fig. 2).
Broken DNA ends are recognized by signaling molecules, which then initiate the DNA repair processes. We selected a few signaling molecules previously shown to be involved in DSB, including ataxia-telangiectasia mutated (ATM); ataxia telangiectasia, Rad3-related protein (ATR); and p53 to test for their requirement in scAAV TR recombination. The ATM-mutant 1 cell line carries an unknown mutation in ATM, and the ATM-mutant 2 cell line carries a C3T point mutation at nucleotide 103. The efficiency of circularization in these cells was 23% and 16% of wt levels, respectively, suggesting that ATM plays an important role (Fig. 2B, left panel, in yellow). In contrast, cells overexpressing ATR-wt and ATR-kinase dead (ATR-kd) proteins showed no significant differences in circu-larization, suggesting that ATR is not essential for this type of recombination (Fig. 2B, left panel, in blue).
p53 protein is another central signaling molecule believed to play a role in DNA repair that also functions as a transcription factor controlling cell cycle and apoptosis. In SAOS2 cells (p53⫺/⫺) and U2OS cells (p53⫹/⫹), we observed no difference
in circularization efficiency (data not shown). Since these two cell lines had different genetic backgrounds, we also tested isogenic cell lines, HCT116 p53⫹/⫹ and HCT116 p53⫺/⫺,
which also suggested that p53 is not required (data not shown). Since p53 is an ATM substrate (phosphorylation at Ser15), this suggested that scAAV TR recombination utilizes an ATM-dependent, p53-independent pathway. Interestingly, since HCT116 cells are mismatch repair deficient, these data also suggested that mismatch repair is not part of the scAAV TR recombination pathway.
Next, we tested the roles of NBS1 and Mre11, which are part of the MRN complex and are believed to be involved in both HR and NHEJ recombination pathways. As shown in Fig. 2B (right panel, in yellow), the efficiency of scAAV genome cir-cularization was 53% and 64%, respectively. We also tested two RecQ helicase family members, Bloom (BLM) and Warner (WRN), to assess the contribution of their DNA-unwinding functions. The efficiency of circularization in BLM and WRN cells was 62% and 64%, respectively (Fig. 2A, right panel, in yellow). These results indicated that the absence of NBS1, Mre11, BLM, or WRN partially inhibits circularization, which suggests that the MRN complex and the two RecQ helicases contribute to TR recombination.
The next group of molecules tested performs various DNA coordinating functions at sites of recombination and repair in the HR pathway (XRCC2, XRCC3, and Rad51c) or in NHEJ (Ku80, DNA-PKCS, and XRCC4). Cells deficient in XRCC2
and XRCC3, as well as Ku80 and XRCC4, were competent in scAAV genome circularization at levels similar to those of the wt cells, while the Rad51c-deficient cell line was marginally less effective (Fig. 2B, right panel, in blue). This suggests that none of these proteins are absolutely required for AAV TR recom-bination. DNA-PKCS protein, previously identified as an
im-portant factor in rAAV circularization and integration in vivo, was also tested using our reporter system in DNA-PKCS
-defi-cient CHO and human glioblastoma cell lines. Interestingly, the absence of DNA-PKCSin human MO59J cells resulted in
a slightly higher percentage of circularization than in the anal-ogous DNA-PKCS-expressing cell line (MO59K) (Fig. 2B, right
panel, *DNA-PKCS-human). Similarly, in DNA-PKCS
-defi-cient CHO cells (V3-3), circularization was not significantly reduced (Fig. 2B, right panel, DNA-PKCS-CHO). These
re-sults using the circularization-dependent GFP vector assay in vitro were inconsistent with our study (see below) and other in vivo studies using DNA-PKCS-deficient mice (50, 51),
suggest-ing that there is an alternate pathway for AAV genome circu-larization available in the DNA-PKCS-deficient cultured cells
tested in this study (see Discussion).
Finally, we tested the requirements for two mammalian DNA ligases, one involved in HR (DNA ligase I) and the other in NHEJ (DNA ligase IV). In both deficient cell lines, the efficiency of genome circularization was comparable to that of the control cells, suggesting that additional ligases are available to complete the recombination process (Fig. 2B, right panel, in blue). In conclusion, of the 19 DNA repair-deficient cell lines tested (Table 1), ATM, Mre11, NBS1, BLM, and WRN pro-teins contributed to efficient scAAV genome circularization (Fig. 2A, highlighted in yellow).
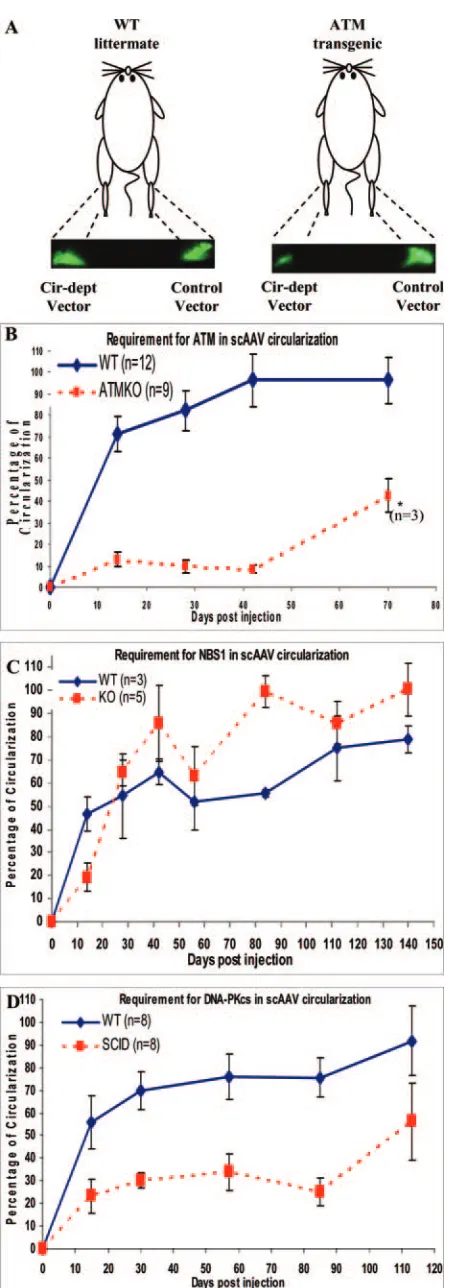
Requirement for DNA repair proteins in scAAV circulariza-tion in transgenic animals.The in vitro study using recombi-nation-deficient cell lines allowed us to screen a large number of candidate cellular molecules for their involvement in TR recombination. However, the tissue of interest for gene ther-apy usually contains nondividing or slowly dividing cells (e.g., muscle and liver). In addition, the role of these proteins in specific cell types may be either more pronounced or silenced depending on conditions (e.g., B-cell differentiation versus neuron). We therefore tested rAAV genome circularization in vivo, testing specifically in a cell type commonly used for rAAV delivery, namely, muscle, and observed GFP expression over time using a live imaging system. Because adipose can substan-tially block the excitation and emission light for GFP reporter in animal studies, the gastrocnemius muscle was chosen as a target due to its size and lack of adipose tissue. To normalize for animal-to-animal variation, the mice were injected with a control scAAV vector (GFP1) in the right leg and the circu-larization-dependent vector (scAAVGFP-cd) in the left leg (Fig. 3A). A representative image is shown in Fig. 3A (ATM wild-type littermate and transgenic knockout). The percent circularization was determined for each animal by comparing the scAAVGFP-cd vector signal to the control vector GFP1 signal and quantified using Image J software (NIH) [percent circulation ⫽ (GFP-cd signal/GFP-control signal) ⫻ 100%]. The quantification of percent circularization is graphed in Fig. 3B, C, and D. The y axis represents the average value of percent circularization. DNA repair-deficient animals (red dashed line) were plotted against wild-type littermates (blue solid line) (Fig. 3B and C) or background-matched controls (see Materials and Methods; Fig. 3D) over time. The results from the in vivo experiments are also summarized in Table 4.
ATM.Because our cultured cell assays pointed to ATM as the most significant effector of TR recombination, we tested the requirement for this factor in ATM-deficient animals. In wt
on November 8, 2019 by guest
http://jvi.asm.org/
littermates, circularization was 70% complete by day 14, climbed to 90% by 42 days, and plateaued thereafter (Fig. 3B, blue solid line). In the ATM-deficient animals, which were bred from heterozygous siblings, we observed three pheno-types of homozygous pups. One type died within 4 weeks of birth, prior to injection. The second type survived for 42 days postinjection, and the remaining third type survived up to 70 days postinjection. In the injected ATM-deficient animals, only 10% of scAAVGFP-cd vector had circularized at 14 days postinfection. This percentage remained flat till day 42 and then increased to 40% by day 70 (Fig. 3B, red dashed line). We note, however, that this increase may be due to the predispo-sition for lymphoma caused by ATM mutation (six out of nine transgenic mice died by 42 days postinjection as described above, leaving only three mice at the last time point). This would have influenced the analysis because the animals that had the highest circularization signals from the outset ulti-mately lived the longest, probably reflecting the observed vari-ation in expressivity of the mutant phenotype. Regardless, we concluded that ATM was required for efficient circularization of scAAV genomes in the muscle, consistent with our obser-vations in ATM-deficient cell lines.
[image:7.585.52.277.69.713.2]NBS1.In the NBS1-deficient cell lines, circularization was approximately half of that observed in normal cells. As NBS1 deletion is embryonic-lethal, Petrini and coworkers (61) were able to develop an NBS⌬B mouse model, in which only a subset of protein functions were inactivated by truncation of the gene. This mutant protein, although not equivalent, still resembles some of the phenotypes of the NBS1 deficiency seen in human patients. The scAAV circularization efficiency in NBS⌬B mice was approximately half that of the wild-type littermates at 2 weeks postinjection but was not significantly different at later time points (Fig. 3C). ThePvalue of thettest (P⫽ 0.0776) confirms that the result is statistically insignifi-cant. This suggested that either NBS1 is not required for TR recombination or this specific NBS⌬B mutation that carries partial function to escape embryonic lethality is sufficient in this setting. We also noted that the NBS⌬B wt littermates reached only 70% circularization at the end of the study.
FIG. 3. Kinetic study of the requirement for cellular proteins in scAAV genome circularization in vivo. (A) Illustration of direct muscle injection in transgenic and wild-type littermates and representative fluorescence images of GFP expression from the scAAV control vec-tor, GFP1, and cir-dept vector in mouse gastrocnemius using the mac-romolecular imaging system. The image was taken 28 days postinjec-tion. (B) ATM. Equal numbers of vector infectious units were injected into the gastrocnemius of the wt and deficient animals (left leg, scAAVGFP-cd vector; right leg, control GFP1 vector). Animals were imaged at the indicated time points.n⫽12 samples for wt group and n⫽ 9 samples for ATM-knockout group (P value is 0.0016). (C) NBS⌬B. The same experimental protocol as in panel B above was used. The NBS⌬B mouse model carries a different mutation than the NBS1 mutation carried in the human skin fibroblasts (see Discussion and the references).nwas three samples for the wt group and five samples for the NBS⌬B group (Pvalue is 0.0776). (D) DNA-PKCS. The same experimental protocol as in panel B above was used.nwas eight samples for the wt group and eight samples for the SCID group (P value is 0.0001). Animals were imaged at indicated time points. Statistical analysis (two-independent-sample t test) was performed, and error bars represent standard deviations of the means.
on November 8, 2019 by guest
http://jvi.asm.org/
This was different from ATM wt littermates, which reached 90% circularization (compare Fig. 3B to 3C, blue lines), demonstrating the influence of the genetic background and underscoring the importance of using the matched back-ground controls.
DNA-PKCS. In our in vitro experiments, the high
circular-ization efficiency in MO59J (DNA-PKCSdeficient) compared
to MO59K (isogenic cells with wt DNA-PKCS) cells conflicted
with several previous studies of AAV TR recombination in SCID mice. When we tested scAAV circularization in SCID mice, GFP expression from scAAVGFP-cd reached only 50% of that from the control vector, GFP1, by 14 days postinjection, and this difference was maintained through 12 weeks postin-jection (Fig. 3D). This was in general agreement with previous studies using rAAV vector in SCID muscle and liver tissue and highlighted the different requirements for specific DNA repair factors in cultured cells versus animal tissues, a theme that has been documented before with other aspects of rAAV efficiency of transgene delivery (6). Nonetheless, these animal studies demonstrate the importance of the scAAV circularization-de-pendent reagent as a molecular substrate for studying DNA repair both in vitro and in vivo.
DISCUSSION
The ability of the AAV genome to persist as episomes relies exclusively on cellular proteins to convert the linear genome into stable circular and concatemeric structures. It is inferred that these resultant DNA intermediates contribute to long-term gene expression in vivo, now over 6 years in nonhuman primates (41) and 3.7 years in humans (H. Jiang, G. F. Pierce, et al., 8th Ann. Meet. Am. Soc. Gene Therapy, abstr., 1 to 5 June 2005). With this knowledge in hand, the recombination mechanism responsible for circularization and concatemeriza-tion of AAV genomes is still poorly understood. To identify cellular proteins involved in this process, we utilized a novel scAAV GFP reporter system to assay recombination between AAV TRs in DNA repair-deficient cells and transgenic mice. Six different cellular factors were found to contribute signifi-cantly to genome circularization: ATM, Mre11, NBS1, WRN, BLM, and DNA-PKCS. ATM was required both in cultured
cells and in transgenic animals, while NBS1 was required only in cultured cells and DNA-PKCS only in animals. No single
mutation completely eliminated vector genome circularization, suggesting a great deal of redundancy in the ability of various host proteins to act on and use AAV TR structures as recom-bination substrates. This redundancy is consistent with the paramount importance of repairing DSBs in order to maintain the integrity of the host genome (57). With respect to wild-type AAV, recent studies have identified only the circular form of the viral genome persisting in human tissue (5, 48). This infor-mation is complemented by numerous studies showing long-term persistence of AAV vectors as circles and concatemers in vivo (5, 12, 47, 48, 62). The only element in common between wt AAV and rAAV vectors is thecis-acting 145-bp terminal repeat. Since AAV vectors do not carry any viral coding se-quences, the formation of circular DNA structures for both wt and vector is likely to be dependent on host mechanisms.
There are multiple pathways for DNA damage repair in the mammalian cell, principally orchestrated by three proteins of the phosphatidylinositol 3-kinase kinase family of Ser/Thr pro-tein kinases: ATM, ATR, and DNA-PKCS. Our observed
in-hibition of circularization in ATM mutants, and not in ATR mutants, supports the premise that the cell recognizes scAAV TRs as double-strand breaks. While both of these factors are important signaling molecules for DNA damage, ATM is pri-marily involved in repair of DSBs caused by ionizing radiation, while ATR responds to damage by UV irradiation or chemi-cally induced lesions (49).
[image:8.585.43.534.81.242.2]In a previous study, in which UV-irradiated rAAV was used to study the relationship between the AAV genome and cel-lular DNA repair proteins, ATR protein was implicated (24). Infection of p53-deficient cells with the UV-irradiated rAAV resulted in cell death, and the viral genome was processed by ATR but not the ATM/NBS1 pathway (24, 40). This observa-tion under these specific condiobserva-tions is consistent with the role of ATR in the repair of UV-induced DNA damage. In another study, ATM-deficient cells displayed a considerably higher transduction efficiency as well as circular and head-to-tail con-catemeric DNA structures when infected with a conventional single-stranded rAAV vector (46). In trying to reconcile these
TABLE 4. Summary of in vivo experimental results from ATM, NBS⌬B, and SCID mice
Time point and strain of animals
% Circularization (n)
compared to intact vector Conclusion
wt KOa
14 days Circularization in all transgenic animals was less efficient than that in their wt littermates at the early stage of viral infection
ATM 70 (12) 10 (9)
NBS⌬B 45 (3) 20 (5)
SCID 55 (8) 20 (8)
Last day Circularization in ATM transgenic and SCID animals was about half as
efficient as that in their wt littermates. NBS⌬B transgenic animals could convert genomes to circles at least as efficiently as their wt littermates could
ATM (70 days) 90 (12) 40 (4)
NBS⌬B (140 days) 75 (3) 90 (5)
SCID (113 days) 90 (8) 50 (5)
aKO, knockout.
on November 8, 2019 by guest
http://jvi.asm.org/
observations with decreased transduction in our study, we pro-pose three possible explanations. One, the ATM-deficient cell lines used in the two studies were different and may contain secondary mutations that have not yet been characterized. Two, the availability of recombination-independent internal control vectors (GFP1 and GFP2, Fig. 1A) allowed us to nor-malize the amount of virus required to achieve equal numbers of functional genomes per cell (e.g., Fig. 1B). This option was not available to Sanlioglu et al. (46), and as a result, we ob-served higher susceptibility of the two ATM-deficient cell lines than of the corresponding wt control cells (data not shown). Finally, we were fortunate to be able to validate our in vitro observation using transgenic animals deficient only in ATM (Fig. 3B). For these reasons, we feel that ATM does play a critical role in AAV recombination. However, even with these likely explanations we should not lose sight of the fact that AAV vectors delivering single-stranded substrates as primary vectors for the DNA genome may be acted upon in the cell differently than the more recently developed scAAV vectors. For this reason, further characterization with both substrates is warranted. Differences between single-stranded and self-com-plementary rAAVs, if any, will only enlighten the field as to how these dependoviruses interact with cellular DNA repair functions.
Several previous studies also implicated DNA-PKCSin
pro-cessing rAAV genomes in mouse muscle and liver. In SCID mice, lacking DNA-PKCS, circularization of rAAV was
inhib-ited in muscle (14, 50), though not in liver (51). However, chromosomal integration of vector DNA was increased in liver, possibly due to a delay in circularization. We also found that DNA-PKCSwas important for rAAV circularization in SCID
mouse muscle but was not required in deficient cell cultures. DNA-PKCSis a central component of the NHEJ repair
mech-anism, which is the predominant pathway of DSB repair in mammalian cells through all phases of the cell cycle, though its greatest impact is in G0/G1, when HR is not available (9, 21,
42). Therefore, our observation that the DNA-PKCS
pheno-type is observable only in the nondividing cells of the mouse muscle in vivo is consistent with the importance of NHEJ in these cells. In contrast, repair of DSB by HR could compensate for lack of NHEJ in our DNA-PKCS-deficient dividing cell
cultures, providing a logical explanation for why we observed different results with our circularization-dependent assay in vitro versus in vivo.
Two factors from the MRN complex, Mre11 and NBS1, also contributed to AAV TR recombination in cell lines. However, the NBS1 mutation available in transgenic animals did not affect GFP reporter recombination in muscle cells of these animals. The differential requirements for NBS1 in muscle versus cell cultures may again relate to the different repair pathways operating in nondividing versus dividing cells. It also draws attention to the fact that the NBS1 mice required the use of a selective mutant protein in order to escape the embryonic lethality of the true NBS1-knockout phenotype (61). The MRN complex is primarily associated with ATM-initiated HR repair, though it may have an overlapping role in NHEJ initi-ated by the DNA-PK complex (26). Therefore, we would ex-pect to see the greatest effect on scAAV circularization in repair-deficient cell lines and not in the muscle. In addition to its 3⬘-to-5⬘exonuclease activity on dsDNA, Mre11 is an
endo-nuclease that nicks non-base-paired regions of both dsDNA hairpins and stem-loop structures (10). The dependence on Mre11 for efficient AAV genome circularization in our in vitro study suggests that mammalian hairpin endonucleases play an important role in AAV TR recombination, and the MRN complex may serve this role in the context of HR-mediated repair. An important role for Mre11 in AAV TR recombina-tion is also consistent with the previous observarecombina-tion that coin-fection with Ad inhibits circularization of rAAV genomes (11, 13). While the Ad E4 gene products can sequester or degrade both Mre11 and p53, our results suggest that only Mre11 is required for this effect, since p53-deficient cells circularized the rAAV genome efficiently.
The observation that the ATM protein had a profound effect on scAAV circularization in muscle cells is intriguing, since it is primarily associated with HR repair. However, recent find-ings strongly suggest that ATM has an important signaling role in the repair of a subset of DSBs through the NHEJ pathway (26). These putatively complex lesions represent approxi-mately 10% of DSBs generated by ionizing radiation and are repaired with a slower kinetic in association with the Artemis protein. ATM-activated Artemis carries out multiple DNA-processing activities including hairpin nicking, endonucleolytic resection of overhanging 5⬘ and 3⬘ ends, and resolution of DNA flap structures (27). This suggests the possibility that the processing of one or both types of scAAV ends (open or closed hairpin structures) requires the Artemis protein, at least in the context of the NHEJ repair pathway. On the other hand, ATM has many additional downstream signaling targets in response to DNA damage (57), and these may contribute directly or indirectly to efficient AAV TR recombination.
Two additional gene products, WRN and BLM, were found to effect scAAV circularization in deficient cell lines. Both of these proteins belong to the RecQ family of helicases, which preferentially bind to and unwind substrates mimicking DNA replication and recombination intermediates (20, 58). Both WRN and BLM exhibit 3⬘-to-5⬘ helicase activity and strand-annealing activities, promoting the strand exchange step in homologous recombination (28, 29). The WRN protein also has 3⬘-to-5⬘ exonuclease activity. WRN colocalizes with both DSB repair and DNA replication sites and may function in restarting stalled or collapsed replication forks. A recent re-port suggests that WRN does not participate in NHEJ but has an essential role in single-strand annealing (SSA), an alternate form of HR (28, 66). In SSA, the 5⬘DNA ends are unwound or degraded to expose 3⬘single-strand tails, as in classical HR, but instead of forming a synapse with a sister chromatid, the two exposed ends are base paired with one another via small regions of chance homology. The nonhomologous tails are then trimmed off, and the two ends are ligated. Repair by SSA does not require Rad51, which mediates the strand invasion step of classical HR, but does involve the MRN complex (17, 66). This is consistent with the requirements for Mre11 and NBS1, but not for Rad51c, XRCC2, and XRCC3, which are all specific for classical HR, in our circularization assays in defi-cient cell lines. A role for SSA in AAV TR end joining is particularly attractive since rAAV-host chromosome DNA junctions are characterized by just such microhomologies (2 to 6 bp) (30, 44, 67). In the context of rAAV circularization, the homologies within the internal palindromes of each TR could
on November 8, 2019 by guest
http://jvi.asm.org/
serve to coordinate the two ends, giving rise to a “double D” terminal repeat structure previously described in AAV recom-bination intermediates and making up a large component of rAAV circularization junction products (13, 64).
Like WRN, the BLM protein promotes strand exchange and branch migration through combined helicase and strand an-nealing activities (1). It has been specifically associated with mitotic HR through synthesis-dependent strand annealing in
Drosophila melanogaster, wherein an exposed 3⬘tail invades the
duplex DNA on the sister chromatid and is extended by DNA polymerase␦(31). While in meiotic cells these structures are generally resolved by crossover recombination, in mitotic cells the newly synthesized strand is displaced and base paired to the 3⬘ tail of its associated broken DNA end (35). While it is unclear if or how synthesis-dependent strand annealing might contribute to recombination between two AAV TRs, the large-scale net synthesis of DNA from a recombination partner could explain the chromosomal duplications that have been observed in some rAAV integration junctions (33). In a sim-pler model of scAAV circularization, the contributions of both these RecQ helicases may derive from the general resem-blances between the open-end AAV TR and that of a stalled DNA replication fork, or they may act to resolve recombina-tion intermediates.
In summary, we present evidence that recombination be-tween AAV TRs can proceed through multiple pathways, in-cluding NHEJ in quiescent cells and HR in dividing cells. There is suggestive evidence that the HR component may rely on the SSA mechanism. Further, we suggest that similar mech-anisms of recombination contribute to the random integration of rAAV vector DNA at sites of chromosomal DSB repair. Since circular intermediates have been identified in wt AAV infection (5, 48) and from vector delivery (47) and since the TR is the only sequence element shared between these viral DNA substrates, the recombination mechanism occurring at the TR becomes crucial in understanding AAV genome persistency. Similar to other viral vectors having an uncontrolled risk of insertional mutagenesis (18, 36), only a complete characteriza-tion of the mechanisms and kinetics of these recombinacharacteriza-tion pathways will allow an assessment of the potential for rAAV gene therapy genotoxicity. In this study, we described a novel AAV molecular substrate (scAAVGFP-cd) and assayed for specific host DNA recombination proteins involved in AAV persistence both in vitro and in vivo. The data and substrates described in this study should facilitate our working under-standing of rAAV persistence and allow further study of such mechanisms in various target tissues (e.g., brain, heart, lung, and liver) currently being tested for rAAV human gene delivery.
ACKNOWLEDGMENTS
We thank Dale Ramsden and William Kaufmann for helpful dis-cussion.
This paper is dedicated to Jerri Coleman for her tireless contribu-tion to the UNC Gene Therapy Center.
This work was supported in part by NIH grants GM059299, HL051818, and HL066973 awarded to Jude Samulski and NIH grant AI048074 awarded to Douglas McCarty.
REFERENCES
1.Adams, M. D., M. McVey, and J. J. Sekelsky.2003. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science
299:265–267.
2.Barlow, C., S. Hirotsune, R. Paylor, M. Liyanage, M. Eckhaus, F. Collins, Y. Shiloh, J. N. Crawley, T. Ried, D. Tagle, and A. Wynshaw-Boris.1996.
Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell86:159–171.
3.Berns, K. I., and R. M. Linden.1995. The cryptic life style of
adeno-associated virus. Bioessays17:237–245.
4.Blunt, T., N. J. Finnie, G. E. Taccioli, G. C. Smith, J. Demengeot, T. M. Gottlieb, R. Mizuta, A. J. Varghese, F. W. Alt, P. A. Jeggo, et al.1995. Defective DNA-dependent protein kinase activity is linked to V(D)J recom-bination and DNA repair defects associated with the murine scid mutation.
Cell80:813–823.
5.Chen, C. L., R. L. Jensen, B. C. Schnepp, M. J. Connell, R. Shell, T. J. Sferra, J. S. Bartlett, K. R. Clark, and P. R. Johnson.2005. Molecular
character-ization of adeno-associated viruses infecting children. J. Virol.79:14781–14792.
6.Choi, V. W., D. M. McCarty, and R. J. Samulski.2005. AAV hybrid serotypes:
improved vectors for gene delivery. Curr. Gene Ther.5:299–310.
7.Choi, V. W., R. J. Samulski, and D. M. McCarty.2005. Effects of
adeno-associated virus DNA hairpin structure on recombination. J. Virol.79:6801–
6807.
8.Clark, K. R., F. Voulgaropoulou, and P. R. Johnson.1996. A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity
titra-tion of adeno-associated virus vectors. Gene Ther.3:1124–1132.
9.Couedel, C., K. D. Mills, M. Barchi, L. Shen, A. Olshen, R. D. Johnson, A. Nussenzweig, J. Essers, R. Kanaar, G. C. Li, F. W. Alt, and M. Jasin.2004. Collaboration of homologous recombination and nonhomologous end-join-ing factors for the survival and integrity of mice and cells. Genes Dev.
18:1293–1304.
10.D’Amours, D., and S. P. Jackson.2002. The Mre11 complex: at the cross-roads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol.
3:317–327.
11.Duan, D., P. Sharma, L. Dudus, Y. Zhang, S. Sanlioglu, Z. Yan, Y. Yue, Y. Ye, R. Lester, J. Yang, K. J. Fisher, and J. F. Engelhardt.1999. Formation of adeno-associated virus circular genomes is differentially regulated by
adenovirus E4 ORF6 and E2a gene expression. J. Virol.73:161–169.
12.Duan, D., P. Sharma, J. Yang, Y. Yue, L. Dudus, Y. Zhang, K. J. Fisher, and J. F. Engelhardt.1998. Circular intermediates of recombinant adeno-asso-ciated virus have defined structural characteristics responsible for long-term
episomal persistence in muscle tissue. J. Virol.72:8568–8577.
13.Duan, D., Z. Yan, Y. Yue, and J. F. Engelhardt.1999. Structural analysis of
adeno-associated virus transduction circular intermediates. Virology261:8–
14.
14.Duan, D., Y. Yue, and J. F. Engelhardt.2003. Consequences of DNA-dependent protein kinase catalytic subunit deficiency on recombinant adeno-associated virus genome circularization and heterodimerization in muscle
tissue. J. Virol.77:4751–4759.
15.Elson, A., Y. Wang, C. J. Daugherty, C. C. Morton, F. Zhou, J. Campos-Torres, and P. Leder.1996. Pleiotropic defects in ataxia-telangiectasia
pro-tein-deficient mice. Proc. Natl. Acad. Sci. USA93:13084–13089.
16.Ferrari, F. K., X. Xiao, D. McCarty, and R. J. Samulski.1997. New devel-opments in the generation of Ad-free, high-titer rAAV gene therapy vectors.
Nat. Med.3:1295–1297.
17.Griffin, C. S., and J. Thacker.2004. The role of homologous recombination repair in the formation of chromosome aberrations. Cytogenet. Genome
Res.104:21–27.
18.Hacein-Bey-Abina, S., C. Von Kalle, M. Schmidt, M. P. McCormack, N. Wulffraat, P. Leboulch, A. Lim, C. S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P. Fraser, J. I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A. Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E. Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L. E. Leiva, M. Wissler, C. Prinz, T. H. Rabbitts, F. Le Deist, A. Fischer, and M. Cavazzana-Calvo. 2003. LMO2-associated clonal T cell
proliferation in two patients after gene therapy for SCID-X1. Science302:
415–419.
19.Heffernan, T. P., D. A. Simpson, A. R. Frank, A. N. Heinloth, R. S. Paules, M. Cordeiro-Stone, and W. K. Kaufmann.2002. An ATR- and Chk1-depen-dent S checkpoint inhibits replicon initiation following UVC-induced DNA
damage. Mol. Cell. Biol.22:8552–8561.
20.Hickson, I. D.2003. RecQ helicases: caretakers of the genome. Nat. Rev.
Cancer3:169–178.
21.Hinz, J. M., N. A. Yamada, E. P. Salazar, R. S. Tebbs, and L. H. Thompson.
2005. Influence of double-strand-break repair pathways on radiosensitivity
throughout the cell cycle in CHO cells. DNA Repair (Amsterdam)4:782–
792.
22.Jackson, S. P. 2002. Sensing and repairing DNA double-strand breaks.
Carcinogenesis23:687–696.
23.Jeggo, P. A., G. E. Taccioli, and S. P. Jackson.1995. Menage a trois: double
strand break repair, V(D)J recombination and DNA-PK. Bioessays17:949–
957.
on November 8, 2019 by guest
http://jvi.asm.org/
24.Jurvansuu, J., K. Raj, A. Stasiak, and P. Beard.2005. Viral transport of
DNA damage that mimics a stalled replication fork. J. Virol.79:569–580.
25.Khanna, K. K., and S. P. Jackson.2001. DNA double-strand breaks:
signal-ing, repair and the cancer connection. Nat. Genet.27:247–254.
26.Lobrich, M., and P. A. Jeggo.2005. Harmonising the response to DSBs: a
new string in the ATM bow. DNA Repair (Amsterdam)4:749–759.
27.Ma, Y., K. Schwarz, and M. R. Lieber.2005. The Artemis:DNA-PKcs en-donuclease cleaves DNA loops, flaps, and gaps. DNA Repair (Amsterdam)
4:845–851.
28.Machwe, A., E. M. Lozada, L. Xiao, and D. K. Orren.2006. Competition between the DNA unwinding and strand pairing activities of the Werner and
Bloom syndrome proteins. BMC Mol. Biol.7:1.
29.Machwe, A., L. Xiao, J. Groden, S. W. Matson, and D. K. Orren.2005. RecQ family members combine strand pairing and unwinding activities to catalyze
strand exchange. J. Biol. Chem.280:23397–23407.
30.McCarty, D. M., S. M. Young, and R. J. Samulski.2004. Integration of adeno-associated virus (Aav) and recombinant Aav vectors. Annu. Rev.
Genet.38:819–845.
31.McVey, M., J. R. Larocque, M. D. Adams, and J. J. Sekelsky.2004. Forma-tion of deleForma-tions during double-strand break repair in Drosophila DmBlm
mutants occurs after strand invasion. Proc. Natl. Acad. Sci. USA101:15694–
15699.
32.Miller, D. G., L. M. Petek, and D. W. Russell.2004. Adeno-associated virus
vectors integrate at chromosome breakage sites. Nat. Genet.36:767–773.
33.Miller, D. G., E. A. Rutledge, and D. W. Russell.2002. Chromosomal effects
of adeno-associated virus vector integration. Nat. Genet.30:147–148.
34.Miller, D. G., G. D. Trobridge, L. M. Petek, M. A. Jacobs, R. Kaul, and D. W. Russell.2005. Large-scale analysis of adeno-associated virus vector
integra-tion sites in normal human cells. J. Virol.79:11434–11442.
35.Moens, P. B., R. Freire, M. Tarsounas, B. Spyropoulos, and S. P. Jackson.
2000. Expression and nuclear localization of BLM, a chromosome stability protein mutated in Bloom’s syndrome, suggest a role in recombination
dur-ing meiotic prophase. J. Cell Sci.113:663–672.
36.Nam, C. H., and T. H. Rabbitts.2006. The role of LMO2 in development and in T cell leukemia after chromosomal translocation or retroviral insertion.
Mol. Ther.13:15–25.
37.Nghiem, P., P. K. Park, Y. Kim, C. Vaziri, and S. L. Schreiber.2001. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal
pre-mature chromatin condensation. Proc. Natl. Acad. Sci. USA98:9092–9097.
38.Nghiem, P., P. K. Park, Y. S. Kim, B. N. Desai, and S. L. Schreiber.2002. ATR is not required for p53 activation but synergizes with p53 in the
replication checkpoint. J. Biol. Chem.277:4428–4434.
39.Qing, K., X. S. Wang, D. M. Kube, S. Ponnazhagan, A. Bajpai, and A. Srivastava.1997. Role of tyrosine phosphorylation of a cellular protein in adeno-associated virus 2-mediated transgene expression. Proc. Natl. Acad.
Sci. USA94:10879–10884.
40.Raj, K., P. Ogston, and P. Beard.2001. Virus-mediated killing of cells that
lack p53 activity. Nature412:914–917.
41.Rivera, V. M., G. P. Gao, R. L. Grant, M. A. Schnell, P. W. Zoltick, L. W. Rozamus, T. Clackson, and J. M. Wilson.2005. Long-term pharmacologi-cally regulated expression of erythropoietin in primates following
AAV-mediated gene transfer. Blood105:1424–1430.
42.Rothkamm, K., I. Kruger, L. H. Thompson, and M. Lobrich.2003. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol.
Cell. Biol.23:5706–5715.
43.Russell, D. W.2003. AAV loves an active genome. Nat. Genet.34:241–242. 44.Rutledge, E. A., and D. W. Russell.1997. Adeno-associated virus vector
integration junctions. J. Virol.71:8429–8436.
45.Samulski, R. J., X. Zhu, X. Xiao, J. D. Brook, D. E. Housman, N. Epstein, and L. A. Hunter.1991. Targeted integration of adeno-associated virus
(AAV) into human chromosome 19. EMBO J.10:3941–3950.
46.Sanlioglu, S., P. Benson, and J. F. Engelhardt.2000. Loss of ATM function enhances recombinant adeno-associated virus transduction and integration
through pathways similar to UV irradiation. Virology268:68–78.
47.Schnepp, B. C., K. R. Clark, D. L. Klemanski, C. A. Pacak, and P. R. Johnson.2003. Genetic fate of recombinant adeno-associated virus vector
genomes in muscle. J. Virol.77:3495–3504.
48.Schnepp, B. C., R. L. Jensen, C. L. Chen, P. R. Johnson, and K. R. Clark.
2005. Characterization of adeno-associated virus genomes isolated from
human tissues. J. Virol.79:14793–14803.
49.Shiloh, Y.2003. ATM and related protein kinases: safeguarding genome
integrity. Nat. Rev. Cancer3:155–168.
50.Song, S., P. J. Laipis, K. I. Berns, and T. R. Flotte.2001. Effect of DNA-dependent protein kinase on the molecular fate of the rAAV2 genome in
skeletal muscle. Proc. Natl. Acad. Sci. USA98:4084–4088.
51.Song, S., Y. Lu, Y. K. Choi, Y. Han, Q. Tang, G. Zhao, K. I. Berns, and T. R. Flotte.2004. DNA-dependent PK inhibits adeno-associated virus DNA
in-tegration. Proc. Natl. Acad. Sci. USA101:2112–2116.
52.Summerford, C., and R. J. Samulski.1999. Viral receptors and vector pu-rification: new approaches for generating clinical-grade reagents. Nat. Med.
5:587–588.
53.Tambini, C. E., A. M. George, J. M. Rommens, L. C. Tsui, S. W. Scherer, and J. Thacker.1997. The XRCC2 DNA repair gene: identification of a
posi-tional candidate. Genomics41:84–92.
54.Tebbs, R. S., Y. Zhao, J. D. Tucker, J. B. Scheerer, M. J. Siciliano, M. Hwang, N. Liu, R. J. Legerski, and L. H. Thompson.1995. Correction of chromosomal instability and sensitivity to diverse mutagens by a cloned
cDNA of the XRCC3 DNA repair gene. Proc. Natl. Acad. Sci. USA92:
6354–6358.
55.Thacker, J., and R. E. Wilkinson.1995. The genetic basis of cellular recovery from radiation damage: response of the radiosensitive irs lines to
low-dose-rate irradiation. Radiat. Res.144:294–300.
56.Thompson, L. H., K. W. Brookman, N. J. Jones, S. A. Allen, and A. V. Carrano.1990. Molecular cloning of the humanXRCC1gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell.
Biol.10:6160–6171.
57.Thompson, L. H., and D. Schild.2002. Recombinational DNA repair and
human disease. Mutat. Res.509:49–78.
58.van Brabant, A. J., R. Stan, and N. A. Ellis.2000. DNA helicases, genomic instability, and human genetic disease. Annu. Rev. Genomics Hum. Genet.
1:409–459.
59.van Gent, D. C., J. H. Hoeijmakers, and R. Kanaar.2001. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet.
2:196–206.
60.Weitzman, M. D.2005. Functions of the adenovirus E4 proteins and their
impact on viral vectors. Front. Biosci.10:1106–1117.
61.Williams, B. R., O. K. Mirzoeva, W. F. Morgan, J. Lin, W. Dunnick, and J. H. Petrini.2002. A murine model of Nijmegen breakage syndrome. Curr. Biol.
12:648–653.
62.Xiao, X., J. Li, and R. J. Samulski.1996. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus
vector. J. Virol.70:8098–8108.
63.Xiao, X., J. Li, and R. J. Samulski.1998. Production of high-titer recombi-nant adeno-associated virus vectors in the absence of helper adenovirus.
J. Virol.72:2224–2232.
64.Xiao, X., W. Xiao, J. Li, and R. J. Samulski.1997. A novel 165-base-pair
terminal repeat sequence is the solecisrequirement for the adeno-associated
virus life cycle. J. Virol.71:941–948.
65.Xu, Y., T. Ashley, E. E. Brainerd, R. T. Bronson, M. S. Meyn, and D. Baltimore.1996. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic
lymphoma. Genes Dev.10:2411–2422.
66.Yan, H., J. McCane, T. Toczylowski, and C. Chen.2005. Analysis of the Xenopus Werner syndrome protein in DNA double-strand break repair.
J. Cell Biol.171:217–227.
67.Yang, C. C., X. Xiao, X. Zhu, D. C. Ansardi, N. D. Epstein, M. R. Frey, A. G. Matera, and R. J. Samulski.1997. Cellular recombination pathways and viral terminal repeat hairpin structures are sufficient for adeno-associated virus
integration in vivo and in vitro. J. Virol.71:9231–9247.
68.Zentilin, L., A. Marcello, and M. Giacca. 2001. Involvement of cellular double-stranded DNA break binding proteins in processing of the
recombi-nant adeno-associated virus genome. J. Virol.75:12279–12287.
69.Zolotukhin, S., B. J. Byrne, E. Mason, I. Zolotukhin, M. Potter, K. Chesnut, C. Summerford, R. J. Samulski, and N. Muzyczka. 1999. Recombinant adeno-associated virus purification using novel methods improves infectious
titer and yield. Gene Ther.6:973–985.