0022-538X/06/$08.00⫹0 doi:10.1128/JVI.80.9.4491–4500.2006
Copyright © 2006, American Society for Microbiology. All Rights Reserved.
DNA Binding Activity of the Herpes Simplex Virus Type 1
Origin Binding Protein, UL9, Can Be Modulated by
Sequences in the N Terminus: Correlation between
Transdominance and DNA Binding
Soma Chattopadhyay and Sandra K. Weller*
Department of Molecular, Microbial, and Structural Biology, University of Connecticut Health Center, Farmington, Connecticut 06030
Received 11 November 2005/Accepted 7 February 2006
UL9, the origin binding protein of herpes simplex virus type 1, is a member of the SF2 family of helicases. Cotransfection of cells with infectious viral DNA and plasmids expressing either full-length UL9 or the C-terminal DNA binding domain alone results in the drastic inhibition of plaque formation which can be partially relieved by an insertion mutant lacking DNA binding activity. In this work, C-terminally truncated mutants which terminate at or near residue 359 were shown to potentiate plaque formation, while other C-terminal truncations were inhibitory. Thus, residues in the N-terminal region appear to regulate the inhibitory properties of UL9. To identify which residues were involved in this regulation, a series of N-terminally truncated mutants were constructed which contain the DNA binding domain and various N-terminal extensions. Mutants whose N terminus is either at residue 494 or 535 were able to bind the origin efficiently and were inhibitory to plaque formation, whereas constructs whose N terminus is at residue 304 or 394 were defective in origin binding activity and were able to relieve inhibition. Since UL9 is required for viral infection at early but not late times and is inhibitory to infection when overexpressed, we propose that the DNA binding activities of UL9 are regulated during infection. For infection to proceed, UL9 may need to switch from a DNA binding to a non-DNA binding mode, and we suggest that sequences residing in the N terminus play a role in this switch.
Replication of the 152-kb double-stranded genome of her-pes simplex virus type 1 (HSV-1) in cell culture requires at least seven essential virus-encoded proteins: a highly proces-sive heterodimeric DNA polymerase (UL30/UL42), a hetero-trimeric helicase-primase complex (UL5/UL8/UL52), a single-stranded DNA binding protein (UL29), and an origin binding protein (UL9) (reviewed in references 5 and 20). UL9 is a multifunctional 94-kDa protein exhibiting the following activities: DNA-stimulated nucleotide triphosphatase, 3⬘-to-5⬘ DNA heli-case on partially double-stranded substrates, ability to form dimers in solution, and cooperative origin-specific DNA bind-ing (reviewed in reference 20). The N-terminal 534 residues define a domain containing seven conserved motifs, which are characteristic of the superfamily II helicases. Mutations within five of the seven conserved motifs inactivated the function of UL9 in an in vivo complementation assay (21) and also inac-tivated the helicase activity (19). These results suggest that the helicase domain is essential for biological activity.
The HSV-1 genome contains three origins of replication, a single copy of OriLand two copies of OriS, and each contains
high-affinity binding sites for UL9 (29, 30, 32–34). The 75-bp minimal OriScontains three closely related 11-bp palindromic
sequences, two of which (box I and box II) have been shown to be essential for efficient DNA replication (33). UL9 binds specifically to box I and box II in a highly cooperative manner
which may be important for distortion within the origins of replication during the initiation of HSV DNA synthesis (7, 11, 14). The ability of UL9 to bind specifically to the origin has been localized to the C-terminal one-third (residues 564 to 832) (1, 6). The N terminus of UL9 has been shown to interact with UL8 and UL42 (22, 23). Residues in the N-terminal region are also required both for dimerization and cooperative binding of UL9 at the origins (7, 11, 14). We suggest, therefore, that the N terminus contains residues important for influencing the nature of UL9-binding at the origins of replication. This hypothesis will be tested in this study.
Although many details of the mechanism of HSV DNA replication remain unclear, several lines of evidence support the model that HSV-1 replication proceeds in two stages (4, 20, 26). DNA replication very likely initiates in an origin-depen-dent manner; however, at later times, replication apparently proceeds by an origin-independent manner, perhaps by a roll-ing circle and/or recombination-driven mechanism. The anal-ysis of temperature-sensitive mutants of UL9 indicates that UL9 is required only during the first 6 h of lytic infection (4, 25), suggesting that UL9 is not required continuously for DNA replication. It has also been reported that the overexpression of wild-type UL9 is inhibitory to HSV-1 infection. Malik et al. demonstrated that infection of cells containing high copy num-bers of the UL9 gene resulted in a severely decreased efficiency of plaque formation (15). Furthermore, cotransfection of cells with infectious DNA and plasmids containing wild-type and mutant versions of UL9 also resulted in the transdominant inhibition of plaque formation (17, 31). Inhibitory effects of UL9 have also been observed in insect cells infected with recombinant viruses expressing the seven replication proteins
* Corresponding author. Mailing address: Department of Molecu-lar, Microbial and Structural Biology, MC3205, University of Connect-icut Health Center, 263 Farmington Avenue, Farmington, CT 06030. Phone: (860) 679-2310. Fax: (860) 679-1239. E-mail: Weller@NSO2 .uchc.edu.
4491
on November 8, 2019 by guest
http://jvi.asm.org/
and an origin (27). The observations that UL9 is only needed at very early times of infection and appears to be inhibitory at later times combined with the observation that UL9 levels do not appreciably diminish during infection (8, 13) suggest that one or more of the activities of UL9 need to be regulated for infection to proceed efficiently. The inhibitory properties of UL9 are mediated at least in part by its ability to bind to the origins of replication, since inhibition can be partially relieved by mutations in the C terminus which abrogate DNA binding (17, 24, 31). According to our model, origin binding activity of UL9 is required at early times; however, progression to the origin-independent later stage of infection likely requires the switch from a DNA binding to a non-DNA binding mode. In this paper we identify residues within the N terminus of UL9 which appear to modulate DNA binding activity of UL9 and thus may play a role in this switch.
MATERIALS AND METHODS
Cells, viruses, and antibodies.African green monkey kidney fibroblasts (Vero cells) were purchased from the American Type Culture Collection (Manassas, Va.) and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carls-bad, Calif.), supplemented with 5% fetal bovine serum, penicillin, and strepto-mycin (Invitrogen). The KOS strain of HSV-1 was used to prepare infectious viral DNA. Four UL9 antibodies were used in this study: 17B is a monoclonal antibody that recognizes the N-terminal 33 amino acids of UL9 (16); RH-7 is a polyclonal antibody raised against a glutathioneS-transferase fusion with the C-terminal domain of UL9 (residues 535 to 851) (a kind gift from Daniel Tenney,
Bristol Meyers Squib); KST-1 is a polyclonal antibody directed against the full-length UL9 protein (kindly provided by Deborah S. Parris, Ohio State University, Columbus); R249 is a rabbit polyclonal directed against the C-ter-minal decapeptide of UL9 (generously provided by Mark Challberg, NIH, Be-thesda, MD).
Plasmids.The pCDNA3-UL9-WT plasmid expressing wild-type UL9 under the control of a cytomegalovirus promoter was reported previously (18). The UL9 mutant plasmids which were used in this study and the PCR primers used for generating these plasmids are listed in Tables 1 and 2, respectively. The pCDNA3-UL9-MV(N) plasmid was made by two-step PCR using pCDNA3-UL9-WT as a template. The two internal primers were MV-forward and MV-reverse, and the two external primers were UL9-start-BamHI and UL9-end-EcoRI. The pCDNA3-UL9-1-359-MV plasmid was generated using UL9-start-BamHI as the downstream primer and oligo I as the upstream primer. The pCDNA3-UL9-1-359⫹3FS plasmid was generated using UL9-start-BamHI as the downstream primer and oligo II as the upstream primer. Plasmids UL9-1-450-WT, UL9-1-354MV, UL9-MV (renamed UL9-MV-FS in this paper), and UL9-1-321-WT were generated previously (18). The pCDNA3-UL9-WT plasmid was also used as a template in PCR for the generation of plasmids UL9-C, UL9-494-851, UL9-394-851, and UL9-304-851. All fragments for making mutants were generated by step-down PCR using UL9-end-EcoRI as the upstream primer. The downstream primers used to generate the truncations UL9-C, UL9-494-851, UL9-394-851, and UL9-304-851 were oligo III, oligo IV, oligo V, and oligo VI, respectively. Plasmid UL9-N was generated using UL9-start-BamHI as the downstream primer and oligo VII as the upstream primer. All plasmids described above were created by inserting the BamHI- and EcoRI-digested PCR fragments into the pCDNA3 vector.
Transient transfection and Western blot analysis.Subconfluent monolayers of Vero cells in 60-mm2
dishes were transfected with 2g of plasmid DNA con-taining wild-type or mutant versions of the UL9 gene with Lipofectamine Plus reagent (Invitrogen, Calif.) according to the manufacturer’s instructions. After 24 h, concentrated cell lysates were prepared in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer as described previously (18).
Protein expression was examined by Western blot analysis. Concentrated cell lysates were boiled for 5 min and subjected to electrophoresis in 10% SDS– polyacrylamide gels. The resolved proteins were transferred onto enhanced chemiluminescence membranes by electroblotting at 80 V for 2 h. The mem-branes were blocked in 5% nonfat dry milk, followed by incubation with primary antibodies at 4°C for 16 h. After washing with TBST (1.5 M NaCl, 0.2 M Tris, pH 7.5, and 2% Tween), the membranes were incubated with alkaline phosphatase-conjugated secondary antibodies at a dilution of 1:5,000 for 1 h and developed with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate (BCIP), the color detection reagents for alkaline phosphatase, according to the manu-facturer’s instructions (Promega, Madison, Wis.).
Plaque reduction assay.The plaque reduction assay was performed as de-scribed previously (17). In brief, Vero cells in 60-mm2dishes at 70% confluence
[image:2.585.43.284.89.246.2]were cotransfected with wild-type infectious DNA (KOS) and a 10-fold molar excess of UL9 wild-type or mutant plasmids with Lipofectamine Plus, according to the manufacturer’s recommendations. The dishes were overlaid with methyl cellulose and incubated at 37°C for 4 to 5 days, at which point cells were fixed and stained with crystal violet and plaques were counted. Each experiment was repeated three times. The number of plaques observed with infectious DNA alone was normalized to 100, and the normalized plaque numbers were calcu-lated for the experimental samples accordingly. Mean normalized plaque num-bers and standard deviations were calculated from the results of three separate experiments.
TABLE 1. Description of wild-type and mutant UL9 plasmids used in this study
Name of plasmid Part of gene encoded (aa)
WT or mutanta
Oligonucleotide(s) used for construction
UL9-WT 1-851 WT
UL9-MV-FS 1-362 M
UL9-MV(N) 1-851 M MV(F)
and MV(R)b
UL9-1-359-MV 1-359 M Oligo I
UL9-1-359⫹3FS 1-362 M Oligo II
UL9-1-450-WT 1-450 WT UL9-1-354-MV 1-354 M (G354A) UL9-1-321-WT 1-321 WT
UL9-C 535-851 WT Oligo III
UL9-494-851 494-851 WT Oligo IV
UL9-394-851 394-851 WT Oligo V
UL9-304-851 304-851 WT Oligo VI
UL9-N 1-534 WT Oligo VII
a
M, mutant. b
F, forward; R, reverse.
TABLE 2. List of oligonucleotides used in PCR for generating the UL9 mutant constructs
Name of oligonucleotide Sequence of oligonucleotide (5⬘–3⬘)
UL9-start-BamHI...GCCGAGGTGGATCCATGCCTTTCGTGGGGGGC UL9-end-EcoRI...GCCGAGGTGAATTCTTATAGGGTGCTAAAGTTCAC MV-forward ...GTAACCGTGGCCCTAAGCTTCGATCC
MV-reverse ...GGATCGAAGCTTAGGGCCACGGTTACGAC
Oligo I ...GCCGAGGTGAATTCTTAGGGATCGAAGCTTAGGGC
Oligo II...GCCGAGGTGAATTCTTATCAAAGTGCAGGGGGATCGAAGCTTAGGCC Oligo III ...GGTGGATCCATGCATCATCATCATCATCATGATCCCGAGGCGTCG Oligo IV ...AGGTGGATCCATGCGCGTGCGCTTCTGGGGA
Oligo V...AGGTGGATCCATGGAGCTACTGATTTACATG Oligo VI ...AGGTGGATCCATGTCGTCGACGGTCTCCTTCGCG
Oligo VII...GGTGAATTCTTAATGATGATGATGATGATGCCGGCACCGCAGCTC
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.585.44.542.603.725.2]In vitro transcription and translation.The wild-type and mutant UL9 genes were cloned in pCDNA3 under the control of the T7 promoter. In vitro tran-scription and translation (IVTT) reactions were performed with 2g of each plasmid DNA template for the synthesis of RNA and protein using the TNT coupled reticulocyte lysate system according to the manufacturer’s recommen-dation (Promega, Madison, Wis). [35
S]methionine was added in the reaction mix to radiolabel the nascent protein. Synthesized proteins were resolved in SDS-PAGE and exposed to Hyperfilm (Amersham Biosciences, England).
Electrophoretic mobility shift assay (EMSA).The 27-mer oligo 1 (GAAGC GTTCGCACTTCGTCCCAATATA) containing the box I sequence of OriS was annealed to the partially complementary oligo 2 sequence (TATATATATTGG GACGAAGTGCGAACGCT) (29 mer) at a ratio of 1:1 to generate partially double-stranded DNA as the substrate (box I region is underlined). Oligonucle-otides were boiled for 10 min in annealing buffer containing 50 mM Tris-HCl, pH 8, 50 mM NaCl, 1 mM dithiothreitol, and 0.5 mM EDTA and slowly cooled to room temperature. The annealed substrate was 12% native PAGE purified and resuspended in Tris-EDTA, pH 8.0. Purified substrate was end labeled using [␥-32
P]ATP (6,000 Ci/mmol) and polynucleotide kinase (New England Biolabs, Beverly, MA). The labeled substrate was purified using Nuctrap Push Columns (Stratagene, La Jolla, CA) and quantified. DNA binding reaction mixtures (20 l) contained 20 mM Tris-HCl, pH 8, 5 mM MgCl2, 50 mM NaCl, 0.1 mg/ml
bovine serum albumin, 0.5 mM dithiothreitol, 4% glycerol, 0.2g dI-dC (com-petitor DNA), and 20 nM32P-labeled substrate. For this assay, the amount of
IVTT translated protein was normalized such that comparable amounts of pro-teins (measuring the incorporation of35S in a liquid scintillation counter using a
Whatman GF/C glass fiber filter and running in SDS-PAGE) were present in the DNA binding reaction mixtures. Reaction mixtures were incubated at room temperature for 30 min, followed by addition of 2.5l of 10⫻gel loading buffer containing 250 mM Tris-HCl, pH 7.5, 40% glycerol, and 0.2% bromophenol blue. Products were loaded onto a native 4% polyacrylamide gel, and electrophoresis was performed at 250 V for 1.2 h at 4°C. The DNA-protein complexes were visualized and analyzed using a Storm PhosphorImager (Amersham Biosciences) and ImageQuant software (version 2.1).
Immunofluorescence microscopy.Vero cells were grown on glass coverslips and transfected as described above. At 18 h posttransfection, coverslips were washed twice with cold 1⫻phosphate-buffered saline (PBS). The cells were fixed with fresh 4% paraformaldehyde, pH 7.4, in PBS for 15 min at room tempera-ture. The coverslips were washed three times with PBS and then permeabilized with 1% Triton X-100 in PBS for 10 min. After three washes, cells were blocked with 5% normal goat serum (Sigma Chemicals, Steinheim, Germany) in PBS overnight at 4°C. Cells were incubated with primary antibodies diluted in 5% normal goat serum for 1 h at room temperature. After extensive washing with PBS, cells were incubated with 1:200-diluted secondary antibodies for 30 min. After seven washes with PBS, coverslips were mounted in gelatin containing 2.5% diazibicyclooctane to reduce photobleaching.
Fluorescence microscopic images were acquired using an Olympus BX-60 microscope using a 100⫻objective, and images were captured using a charge-coupled device cooled camera (Hammamatsu Corp., Boston, MA). Open Lab image software (Improvision, Inc., Lexington, MA) was used to analyze the images, and Adobe Photoshop 7.0 was used for arranging the images.
RESULTS
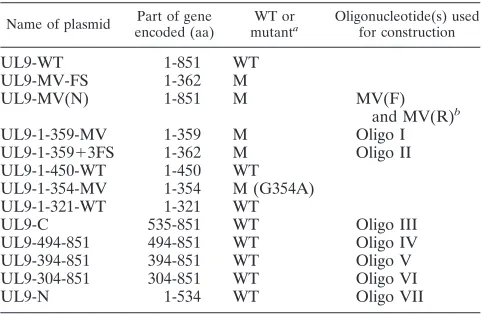
Previously described UL9 mutants were classified as trans-dominant or potentiating according to a plaque reduction as-say in which Vero cells were cotransfected with the HSV-1 infectious DNA and plasmid DNA capable of expressing wild-type or mutant versions of UL9 (18). The number of plaques observed with only HSV-1 infectious DNA was normalized to 100% (17), and plasmids which resulted in 10% or fewer plaques were considered to be transdominant. Cotransfection with wild-type UL9 plasmid was also very inhibitory: the num-ber of plaques was only 7% of the numnum-ber obtained with infectious DNA alone. Several mutants were shown to poten-tiate viral infection or to reverse the inhibition seen with wild-type plasmid. For instance, cotransfection with the motif V mutant (G354A, UL9-MV) consistently resulted in 150 to 200 plaques in this assay (18). We were intrigued by the fact that we were not able to detect full-length UL9 in cells transfected
with this mutant and instead detected an N-terminal 38-kDa protein fragment. We postulated that this mutant generated an unstable form of UL9 which was processed to a 38-kDa form (18); however, subsequent studies have now revealed that, in addition to the point mutation G to A at residue 354, this mutant contains a frameshift mutation 5 amino acids down-stream of the desired MV mutation. The frameshift mutation at residue 359 resulted in the substitution of three amino acids at this point (LHF to PSL) followed by a stop codon (Fig. 1A and 1B). Three additional mutations were engineered to de-termine whether potentiation was due to the G-to-A mutation at residue 354, the LHF-to-PSL frameshift mutation or the production of a truncated version of UL9. A mutant which contains only the desired point mutation G to A at amino acid position 354 (Fig. 1A, no. 3) was constructed and designated UL9-MV(N) for new MV mutation. UL9-1-359 MV expressed a truncated version of UL9 (residues 1 to 359) in which G354 was replaced by alanine (Fig. 1A, no. 4). UL9-1-359⫹3FS, which contains the frameshift mutation and three changed amino acids (PSL) (Fig. 1A, no. 5) also expressed a truncated version of UL9. This mutant lacked the G-to-A substitution found in the original UL9-MV mutant. The presence of the desired mutations was confirmed by sequencing.
Stable mutant proteins can be detected in Vero cell lysates
after transfection.To determine whether the new mutant
con-structs [UL9-MV(N), UL9-1-359 MV, and UL9-1-359⫹3FS] were able to express stable protein, cells were transfected and cell lysates were analyzed by Western blotting as described in Materials and Methods. UL9 protein was detected using the 17B monoclonal antibody, which was directed against the N-terminal 33 amino acid (aa) residues of UL9 (16). As shown in Fig. 1C, lane 3, the new MV mutant construct with the G354A point mutation expressed a 94-kDa stable full-length protein that migrated at a similar position to that of the wild-type UL9 protein. The other two mutants, 359 MV and UL9-1-359⫹3FS, expressed comparable amounts of the predicted 38-kDa UL9 fragments (Fig. 1C, lanes 4 and 5, respectively). As previously described, a truncated version of UL9 (residues 1 to 354) with the G354A mutation, designated UL9-1-354MV (18), was able to synthesize a 37-kDa UL9 fragment, consistent with its terminus being several amino acids before the terminus of the UL9-1-359 MV or UL9-1-359⫹3FS construct (Fig. 1A, no. 7, and 1C, lane 7). These results indicate that the MV (G354A) mutation by itself is not responsible for the formation of the 38-kDa N-terminally truncated protein. Contrary to our previous report suggesting that the 38-kDa protein was a pro-cessed form of the motif V mutant (18), it now appears that the 38-kDa form of MV protein was generated as a result of premature termination.
Potentiation correlates with particular truncated mutants
of UL9.Next we asked whether any of the new constructs could
potentiate infection, as seen with the original UL9-MV-FS mutation. Plaque reduction assays were carried out as described in Materials and Methods using a 1:10 molar ratio of infectious DNA to plasmid DNA. The new MV mutant, UL9-MV (N), which does not complement the null virus, resembled wild-type UL9, producing less than 10% plaques (Fig. 1D); therefore, it is clear that the G354A mutation alone cannot potentiate viral infection. This mutant resembles mutants in motifs I, Ia, II, and VI in being strongly inhibitory to viral infection (19).
on November 8, 2019 by guest
http://jvi.asm.org/
We next asked whether potentiation was merely due to the expression of a truncated version of UL9. Previous work indi-cated that proteins expressing only the C-terminal one-third of UL9 are transdominant, and we wondered whether all mutants that expressed part or all of the N-terminal region but lacked the C-terminal one-third would be able to potentiate viral infection. The plaque reduction assay was carried out with the C-terminally truncated versions of UL9 mentioned above. In-terestingly, both mutants which produce a 38-kDa protein, UL9-1-359-MV and UL9-1-359⫹3FS, were able to relieve in-hibition almost as efficiently as the original UL9-MV-FS mu-tant (Fig. 1D). Previously engineered constructs which produce smaller 354-MV, UL9-1-321-WT) or larger (UL9-1-450) forms of truncated UL9 protein were tested, and inter-estingly, each of these turned out to be transdominant (Fig. 1D). Thus, it appears that merely deleting the C-terminal region is not sufficient to potentiate viral infection; instead, there appears to be a region within the N terminus between resides 354 and 363 that drastically affects the ability of UL9 to inhibit viral infection but only in a context-specific fash-ion. It is possible that this domain is hidden or masked in the context of the residues 1 to 450 or full-length versions of UL9 but is exposed in constructs which terminate at or near
359. Since the residues 1 to 354 mutation is inhibitory to infection, it appears that the five amino acids between 354 and 359 are essential for the ability of the longer fragments to abrogate inhibition.
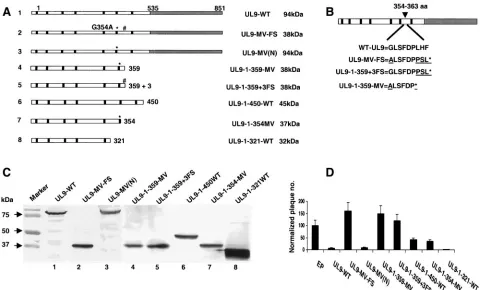
[image:4.585.52.532.69.359.2]The observation described above, that residues 354 to 363 are correlated with potentiation, supports the suggestion that this region of UL9 may be important for regulating UL9 ac-tivity in some way. We were intrigued by the possibility that potentiating mutations may decrease the ability of UL9 to bind DNA, leading to relief of the inhibition exerted by wild-type and mutant versions of UL9 mutants. The fact that potentiat-ing mutants only express a portion of the N-terminal region and do not have a DNA binding domain makes it impossible to test their DNA binding ability directly. Several additional UL9 constructs were therefore engineered which contain an intact C-terminal domain (535 to 851 aa) and various N-terminal extensions (shown in Fig. 2A). Since deletion of residues 292 to 404 also partially relieved the inhibition exhibited by wild-type (WT) UL9 (17), we were interested to determine whether mutants truncated between residues 292 and 404 could alter the inhibitory activity of the C-terminal domain of UL9 in plaque reduction assays and whether inhibition was related to the ability of UL9 to bind the origin.
FIG. 1. Mutations ending at or around 359 are potentiating. (A) Schematic diagram of UL9 truncations. The N-terminal domain of UL9 is depicted as an open box, and the C terminus is depicted as a gray box. The N-terminal seven conserved motifs of superfamily II helicases are depicted as black boxes. An asterisk points out the position of the MV (G354A) mutation, and the position of the frame shift (FS) mutation (at 359 aa) has been marked by a pound sign (#). The size of the protein synthesized from each truncation is indicated at the right side of the figure. (B) Amino acid sequences present between residues 354 to 363 of the wild-type and mutated versions of UL9. The mutant residues are underlined. Sequencing was repeated three times to confirm the presence of these mutations. (C) The 17B antibody was used to detect truncated UL9 proteins in concentrated cell lysates from Vero cells transfected with the plasmids used in the plaque reduction assay shown in panel D. Prestained protein markers are depicted on the left. (D) Plaque reduction assays were performed by cotransfecting infectious viral DNA and the plasmid expressing the indicated protein. EP, empty plasmid. Error bars represent standard deviations calculated from the results of three independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
N-terminal truncations initiating between residues 304 to
394 can also relieve inhibition. To examine whether stable
proteins of the expected size could be detected, Vero cells were transfected with the mutant constructs shown in Fig. 2A (UL9-C/535-851, UL9-494-851, UL9-394-851, and UL9-304-851), and concentrated cell extracts were examined by Western blot analysis. The RH-7 polyclonal antibody that can recognize C-terminal amino acids of UL9 was used for detection. Most of the constructs expressed similar amounts of full-length (WT) or truncated versions of UL9 except UL9-C/535-851, which expressed much higher levels of protein in transfected cells (Fig. 2B, lane 2). The sizes corresponded to the predicted molecular masses of each construct. In the case of the mutant protein UL9-494-851, two bands were detected which appear to reflect the presence of an in-frame methionine at residue 571 downstream from the major open reading frame starting at 494 (Fig. 2B, lane 3).
Next we asked whether constructs (Fig. 2A) expressing WT UL9, C/535-851, 494-851, 394-851, and
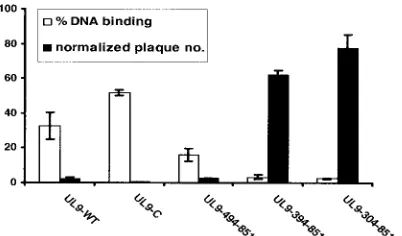
[image:5.585.123.466.67.415.2]UL9-304-851 could inhibit plaque formation in a plaque reduction assay (Fig. 2C). The number of plaques observed with HSV-1 infectious DNA was normalized to 100% and compared with the number of plaques obtained when plasmids expressing wild-type or mutant versions of UL9 were included. Figure 2C shows that, consistent with previous reports (24, 28, 31), the C-terminal fragment (UL9-C) is even more inhibitory than wild-type UL9. A truncation containing an extra 41 amino acid residues at the N terminus (UL9-494-851) was also very inhib-itory. Interestingly, the two constructs which contained an ad-ditional 141 or 231 aa at the N terminus, UL9-394-851 and UL9-304-851, respectively, were able to partially relieve the inhibitory effects exhibited by C-terminal UL9 and by wild-type UL9 itself, resulting in 63 and 78 normalized plaques, respec-tively (Fig. 2C). This supports previous studies on a naturally occurring N-terminal truncation, OBPC, which is encoded from a separate mRNA (UL8.5) overlapping the C terminus of UL9. OBPC (aa 365 to 851) is less efficient in the inhibition of plaque formation than either UL9-WT or UL9-C (2, 3). Thus,
FIG. 2. Inhibitory activity of the C terminus of UL9 protein in HSV-1 replication is relieved in the presence of residues 304 to 394. (A) A schematic representation of the truncated UL9 proteins is shown. The N terminus of the UL9 protein is represented by the black box. The C terminus of the protein is depicted by three black lines, and dashed lines represent the deleted amino acid sequences. The size of the protein synthesized from each truncation is indicated on the right. (B) The RH-7 antibody was used to detect UL9 truncations in concentrated cell lysates from Vero cells transfected with the plasmids used in plaque reduction assay shown in panel C. Wild-type UL9 and mutated UL9 proteins expressed in Vero cells are marked by asterisks. (C) Plaque reduction assays were performed by cotransfecting infectious viral DNA and the plasmid expressing the indicated protein. EP, empty plasmid. Error bars represent standard deviations calculated from the results of three independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
despite the fact that all the constructs in this experiment con-tain the same inhibitory C-terminal DNA binding domain (535 to 851), there appears to be something unusual about the constructs whose N termini are at 304, 365, or 394. These results are consistent with the suggestion of the existence of a domain within the N-terminal region which, when exposed, can relieve the inhibitory effects of the C-terminal DNA-binding domain. When this domain of the N-terminal region is present either within full-length UL9 or within constructs containing upstream or downstream residues, this domain is apparently masked.
Mutants UL9-394-851 and UL9-304-851 are severely
defec-tive in their ability to bind to ori-DNA.Previous reports that
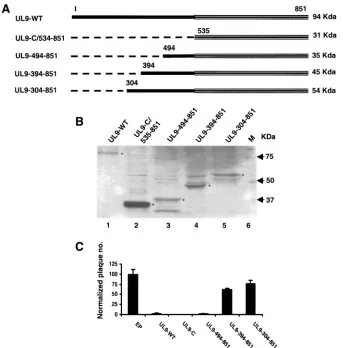
the inhibition of UL9 could at least be partially relieved by mutations in the DNA binding domain (17) prompted us to ask whether the inhibitory effects of UL9 in viral plaque formation shown in Fig. 2C were related to the origin binding ability of UL9. Wild-type and mutant proteins were synthesized in vitro using the TNT coupled reticulocyte lysate system as described in Materials and Methods. [35S]methionine incorporation was
measured, and aliquots containing equivalent numbers of counts were subjected to SDS-PAGE. Figure 3A shows that similar amounts of proteins migrating with the expected sizes could be
detected by autoradiography. In the case of the mutant protein UL9-494-851, two bands were detected which, as mentioned above, appear to reflect the presence of an in-frame methio-nine at residue 571 downstream from the major open reading frame at 494 (Fig. 3A, lane 3). Figure 3A, lane 6, shows that no labeled proteins could be detected when empty plasmid DNA was used in the IVTT reaction. An electrophoretic mobility shift assay (EMSA) was performed by mixing equivalent amounts of IVTT proteins and [␥-32P]ATP-labeled, partially
double-stranded DNA containing the box I sequence of HSV-1 oriS. As observed in Fig. 3B, lanes 6 and 7, UL9-C was able to shift the box I oligonucleotide very efficiently, and wild-type UL9 (Fig. 3B, lanes 2 and 3) also exhibits strong DNA binding ability with slightly reduced efficiency compared to the C ter-minus. The IVTT reaction mixture containing empty plasmid was also included in the DNA binding assay as a control (Fig. 3B, lanes 4 and 5) and exhibited no mobility shift, indicating that the mobility shift seen with wild-type and C-terminal frag-ments was not due to other proteins present in rabbit reticu-locyte lysates. All other mutant proteins displayed reduced DNA binding efficiencies (Fig. 3B, lanes 8 to 13). The UL9-494-851 fragment exhibited partial DNA binding activity (16.1%) (lanes 8 and 9); in this case, the two shifted bands likely
rep-FIG. 3. The ability to bind the origin of replication correlates with inhibition in the plaque reduction assay. (A) SDS-polyacrylamide gel electrophoresis of in vitro-synthesized wild-type UL9 and truncated proteins. Synthesis was performed in rabbit reticulocyte lysates containing 2 g of each plasmid DNA according to the procedure described in Materials and Methods. EP, reaction was carried out using empty plasmid as a control. (B) Origin DNA binding affinity was determined by an EMSA in a 30-min reaction, using a partially double-stranded DNA (containing box I ori sequence of HSV-1 replication) as the substrate. Lane 1 represents the control reaction in the absence of proteins. For each experimental sample, two different protein concentrations were used for the binding assay. The actual binding efficiency is defined as 100⫻[protein-DNA complex/(protein-DNA⫹free DNA)]. Squares indicate the areas of the image that have been used to calculate the binding efficiency of these proteins.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.70.527.67.365.2]resent DNA binding by the two open reading frames seen in the IVTT reaction. The most interesting observation was that the mutants UL9-394-851 and UL9-304-851 showed a severe defect in DNA binding activity (3.3% and 2.4%, respectively) (lanes 10 to 13) compared to UL9-C. A few slow-migrating DNA-protein complexes were observed in reaction mixtures containing either UL9-394-851 (lanes 10 and 11) or UL9-304-851 (lanes 12 and 13), which may represent aggregated forms of these proteins or higher oligomeric complexes of those proteins.
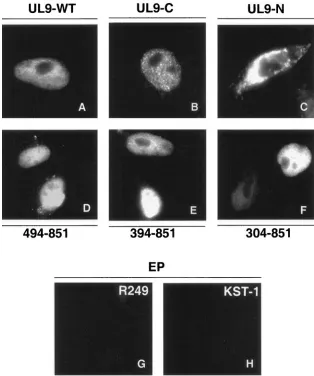
It is possible that the defect in DNA binding exhibited by UL9-394-851 and UL9-304-851 is due to structural alteration or misfolding of the mutant proteins. To determine whether these proteins were folded properly, we tested the mutant proteins for their ability to perform other functions, including nuclear transport and protein-protein interactions. First, we took advantage of the fact that the nuclear localization signal (NLS) of UL9 has been mapped to the extreme C terminus
[image:7.585.134.448.69.445.2](residue 744 to 851) (16). We reasoned that if the mutant proteins were misfolded, they might be expected to mislocalize in the cytoplasm, while properly folded proteins would be expected to enter the nucleus. Immunofluorescence micros-copy was performed as described in Materials and Methods. As expected, the wild-type and C-terminal DNA binding do-mains localized efficiently to the nucleus (Fig. 4A and B, re-spectively). Mutant proteins UL9-494-851, UL9-394-851, and UL9-304-851 were also able to localize efficiently to the nu-cleus (Fig. 4D, 4E, and 4F, respectively), while the N-terminal fragment (residues 1 to 534) was localized within the cytoplasm (Fig. 4C). These results indicate that the NLS-containing C termini of UL9-394-851 and UL9-304-851 are not grossly mis-folded, since they can be transported to the nucleus. Since the NLS resides within the C terminus, this experiment only indi-cated that the C terminus is still functional. To rule out that the N-terminal portions (residues 394 to 534 or residues 304 to 534) are not misfolded in these truncated proteins, we tested
FIG. 4. Intracellular localization of UL9 in cells transfected with wild-type and mutant versions of UL9. Vero cells were transfected with UL9-WT (A), UL9-C (B), UL9-N (C), plasmid UL9-494-851 (D), plasmid UL9-349-851 (E), and plasmid UL9-304-851 (F). Panels A, B, D, E, and F were stained with R249 antibody (recognizes the C terminus of UL9). Panel C was stained with KST-1 (directed against the whole UL9 protein), indicating that, in the absence of NLS, fragmented UL9 protein localizes in the cytoplasm. Panels G and H contain the images of Vero cells transfected with empty plasmid (pCDNA3 vector) and stained with R249 and KST-1 antibodies, respectively, which can be considered background staining.
on November 8, 2019 by guest
http://jvi.asm.org/
whether UL9-394-851 and UL9-304-851 could still participate in protein-protein interactions. The dimerization domain of UL9 was previously mapped to the N-terminal 1 to 534 residues (7). We have recently mapped at least one dimerization domain to a region between residues 304 and 534 (S. Chattopadhyay and S. K. Weller, unpublished data). Both 394-851 and UL9-304-851 are still able to form dimers with full-length UL9, as assessed by coimmunoprecipitation experiments, indicating that the N-terminal portions of these truncation mutants are folded properly. In summary, these experiments show that the inabil-ity of UL9-394-851 and UL9-304-851 to bind DNA is most likely not due to gross misfolding. We suggest instead that the behavior of these mutants can be explained by the existence of a domain which may act to inhibit DNA binding when exposed.
Mutants, which are impaired in DNA binding, also fail to
inhibit viral infection. To determine whether alterations in
DNA binding correlate with the inhibitory properties of UL9, the results of the plaque reduction assay and DNA binding assay were plotted. Figure 5 shows that the C terminus and wild-type UL9 protein were both strongly inhibitory in the plaque reduction assay and both are efficient in DNA binding ability. On the other hand, mutants 394-851 and UL9-304-851 resulted in an abrogation of inhibition as well as in DNA binding. Thus, relief of inhibition correlates very well with the inability of UL9 protein to form stable complexes with origin DNA.
DISCUSSION
We and others have proposed that HSV-1 replication occurs first in an origin-dependent stage, followed by an origin-inde-pendent stage; however, the mechanism for progression from one stage to the other is not known. The observation that UL9 levels do not change dramatically during infection (8, 13) sug-gests that one or more of the activities of UL9 may be regu-lated for infection to proceed. In this study, we have used potentiating and inhibitory mutants of UL9 to further our understanding of how UL9 might be regulated during infec-tion. We reasoned that the identification of residues which modulate the DNA binding activity of UL9 might be helpful in
understanding the nature of the switch from origin-dependent to origin-independent HSV DNA replication.
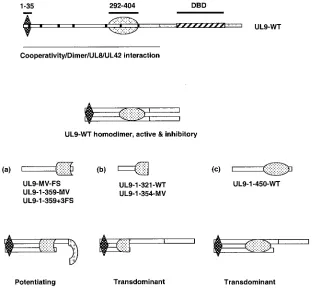
A series of C-terminally truncated UL9 mutant constructs were examined, and some were found to be inhibitory to viral infection, while others were potentiating. The potentiating mu-tants terminated within a region at or near residue 359, while mutant constructs which terminated at either aa 321, 354, or 450 were inhibitory. These results suggest that a domain within the N-terminal region of UL9 is able to reverse the inhibitory properties of UL9 in a context-specific manner. We propose that potentiating mutants encode a 38-kDa fragment exposing a domain which negatively regulates the DNA binding activity of full-length UL9 intrans. We hypothesize further that the potentiating activity of the 38-kDa protein may reflect its abil-ity to form a heterodimer with full-length UL9. Coimmuno-precipitation experiments demonstrated that all C-terminal truncation mutants are able to form heterodimers with wild-type UL9 (Chattopadhyay and Weller, unpublished). We sug-gest that when the 38-kDa species enters into a heterodimer, a region is exposed that can interact with the C-terminal domain, resulting in masking or inhibition of its DNA binding activity (Fig. 6a). We suggest that even though they can still form heterodimers, the N-terminal fragments that terminate at aa 321, 354, or 450 do not affect DNA binding because the region needed for the abrogation of DNA binding is either not present (in the constructs ending at 321 or 354) (Fig. 6b) or masked by a region of the protein ending at 450 (Fig. 6c).
We previously showed that the deletion of residues 1 to 35 and 292 to 404 in the N terminus can also partially relieve inhibition (17); however, the mechanism of relief was not un-derstood. This finding together with the observation that a mutation in motif IV (303 aa) exhibited decreased DNA bind-ing ability along with relief of inhibition (19) lead us to hy-pothesize that residues in the N terminus can affect DNA binding activity directly. To test this hypothesis, a series of constructs that contain the entire C-terminal DNA binding domain and varying lengths of the N terminus were examined. Our results demonstrate that constructs whose N terminus is at residue 304 or 394 exhibited diminished ability to bind to the origin, and these mutants were able to relieve inhibition, whereas mutants whose N-terminal residue is at aa 494 or 535 were able to bind the origin efficiently and were inhibitory to plaque formation. Interestingly, Baradaran et al. showed that the naturally occurring OBPC protein (UL8.5) whose N ter-minus is at aa 365 could also relieve inhibition compared to the C terminus alone (3). Collectively, these results suggest that residues within the N-terminal region can affect DNA binding activity and modulate the inhibitory properties of the C-termi-nal domain. These results support the suggestion that inhibi-tion is caused by UL9 remaining bound to the origin. This inactive UL9-origin complex may prevent rolling circle and/or recombination-dependent replication.
[image:8.585.65.266.69.187.2]The inhibitory and potentiating properties of various mutants examined in this and previous studies as well as the inhibition of HSV infection by overexpression of wild-type UL9 may be somewhat artificial, as these conditions might not be expected to be produced during normal infection; however, our results raise important questions about the regulation of UL9 activity during the course of infection. The observation that sequences within the N terminus communicate with the C-terminal DNA
FIG. 5. The inhibition of plaque formation is correlated with DNA binding ability. The normalized plaque numbers (black bars) and DNA binding efficiencies (white bars) were plotted for the wild-type and truncated UL9 proteins. Error bars represent standard deviations cal-culated from the results of three independent experiments. In some cases, error bars are too small to see.
on November 8, 2019 by guest
http://jvi.asm.org/
binding domain may reflect events that occur during the tran-sition between the origin-dependent initial stages of replica-tion and origin-independent later stages. This regulareplica-tion could be mediated by functions performed by the N-terminal region, such as dimerization, ability to form higher-order structures, or interaction with other proteins such as UL8 or UL42. A pre-cedent for regulation of a viral origin binding protein regulated during infection exists in simian virus 40 (SV40). During SV40 DNA replication, large-T antigen activity is regulated through phosphorylation (10).
Several possible mechanisms for how UL9 may be regulated can be considered. Although protein levels of UL9 apparently do not change dramatically during infection (8, 13), it is possible that a fraction of UL9, for instance the DNA-bound fraction, is de-graded at later times postinfection. If this were the case, total UL9 levels would not decrease appreciably, but origins could be cleared to allow the transition to UL9-independent DNA repli-cation. Alternatively, as with SV40 T antigen, regulation of UL9 may be mediated through phosphorylation. Interestingly, it has been reported that UL9 is phosphorylated 4 to 8 h after infection, and virus-encoded proteins or virus-induced cellular proteins enhance this phosphorylation (12). Lehman et al. have reported that NFB42, a ubiquitin ligase, which is enriched in the nervous system, binds to phosphorylated UL9. As a result, they speculate that UL9 becomes polyubiquitinated and undergoes degradation by the 26S proteasomal pathway (9). The ability of a ubiquitin ligase to bind and potentially degrade
UL9 during either lytic or latent infection could be important in its regulation. It will be of interest to determine if phosphor-ylation and/or the presence of a PEST sequence in the N terminus of UL9 (18) plays a role in regulation of the levels of protein and/or its activity during HSV-1 infection. It will also be interesting to determine whether OBPC, the product of the UL8.5 RNA expressed with delayed-early kinetics (3), plays a role in regulation of UL9 activity.
In summary, we have demonstrated that N-terminal residues of UL9 can regulate its origin DNA binding ability, and we propose that this type of regulation may play an important role during infection for the transition between origin-dependent and origin-independent replication. The mechanism of down regulation of DNA binding is not clear, but it is possible that phosphorylation, degradation, and/or conformational changes to the protein could affect its DNA binding activities. Confor-mational changes may be mediated by protein-protein interac-tions between UL9 and itself or with other viral and cellular proteins. Such interactions may serve to expose a regulatory domain in the N terminus which results in clearing UL9 from the origin, allowing replication to occur in an origin-indepen-dent manner.
ACKNOWLEDGMENTS
[image:9.585.137.451.69.357.2]We thank Shlomo Eisenberg and the members of our laboratory for helpful comments on the manuscript. We are grateful to Mark Chall-berg, Daniel Tenney, and Deborah Parris for providing the antibodies FIG. 6. Model for potentiation and transdominance of HSV-1 replication. A schematic diagram of UL9 is shown at the top. The two potential regulatory regions 1 to 35 and 292 to 404 are depicted by diamond and oval shapes, respectively. The DNA binding domain (DBD) is marked by black diagonal stripes in the C terminus. For the potentiating mutants (a), we propose that the truncated protein can form a heterodimer causing a conformational change such that the DNA binding domain is masked and DNA binding is inhibited. In the case of transdominant mutants, the region required for this negative effect is either not present (b) or not able to cause this conformational change (c).
on November 8, 2019 by guest
http://jvi.asm.org/
used in this study. We thank B. Marintcheva for generating UL9-WT, UL9-1-450-WT, UL9-1-354-MV, and UL9-1-321-WT plasmids.
This work was supported by Public Health Service grant AI-21747 from the National Institutes of Health.
REFERENCES
1.Arbuckle, M. I., and N. D. Stow.1993. A mutational analysis of the DNA-binding domain of the herpes simplex virus type 1 UL9 protein. J. Gen. Virol.74(Pt 7):1349–1355.
2.Baradaran, K., C. E. Dabrowski, and P. A. Schaffer.1994. Transcriptional analysis of the region of the herpes simplex virus type 1 genome containing the UL8, UL9, and UL10 genes and identification of a novel delayed-early gene product, OBPC. J. Virol.68:4251–4261.
3.Baradaran, K., M. A. Hardwicke, C. E. Dabrowski, and P. A. Schaffer.1996. Properties of the novel herpes simplex virus type 1 origin binding protein, OBPC. J. Virol.70:5673–5679.
4.Blumel, J., and B. Matz.1995. Thermosensitive UL9 gene function is re-quired for early stages of herpes simplex virus type 1 DNA synthesis. J. Gen. Virol.76(Pt 12):3119–3124.
5.Challberg, M.1996. Herpesvirus DNA replication. Cold Spring Harbor Press, Cold Spring Harbor, N.Y.
6.Deb, S., and S. P. Deb.1991. A 269-amino-acid segment with a pseudo-leucine zipper and a helix-turn-helix motif codes for the sequence-specific DNA-binding domain of herpes simplex virus type 1 origin-binding protein. J. Virol.65:2829–2838.
7.Elias, P., C. M. Gustafsson, O. Hammarsten, and N. D. Stow.1992. Struc-tural elements required for the cooperative binding of the herpes simplex virus origin binding protein to oriS reside in the N-terminal part of the protein. J. Biol. Chem.267:17424–17429.
8.Elias, P., M. E. O’Donnell, E. S. Mocarski, and I. R. Lehman.1986. A DNA binding protein specific for an origin of replication of herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA83:6322–6326.
9.Eom, C. Y., and I. R. Lehman.2003. Replication-initiator protein (UL9) of the herpes simplex virus 1 binds NFB42 and is degraded via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA100:9803–9807. 10.Fanning, E.1994. Control of SV40 DNA replication by protein
phosphory-lation: a model for cellular DNA replication? Trends Cell Biol.4:250–255. 11.Hazuda, D. J., H. C. Perry, and W. L. McClements. 1992. Cooperative interactions between replication origin-bound molecules of herpes simplex virus origin-binding protein are mediated via the amino terminus of the protein. J. Biol. Chem.267:14309–14315.
12.Isler, J. A., and P. A. Schaffer.2001. Origin binding protein-containing protein-DNA complex formation at herpes simplex virus type 1 oriS: role in oriS-dependent DNA replication. J. Virol.75:6808–6816.
13.Isler, J. A., and P. A. Schaffer.2001. Phosphorylation of the herpes simplex virus type 1 origin binding protein. J. Virol.75:628–637.
14.Koff, A., J. F. Schwedes, and P. Tegtmeyer.1991. Herpes simplex virus origin-binding protein (UL9) loops and distorts the viral replication origin. J. Virol.65:3284–3292.
15.Malik, A. K., R. Martinez, L. Muncy, E. P. Carmichael, and S. K. Weller.
1992. Genetic analysis of the herpes simplex virus type 1 UL9 gene: isolation of a LacZ insertion mutant and expression in eukaryotic cells. Virology
190:702–715.
16.Malik, A. K., L. Shao, J. D. Shanley, and S. K. Weller.1996. Intracellular localization of the herpes simplex virus type-1 origin binding protein, UL9. Virology224:380–389.
17.Malik, A. K., and S. K. Weller.1996. Use of transdominant mutants of the origin-binding protein (UL9) of herpes simplex virus type 1 to define func-tional domains. J. Virol.70:7859–7866.
18.Marintcheva, B., and S. K. Weller.2003. Existence of transdominant and potentiating mutants of UL9, the herpes simplex virus type 1 origin-binding protein, suggests that levels of UL9 protein may be regulated during infec-tion. J. Virol.77:9639–9651.
19.Marintcheva, B., and S. K. Weller.2001. Residues within the conserved helicase motifs of UL9, the origin-binding protein of herpes simplex virus-1, are essential for helicase activity but not for dimerization or origin binding activity. J. Biol. Chem.276:6605–6615.
20.Marintcheva, B., and S. K. Weller.2001. A tale of two HSV-1 helicases: roles of phage and animal virus helicases in DNA replication and recombination. Prog. Nucleic Acid Res. Mol. Biol.70:77–118.
21.Martinez, R., L. Shao, and S. K. Weller.1992. The conserved helicase motifs of the herpes simplex virus type 1 origin-binding protein UL9 are important for function. J. Virol.66:6735–6746.
22.McLean, G. W., A. P. Abbotts, M. E. Parry, H. S. Marsden, and N. D. Stow.
1994. The herpes simplex virus type 1 origin-binding protein interacts spe-cifically with the viral UL8 protein. J. Gen. Virol.75(Pt 10):2699–2706. 23.Monahan, S. J., L. A. Grinstead, W. Olivieri, and D. S. Parris.1998.
Inter-action between the herpes simplex virus type 1 origin-binding and DNA polymerase accessory proteins. Virology241:122–130.
24.Perry, H. C., D. J. Hazuda, and W. L. McClements.1993. The DNA binding domain of herpes simplex virus type 1 origin binding protein is a transdomi-nant inhibitor of virus replication. Virology193:73–79.
25.Schildgen, O., S. Graper, J. Blumel, and B. Matz.2005. Genome replication and progeny virion production of herpes simplex virus type 1 mutants with temperature-sensitive lesions in the origin-binding protein. J. Virol.79:7273– 7278.
26.Severini, A., A. R. Morgan, D. R. Tovell, and D. L. Tyrrell.1994. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology200:428–435.
27.Skaliter, R., and I. R. Lehman.1994. Rolling circle DNA replication in vitro by a complex of herpes simplex virus type 1-encoded enzymes. Proc. Natl. Acad. Sci. USA91:10665–10669.
28.Stow, N. D.1992. Herpes simplex virus type 1 origin-dependent DNA rep-lication in insect cells using recombinant baculoviruses. J. Gen. Virol.73(Pt 2):313–321.
29.Stow, N. D.1982. Localization of an origin of DNA replication within the TRS/IRS repeated region of the herpes simplex virus type 1 genome. EMBO J.1:863–867.
30.Stow, N. D.1985. Mutagenesis of a herpes simplex virus origin of DNA replication and its effect on viral interference. J. Gen. Virol.66(Pt 1):31–42. 31.Stow, N. D., O. Hammarsten, M. I. Arbuckle, and P. Elias.1993. Inhibition of herpes simplex virus type 1 DNA replication by mutant forms of the origin-binding protein. Virology196:413–418.
32.Stow, N. D., and E. C. McMonagle.1983. Characterization of the TRS/IRS origin of DNA replication of herpes simplex virus type 1. Virology130:427– 438.
33.Weir, H. M., and N. D. Stow.1990. Two binding sites for the herpes simplex virus type 1 UL9 protein are required for efficient activity of the oriS repli-cation origin. J. Gen. Virol.71(Pt 6):1379–1385.
34.Weller, S. K., A. Spadaro, J. E. Schaffer, A. W. Murray, A. M. Maxam, and P. A. Schaffer.1985. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol. Cell. Biol.5:930–942.