0022-538X/11/$12.00 doi:10.1128/JVI.02308-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
The Transmembrane Domain Sequence Affects the Structure and
Function of the Newcastle Disease Virus Fusion Protein
䌤
Kathryn A. Gravel, Lori W. McGinnes, Julie Reitter,† and Trudy G. Morrison*
Department of Microbiology and Physiological Systems/Program in Immunology and Virology, University of Massachusetts Medical School, Worcester, Massachusetts 01655
Received 4 November 2010/Accepted 17 January 2011
The role of specific sequences in the transmembrane (TM) domain of Newcastle disease virus (NDV) fusion (F) protein in the structure and function of this protein was assessed by replacing this domain with the F protein TM domains from two other paramyxoviruses, Sendai virus (SV) and measles virus (MV), or the TM domain of the unrelated glycoprotein (G) of vesicular stomatitis virus (VSV). Mutant proteins with the SV or MV F protein TM domains were expressed, transported to cell surfaces, and proteolytically cleaved at levels comparable to that of the wild-type protein, while mutant proteins with the VSV G protein TM domain were less efficiently expressed on cell surfaces and proteolytically cleaved. All mutant proteins were defective in all steps of membrane fusion, including hemifusion. In contrast to the wild-type protein, the mutant proteins did not form detectable complexes with the NDV hemagglutinin-neuraminidase (HN) protein. As determined by binding of conformation-sensitive antibodies, the conformations of the ectodomains of the mutant proteins were altered. These results show that the specific sequence of the TM domain of the NDV F protein is important for the conformation of the preactivation form of the ectodomain, the interactions of the protein with HN protein, and fusion activity.
Infection of cells by enveloped viruses requires fusion of the viral membrane with a cellular membrane in order to deliver the viral genome to cell interiors. Membrane fusion during paramyxovirus infection is mediated by the fusion protein (F), which is classed as a type 1 fusion protein (10, 15, 23, 30, 67). Current models of type 1 fusion propose that these proteins are synthesized and delivered to cell surfaces in a metastable form (30, 67, 68). Upon the activation of fusion, the molecules are thought to undergo dramatic conformational changes that ultimately result in fusion of the viral and cellular membranes (10, 13, 23, 57, 67, 68). Activation of these conformational changes is accomplished by acidic pH or by receptor binding (16, 17, 29, 30, 68).
Several domains in class 1 fusion proteins are key to mem-brane merger. These proteins, which fold as trimers (10), are synthesized as inactive precursors that are cleaved into two subunits, F1and F2in the case of paramyxoviruses (reviewed in reference 29). The sequence at the new amino terminus gen-erated by this cleavage is the fusion peptide (FP). This domain inserts into the target membrane upon the onset of fusion, anchoring the protein to the target membrane (reviewed in references 10, 29, and 30). Type 1 fusion proteins also contain two heptad repeat domains (HR1 and HR2, or HRA and HRB in the case of paramyxoviruses), which have a strong affinity and form a stable six-stranded coiled coil (reviewed in refer-ence 30). It is thought that these two domains are not
associ-ated prior to the onset of fusion but are complexed in the postfusion F protein. Indeed, this proposal is supported by two different crystal structures of paramyxovirus F proteins. One structure, proposed to be the prefusion form of the protein, does not contain the HR1-HR2 complex (73), while the other structures, proposed to represent the postfusion form of the protein, do contain this complex (7, 60, 72). These structures lend support to the model that refolding of the F protein with the formation of the HR1-HR2 complex pulls the target and effector membranes together (30, 40, 48).
The cytoplasmic domain (CT) and transmembrane (TM) domains of paramyxovirus F proteins are not represented in crystal structures but may also have roles in fusion. The cyto-plasmic domain has been implicated in the fusion activity of some paramyxovirus F proteins, since mutations in the CT domain or extensions of this domain can affect fusion activity (1, 5, 52, 63, 66). The role of the TM domain in the structure and function of paramyxovirus F proteins has not, however, been extensively investigated.
TM domains of viral fusion proteins are clearly necessary to anchor the protein in membranes. They are also necessary for complete fusion, as the replacement of a TM domain (and the CT domain) with a glycosylphosphatidylinositol (GPI) anchor results in proteins that cannot complete membrane merger (for examples, see references 4, 24, and 62). It has been sug-gested that the specific TM domain sequence is not critical, since complete replacement of a TM domain with that of another viral fusion protein often has no effect on the activity of the protein. The TM domain of influenza hemagglutinin (HA) has been replaced with that of Sendai virus (SV) or the polyimmunoglobulin receptor, and the mutant HA proteins were fully functional (39, 49). The TM domain of the vesicular stomatitis virus (VSV) glycoprotein (G) has been replaced with that of herpes simplex virus 1 (HSV-1) gB and CD4, adenovi-* Corresponding author. Mailing address: Department of
Microbi-ology and Physiological Systems/Program in ImmunMicrobi-ology and Virol-ogy, University of Massachusetts Medical School, Worcester, MA 01655. Phone: (508) 856-6592. Fax: (508) 856-5920. E-mail: trudy [email protected].
† Present address: Department of Chemistry, Holy Cross College, Worcester, MA.
䌤Published ahead of print on 26 January 2011.
3486
on November 7, 2019 by guest
http://jvi.asm.org/
rus E3-11.6 K protein, or HSV-1 gD with no effect on the function of the protein (44). It has been reported that replace-ment of the TM domain of HIV gp160 with a foreign sequence has no effect on infectivity (69). In contrast, there are reports of point mutations in TM domains of some viral fusion pro-teins, including the VSV G protein and the influenza HA protein, that do affect fusion (9, 39, 45, 62). However, in most of these cases, pore formation was inhibited, not hemifusion (4, 39, 41). These combined results have led to the proposal that there is significant latitude in the precise TM domain sequence that is functional and that TM domains participate in pore formation but not in the earlier stages of fusion (4, 39, 41).
In contrast to that of most type 1 fusion proteins, the acti-vation of most paramyxovirus F proteins requires coexpression of the attachment protein (reviewed in references 29 and 58), and activation is linked, in poorly understood ways, to receptor binding (29, 58) as well as to virus-specific interactions between the attachment protein and the F protein (18, 58). This addi-tional requirement for the activity of paramyxovirus F proteins may further impact the effect of alterations in the F protein TM domain on membrane fusion.
To explore the role of a specific Newcastle disease virus (NDV) F protein TM domain sequence, this TM domain was replaced with the F protein TM domains from two other paramyxoviruses, Sendai virus and measles virus (MV), as well as the TM domain of the unrelated VSV G protein. The F proteins are closely related to the NDV F protein, with likely very similar structures and similar activities, while the structure and function of the G protein is significantly different (47). The mutant F proteins with foreign TM domains were expressed and efficiently delivered to cell surfaces, but they were defec-tive in all steps in fusion. Furthermore, the mutant F proteins did not form a detectable complex containing the NDV hem-agglutinin-neuraminidase (HN) protein and thus were likely defective in fusion activation. The conformations of these mu-tant proteins were also altered, as determined by the binding of two different conformation-sensitive antibodies. These results suggest that the structure and function of the ectodomain of the NDV F protein are influenced by specific sequences in the TM domain and that a TM domain from a closely related F protein is not an effective substitute.
MATERIALS AND METHODS
Cells and plasmids.COS-7 cells obtained from the American Type Culture Collection were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with nonessential amino acids, vitamins, penicillin-streptomycin, and 10% fetal calf serum. Both wild-type and mutant NDV F genes and wild-type HN protein genes were expressed in COS-7 cells using pSVL (52, 53) or the pCAGGS vector (43). The cDNAs were inserted into the pSP6 vector for gen-eration of mRNA for cell-free translation (37). Sequences encoding the MV F protein, the SV F protein, and the VSV G protein were obtained from M. Oldstone, D. Nayak, and M. Schmidt, respectively.
To construct NDV F proteins with the TM domain sequences of MV and SV F and VSV G, an Eco47III site was inserted at the sequence encoding the junction of the ectodomain and the TM domain of the NDV F protein by PCR. The changes introduced did not change the coding sequence of the DNA. This site and a naturally occurring SphI site at the junction of the TM domain and the CT domain were used to excise the NDV TM domain sequences and to insert sequences encoding the MV and SV F or VSV G protein TM sequences obtained by PCR of plasmids encoding these glycoproteins, using appropriate primers. Chimera F protein genes were sequenced in their entirety to verify the insertion of the sequences and to verify that no other changes in the F protein were
present. FLAG sequences were added to the ends of the coding sequences of the NDV F protein gene and the SV F protein gene by standard PCR protocols.
Antibodies. Anti-F protein monoclonal antibody (anti-Fu1a) was obtained from M. Peeples (42). Anti-HN monoclonal antibodies were a mixture of three different anti-HN antibodies obtained from R. Iorio (19, 20). Anti-HR2 antibody and anti-HR1 antibody were raised against glutathioneS-transferase (GST) fusion proteins encoding amino acids 449 to 503 or 132 to 173, respectively, of the NDV F protein as previously described (12, 34, 50, 53). Anti-HN-AS anti-bodies were raised against amino acids 117 to 515 of the NDV HN protein ectodomain expressed as a type E fusion protein as previously described (35). Anti-NDV was raised against a purified stock of egg-grown NDV (36) that had been UV inactivated.
Transfections.Transfections using Lipofectamine (Invitrogen) were done es-sentially as recommended by the manufacturer. COS-7 cells were plated at 3⫻ 105
cells per 35-mm plate. Twenty to 24 h later, the cells were transfected. For each 35-mm plate, a mixture of DNA (0.5 to 0.75g, depending upon the experiment) in 0.1 ml of Opti-MEM (Invitrogen) and 7l of transfection reagent in 0.2 ml of Opti-MEM was incubated at room temperature for 45 min and then diluted with 0.7 ml of Opti-MEM and added to a plate previously washed twice with 2 ml of Opti-MEM. Cells were incubated for 5 h, DNA-Lipofectamine complexes were removed, and then 2 ml of supplemented DMEM medium was added.
Flow cytometry.Cells transfected with 0.75g of plasmid DNA per 35-mm plate for 48 h were removed from plates with trypsin (50g/ml), washed in FACS buffer (phosphate-buffered saline [PBS] containing 1% bovine serum albumin [BSA] and 0.02% azide), and incubated with anti-NDV antibody (1:2,500 dilu-tion) for 1 h on ice. After the cells were washed three times with FACS buffer, they were incubated for 1 h on ice with goat anti-rabbit IgG coupled to Alexa Fluor 488 dye (Molecular Probes) (1:1,400 dilution). After three washes in FACS buffer, cells were resuspended in PBS containing 2% paraformaldehyde and subjected to flow cytometry (University of Massachusetts Medical School Flow Cytometry Facility). The background was cells transfected with vector alone and incubated with both primary and secondary antibodies.
Syncytium formation.COS-7 cells were cotransfected with cDNAs encoding wild-type or mutant F protein genes (0.75g/plate) in pSVL and the wild-type HN protein gene (0.75g/plate) in pSVL, using Lipofectin in place of Lipo-fectamine. After 72 h, the numbers of nuclei in 40 fusion areas were counted to determine the average size of syncytia, as previously described (51). Values obtained after transfection of the vector alone were subtracted.
Content mixing.Content mixing was measured using modifications of a protocol previously described (38). Briefly, a plasmid encoding atet -respon-sive transcriptional activator, tTA (Clontech), was transfected (1g/35-mm plate) with pCAGGS-HN (0.75g/35-mm plate) and pCAGGS-F-tmNDV or pCAGGS-F-tmMV, pCAGGS-F-tmSV, or pCAGGS-F-tmG DNAs (0.75g/ 35-mm plate) into COS-7 cells. A separate population of COS-7 cells was trans-fected with a plasmid encoding the-galactosidase protein under the control of thetet-responsive transcriptional activator (Clontech) (1g/35-mm plate). After 5 h, cells transfected with the plasmid encoding the-galactosidase protein were removed from the plate with trypsin and added on top of the HN and F protein-expressing cells. At 40 h posttransfection, when fusion was evident in the con-trols, the monolayers were lysed (Promega cell lysis buffer), and extracts were assayed for-galactosidase activity. Activity due to background fusion typical of COS-7 cells was measured after the transfection of cells with comparable amounts of vector alone. The values obtained were subtracted from the values obtained for cells expressing wild-type or mutant F protein and wild-type HN protein.
Hemifusion.Guinea pig red blood cells (RBCs) (Bio-Link) were washed in PBS, resuspended in PBS, and incubated with 15g/ml of R18, a fluorescently labeled lipid that inserts into viral and cellular membranes (Molecular Probes), for 30 min at room temperature in the dark. Three volumes of complete media (DMEM with 10% fetal calf serum) were added, and incubation was continued for 30 min. The RBCs were washed 5 times in ice-cold PBS, resuspended in PBS containing CaCl2(0.01%), and added to transfected cells grown on coverslips for
48 h posttransfection that had been washed in PBS with CaCl2. Transfected cells
were incubated with labeled RBCs for 30 min on ice. Cells were washed with ice-cold PBS containing CaCl2and then incubated for 60 min at 37°C. Cells were
washed in cold PBS containing CaCl2and immediately visualized, counted, and
photographed using a Nikon fluorescence microscope.
Polyacrylamide gel electrophoresis and Western blotting.Samples were mixed with 25l of gel sample buffer (125 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate, and 10% glycerol) in the presence or absence of 1 M-mercaptoethanol, and proteins were separated in 8% SDS-polyacrylamide gels. After electropho-resis, the gels were equilibrated in transfer buffer (25 mM Tris-HCl [pH 8.2], 192
on November 7, 2019 by guest
http://jvi.asm.org/
mM glycine, and 12% methanol) and transferred for 12 to 15 h to Immobilon-P (Millipore) membranes. The membranes were blocked in PBS containing 0.5% Tween 20 and 10% nonfat dried milk overnight at 4°C. Membranes were washed in PBS-Tween 20 and incubated with anti-HR2 antibody diluted in PBS-Tween 20 and 0.5% nonfat milk for 1 h at room temperature. Membranes were washed and then incubated with anti-rabbit IgG coupled to horseradish peroxidase (1:10,000 dilution in PBS-Tween 20) (Amersham Biosciences) for 1 h at room temperature. Membranes were washed extensively, and bound antibody was detected using an enhanced chemiluminescence (ECL) Western blotting detec-tion reagent system (Amersham Biosciences).
Alternatively, blots previously incubated in PBS-Tween 20 and 10% nonfat dried milk were incubated with anti-FLAG coupled to horseradish peroxidase (HRP) (Sigma) for 1 h at room temperature. Membranes were washed exten-sively, and bound antibody was detected by ECL (Amersham).
Immunofluorescence.COS-7 cells were plated in 35-mm plates containing glass coverslips and transfected as described above. After 48 h, the cells were washed twice with ice-cold IF buffer (PBS containing 1% bovine serum albumin, 0.02% sodium azide, and 0.01% CaCl2) and incubated at 4°C in IF buffer
containing anti-F monoclonal antibody (diluted 1:100) for 1 h. Cells were washed three times with ice-cold IF buffer and incubated with IF buffer containing Alexa Fluor 488-labeled anti-mouse IgG (Molecular Probes) for 1 h on ice. Cells were washed in ice-cold IF buffer, fixed with 2% paraformaldehyde, and mounted for microscopy in IF buffer containing Vectashield (Vector Laboratories).
Images were acquired using Openlab software. All images within an experi-ment were acquired using the same exposure times. Digital images were opened in Adobe Photoshop using the same settings. Quantification of surface immu-nofluorescence was accomplished by determining the mean fluorescence inten-sities for cells, using Adobe Photoshop as described previously (22, 61). Individ-ual cells were outlined manIndivid-ually using the lasso tool on immunofluorescence images opened in Photoshop. The mean fluorescence of the selected areas was determined with the histogram submenu. Background fluorescence values were obtained from empty-vector-transfected cells incubated with both primary and secondary antibodies and were subtracted from the values obtained from cells expressing the viral proteins. The mean fluorescence intensity of 20 or more individual cells was determined, and the values were averaged.
Coimmunoprecipitation.After a wash in ice-cold PBS buffer (145 mM NaCl, 7.6 mM K2HPO4, and 2.4 mM KH2PO4[pH 7.2]), COS-7 cells were lysed in
ice-cold TNE buffer (25 mM Tris-HCl [pH. 7.4], 15 mM NaCl, and 5 mM EDTA) containing 1% Triton X-100, 2.5 mg/mlN-ethylmaleimide (NEM), and 2g/ml DNase I. After incubation on ice for 30 min, cell lysates were homogenized by passing them through a 25-gauge needle 5 times. Extracts were incubated with primary antibodies for 2 h on ice, and then Pansorbin cells, which had been prewashed twice in TNE buffer containing 1% Triton X-100 and 5 mg/ml and then 1 mg/ml of BSA, were added, and the mixture was incubated for 2 h with constant mixing on ice. The immune complexes were washed three times in TNE buffer containing 0.5% Triton X-100 and then resuspended in gel sample buffer containing 6% SDS. All steps were accomplished on ice in a cold room.
Cell-free protein synthesis.Transcription and translation reactions were done as recommended by the Promega Protocols and Application Guide (46a). Tran-scription reaction mixtures (50l) contained 5 g of linearized DNA and transcription buffer (Promega), dithiothreitol (DTT) (10 mM), GTP (0.05 mM), UTP, ATP, CTP (0.5 mM each), RNasin (1l), CAP analogue (0.5 mM), and SP6 polymerase (0.8 U/l) and were incubated for 40 min at 37°. RNA tran-scripts were isolated using an RNeasy RNA cleanup kit (Qiagen) as recom-mended by the manufacturer.
Cell-free translation reactions (25l) contained rabbit reticulocyte extract (17.5l; Promega), amino acids (20M each) minus methionine, [35
S]methio-nine (10Ci; New England Nuclear), RNasin (1g/l), potassium acetate (96 mM), and 0.2g of mRNA transcripts and were incubated for 1 h at 30°. Some reaction mixtures contained dog pancreas membranes, a generous gift from Reid Gilmore, and the peptide glycosylation inhibitor NYT, also obtained from R. Gilmore.
RESULTS
Expression of mutant F proteins. The TM domain of the
NDV F protein was replaced with the TM domains of two other paramyxovirus F proteins, those of measles virus and Sendai virus, and with the TM domain of the unrelated G protein from VSV. The amino terminus of the TM domains of all glycoproteins was defined as the first hydrophobic amino
acid just carboxyl terminal to a serine- and threonine-rich sequence common to these glycoproteins (Fig. 1). The carboxyl terminus of the NDV F protein TM domain was defined as an alanine followed by a cysteine residue, since alanine is neutral with respect to its tendency to be found in a transmembrane helix (65, 70) and since cysteine residues partition more favor-ably at water-lipid interfaces than the center of a bilayer (31). The end of the MV F protein TM domain was defined as a cysteine followed by a charged residue (R), while the ends of the SV F and VSV G protein TM domains were defined by charged residues (R and K, respectively). Using these criteria, the NDV F protein TM domain is 21 amino acids in length, while the MV F protein TM domain is 22 residues long and the TM domains of the SV F and VSV G proteins are 23 residues long.
Two sets of NDV F protein TM mutants were made. In one set, the NDV 21 amino acid TM domain was replaced with the 22-amino-acid TM domain of the MV F protein or the 23-amino-acid TM domains of the SV F and VSV G proteins (F-tmMV, F-tmSV, and F-tmVSV, respectively) (Fig. 1). In the other set of mutants, the 21-amino-acid TM domain of the NDV F protein was replaced with the amino terminal 21 amino acids from the other TM domains (F-tmMV21, F-tmSV21, and F-tmVSV21) (Fig. 1).
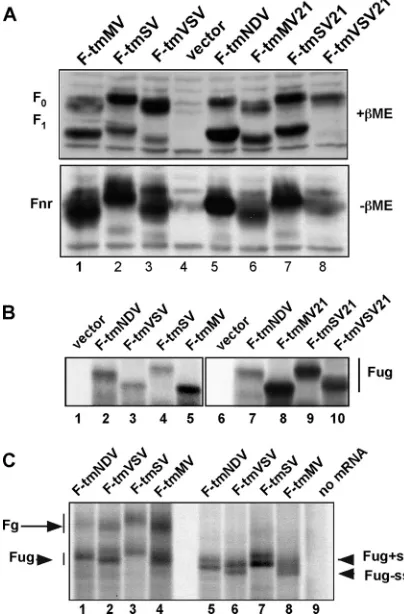
All mutant proteins were expressed at wild-type levels (Fig. 2A). The F-tmMV, F-tmMV21, and F-tmSV21 proteins were cleaved with efficiencies very similar to that of the wild type (Table 1), while the F-tmSV protein was cleaved at a slightly lower efficiency. The F-tmVSV protein and, in particular, the F-tmVSV21 protein were significantly less efficiently cleaved (Table 1), suggesting that the conformations of the NDV F protein ectodomains of these proteins were altered by the foreign TM domain.
The migrations of some of the mutant proteins on poly-FIG. 1. Wild-type and mutant protein TM domains. The sequences of the wild-type NDV F protein (strain AV; F-tmNDV), MV F pro-tein, SV F propro-tein, and VSV G protein TMs and the junctions of the TM domains with the ectodomains and CT domains are shown at the top. The TM domain sequences of the six NDV F protein mutants are shown at the bottom. In the F-tmMV, F-tmSV, and F-tmVSV mutants, the 21-amino-acid NDV F protein TM domain was replaced with the 22-amino-acid TM domain of the MV F protein or the 23-amino-acid TM domain of the SV F or VSV G protein. In the tmMV21, F-tmSV21, and F-tmVSV21 mutants, the 21-amino-acid NDV F protein TM domain was replaced with the amino-terminal 21 amino acids from the TM domain of the MV F, SV F, or VSV G protein.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.301.540.68.216.2]acrylamide gels in the presence (Fig. 2A, top) or absence (Fig. 2A, bottom) of reducing agent were slightly different than that of the wild-type protein, although the predicted molecular weights of the mutant and wild-type proteins were very similar. Altered migration could be due to differential glycosylation site usage or, perhaps, to differences in signal sequence cleavage. We have previously shown that four of the five potential gly-cosylation addition sites in the NDV F wild-type protein are used (33). Faster migration could be due to a failure to utilize a glycosylation site, while the larger-than-wild-type size of F-tmSV and F-F-tmSV21 could be accounted for by usage of the
carbohydrate addition site not used in the wild-type protein. This question was addressed by comparing the sizes of the unglycosylated forms of the mutant proteins to that of the wild type. If differential glycosylation site usage totally accounted for the differences in sizes, the unglycosylated forms of the proteins should comigrate. Unglycosylated wild-type and mu-tant proteins were synthesized in a rabbit reticulocyte cell-free system devoid of membranes, which is therefore incapable of glycosylating proteins (Fig. 2B). Differences in the migration of the resulting unglycosylated proteins (Fug) on polyacrylamide gels were also observed, indicating that the size differences cannot be totally attributed to differential glycosylation site usage. In addition, representative mRNAs were translated in the re-ticulocyte cell-free system containing dog pancreas membranes to allow for glycosylation (Fig. 2C). The size differences in the residual unglycosylated forms of the protein (Fug) were also seen in the glycosylated products of the reaction (Fg).
In order to directly address signal sequence cleavage, these proteins were synthesized in the reticulocyte cell-free system containing dog pancreas membranes and the peptide glycosyl-ation inhibitor NYT, which blocks the addition of the core oligosaccharide to nascent polypeptides translocated across the membranes but allows for signal sequence cleavage of the translocated molecules. Again, size differences were detected in both the intact, untranslocated F protein (F⫹ss) and the translocated F proteins that are missing their signal sequences (F⫺ss). Thus, aberrant signal sequence cleavage or a lack of cleavage cannot totally account for differences in the sizes of the proteins. Taken together, these results suggest that the differences in migration of the wild-type and mutant F proteins likely relate to differences in conformation of the proteins in solution and differential binding of SDS, although differential glycosylation site usage as well cannot be formally eliminated, since the sizes of the unglycosylated forms of the proteins are different.
[image:4.585.59.261.73.380.2]The surface expression of the mutant proteins was deter-mined by flow cytometry (illustrated in Fig. 3A) and by surface immunofluorescence, using a polyclonal anti-NDV antiserum (Fig. 3B). Table 1 shows the quantifications of these and com-parable results for all mutant proteins. The F-tmMV, F-tmSV,
TABLE 1. Cleavage efficiency and cell surface expression of mutant proteins
Protein Efficiency of cleavagea
Surface expressionb
(% of wild type)
F-tmNDV 57⫾4.9 100
F-tmMV 51⫾3.0 95⫾8
F-tmSV 40⫾1.5 118⫾10
F-tmVSV 34⫾2.5 96⫾9
F-tmMV21 51⫾4.9 85⫾6
F-tmSV21 52⫾6.6 94⫾16
F-tmVSV21 14⫾1.0 61⫾1
a
The efficiency of F protein cleavage is represented as the percentage of total F protein that is F1, as determined by densitometer analysis of Western blots
similar to those shown in Fig. 2A. Values are the averages of at least three separate determinations, with standard deviations.
b
[image:4.585.301.540.550.645.2]Surface expression was determined by flow cytometry as described in the legend to Fig. 3, using anti-NDV antisera. The mean fluorescence intensity of cells expressing mutant proteins was expressed as a percentage of the mean fluorescence intensity of the same number of cells expressing the wild-type protein determined in a parallel transfection in the same experiment. Values are the averages of at least three separate experiments, with standard deviations. FIG. 2. Expression of mutant F proteins. (A) Proteins in cell
ex-tracts of COS-7 cells transfected with cDNAs encoding wild-type or mutant F proteins or empty vector were resolved in 10% polyacryl-amide gels under reducing conditions (top,⫹ME) or nonreducing conditions (bottom,⫺ME), and the F proteins were detected by Western blotting using anti-HR2 antibody. F0, uncleaved F protein; F1,
the cleaved F protein; Fnr, nonreduced form of the protein (F0and
F1⫹F2). F proteins are defined in the legend to Fig. 1. (B) mRNAs
generated by transcription of cDNAs encoding wild-type or mutant F proteins were translated in a rabbit reticulocyte cell-free system in the absence of membranes. The resulting radioactively labeled proteins were resolved on polyacrylamide gels and detected by autoradiogra-phy. Fug, unglycosylated F protein. (C) mRNAs generated by cell-free transcription were translated in a rabbit reticulocyte cell-free system containing dog pancreas membranes. Lanes 5 through 9 show products of reactions that also contained the peptide glycosylation inhibitor NYT. Fg, glycosylated F protein; Fug, unglycosylated F protein; F⫹ss, untranslocated F protein retaining the signal sequence; F-ss, translo-cated but unglycosylated F protein with signal sequence cleaved from the protein. Note that the order of proteins in panel B, lanes 1 through 5, and in panel C is different from the order in panels A and B, lanes 6 through 10.
on November 7, 2019 by guest
http://jvi.asm.org/
F-tmVSV, and F-tmSV21 mutant proteins were expressed at cell surfaces at levels quite comparable to that of the wild-type protein, while the surface expression of the F-tmMV21 protein was 85% of that of the wild-type protein and the surface ex-pression of the F-tmVSV21 protein was 61% of that of the wild-type protein.
Fusion activities of mutant proteins. The cell-cell fusion
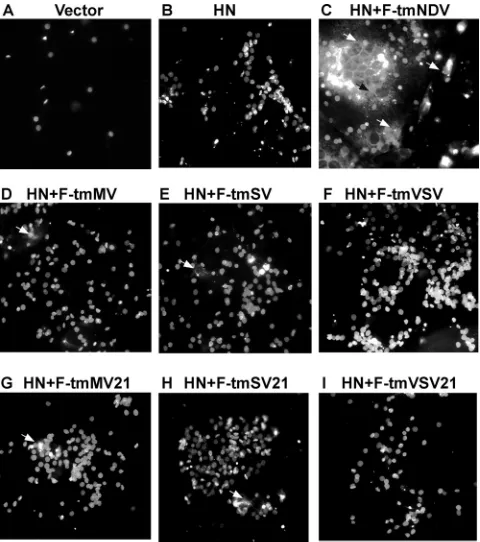
activities of all mutant F proteins were defective as mea-sured by hemifusion, content mixing, and syncytium forma-tion. Hemifusion, the fusion of the outer membranes of target and effector cells, can be detected by the spread of the fluo-rescence-labeled lipid R18 from RBC membranes into mem-branes of cells expressing HN and F-tmNDV, as demonstrated in Fig. 4C by the fluorescence of the underlying cell monolayer (24). That there was no nonspecific fusion of RBC membranes with underlying monolayers in this assay was shown by the total absence of dye transfer between RBCs and cells expressing only HN protein (Fig. 4B). Figure 4D through I shows that there was also virtually no spread of the fluorescence-labeled lipid into underlying cells expressing the mutant F proteins and HN protein during the 1-h course of the experiment. Only rare RBCs in the fields, marked by arrowheads on the figure, showed some spread of R18 fluorescence into the un-derlying cells. Thus, these mutant proteins, under these con-ditions, cannot direct significant amounts of hemifusion.
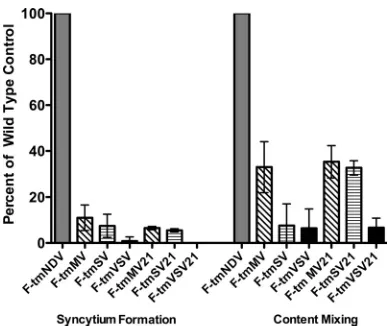
[image:5.585.134.448.71.253.2]All the mutant F proteins also directed only very low levels of syncytium formation (Fig. 5, left). These mutant proteins were also defective to various degrees in the formation of fusion pores (Fig. 5, right), as measured in a reporter assay of content mixing between target and effector cells. While the F-tmSV, F-tmVSV, and F-tm VSV21 mutants directed less then 10% of the levels of pore formation of the wild-type protein, the F-tmMV, F-tmMV21 and F-tmSV21 mutant pro-teins retained approximately 30% of wild-type pore formation. The levels of pore formation may be higher than those of
FIG. 3. Surface expression of wild-type and mutant F proteins. (A) Flow cytometry analyses of the levels of surface expression of three mutant proteins compared to that of the wild-type protein, detected by the binding of anti-NDV antibody after transfection of cells with cDNAs. Cells were processed for flow cytometry at 48 h posttransfection. Each panel shows the numbers of cells expressing the wild-type protein (dashed line), the background (cells transfected with vector and incubated with both primary and secondary antibodies; gray line), and cells expressing a mutant F protein (black line and arrow). (B) Surface expression of wild-type (F-tmNDV) or mutant F proteins (F-tmMV, F-tmSV, and F-tmVSV) detected by immunofluorescence using anti-NDV antibody. All images were acquired at 48 h posttransfection using the same exposures and were processed through Adobe Photoshop using identical settings for all images.
FIG. 4. Hemifusion directed by mutant F proteins. Hemifusion was detected, as described in Materials and Methods and elsewhere in the text, by the spread of fluorescence-labeled lipid R18 from red blood cell membranes to underlying COS-7 cell monolayers. (A) Negative controls transfected with vector alone (pSVL). (B) Cells expressing HN protein alone. (C through I) Monolayers expressing HN protein with wild-type or mutant F proteins (as designated at the top of each panel) and bound fluorescent red blood cells. Positive fusion, present in panel C, is indicated by arrows. The panels show representative fields from multiple experiments. Rare hemifusion events in panels D, E, G, and H are indicated by arrows.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.301.541.353.624.2]hemifusion due to the very different lengths of the two assays (40 h versus 1 h, respectively).
Formation of F and HN protein-containing complexes.That
the mutant proteins were defective in mediating even the ear-liest steps in fusion suggested that these proteins may be de-fective in fusion activation. Activation of paramyxovirus fusion has been linked to an interaction of the F protein with the HN protein (18, 28, 35; reviewed in reference 58). One potential explanation for the fusion-defective phenotypes of these mu-tant proteins is failure to form a functional complex with the NDV HN protein.
We and others previously reported the detection of signifi-cant levels of HN-F protein-containing complexes after Triton X-100 lysis of cells cotransfected with HN and F cDNAs (11, 32, 46, 59, 71). Furthermore, we reported that the levels of these complexes decreased upon HN protein binding to recep-tors (35). We found that these complexes are not reproducibly detected unless lysis was accomplished with Triton X-100 and coimmunoprecipitation was accomplished under strictly con-trolled low-temperature conditions. It is well documented that these conditions do not solubilize liquid-ordered membranes or lipid rafts (14, 55). Indeed, this protocol is used to isolate detergent-resistant membranes (DRMs), a cell fraction thought to represent lipid rafts (56). Since both NDV HN and F pro-teins are detected in DRMs (26, 27), it is possible that the detection of these complexes reflected only the colocalization in DRMs. To determine the functional significance of HN and F protein-containing complexes detected under these condi-tions, we determined the virus specificity of complexes de-tected under these conditions.
It has been reported that Sendai virus glycoproteins are also detected in DRMs (3). Thus, if the NDV HN and F
protein-containing complexes detected after cold Triton X-100 lysis reflect only colocalization in a detergent-resistant membrane fraction, then NDV HN protein should also coimmunoprecipi-tate with the SV F protein due to colocalization in DRMs. To analyze the coprecipitation of NDV HN and SV F proteins, the SV F protein was tagged with the FLAG sequence at its car-boxyl terminus. The FLAG tag was also added to the NDV F protein as a control for the effects of this added sequence. The addition of the FLAG sequence to the end of the NDV F protein had no effect on its cleavage or surface expression, as determined by surface biotinylation (Fig. 6A, lanes 6 and 10), nor on the fusion activity of the protein (data not shown). Similarly, SV F protein with a FLAG sequence at its carboxyl terminus was expressed on cell surfaces at levels comparable to those of the NDV F-FLAG protein (Fig. 6A, lane 12). When NDV F-FLAG was coexpressed with the NDV HN protein, the NDV-F-FLAG was precipitated not only with anti-FLAG (Fig. 6B, lane 3) and anti-F protein monoclonal antibodies (Fig. 6B, lane 4) but also with anti-HN protein antibodies (Fig. 6B, lane 5), demonstrating that a complex containing both the HN protein and NDV-F-FLAG was formed. However, SV-F-FLAG, when coexpressed with the NDV HN protein, was not precip-FIG. 5. Syncytium and pore formation directed by mutant F
[image:6.585.65.259.67.230.2]pro-teins. (Left) Quantification of syncytium formation directed by wild-type or mutant F proteins in the presence of wild-wild-type NDV HN protein. All proteins were expressed using the pSVL vector. Values are expressed as percentages of syncytium formation detected in cells transfected with wild-type F and HN proteins at 72 h posttransfection. Error bars indicate standard deviations. (Right) Quantification of con-tent mixing directed by wild-type or mutant F proteins in the presence of wild-type HN protein as measured by-galactosidase activities. Viral proteins were expressed using the pCAGGs vector. Values are expressed as percentages of content mixing detected in cells trans-fected with wild-type F and HN proteins. Error bars indicate standard deviations.
FIG. 6. Virus specificity of coimmunoprecipitation of HN and F proteins. The cDNAs (plasmids) used for transfection are indicated at the bottoms of the panels. All cDNAs (NDV Fwt, F protein cDNA; HN, HN protein cDNA; NDV-F-FLAG, NDV F cDNA with the FLAG sequence inserted in-frame at the carboxyl terminus of the coding sequence; SV-F-FLAG, SV F cDNA with the FLAG sequence inserted in-frame at the carboxyl terminus of the coding sequence) were inserted into pSVL. (A) Surface expression of FLAG-tagged NDV F and SV F proteins. Surfaces of cells expressing these proteins were biotinylated as previously described (21, 22). After cell lysis, biotinylated molecules were precipitated with NeutrAvidin and the proteins in the precipitate were detected by anti-NDV F (anti-HR2) (left) or anti-FLAG (right) antibody. T, total proteins in cell extracts; B, biotinylated cell surface proteins. (B and C) Coimmunoprecipita-tion of HN protein with FLAG-tagged NDV F or SV F proteins. The antibodies used to precipitate proteins from each extract (IP) are indicated at the top of each lane in both panels. T is the total cell extract and represents 20% of that used in the immunoprecipita-tions. All proteins were electrophoresed in the presence of reducing agent. (B) Detection by Western blotting of NDV-F-FLAG or SV F-F-FLAG protein using a polyclonal antibody specific for the FLAG sequence (anti-FLAG). (C) Detection by Western blotting of HN protein in total extracts or in immunoprecipitates using anti-HN-AS antibody. WB, Western blot.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.300.541.70.241.2]itated with anti-HN protein antibodies (Fig. 6B, lane 12), al-though it was precipitated with anti-FLAG antibody (Fig. 6B, lane 11). Similarly, the NDV HN protein, when coexpressed with NDV F-FLAG, was precipitated with anti-F protein antibody (Fig. 6C, lane 4). The NDV HN protein was not precipitated with anti-FLAG antibody when coexpressed with SV-F-FLAG (Fig. 6C, lane 8). Thus, the NDV HN-F protein-containing complexes detected by coimmunoprecipita-tion after cell lysis in cold Triton X-100 were virus specific.
To determine if the NDV F proteins with the foreign TM domains would coimmunoprecipitate with wild-type NDV HN protein, the mutant F proteins were coexpressed with NDV HN protein and the resulting cell extracts were precipitated with anti-HN protein antibody or anti-F protein antibody (Fig. 7). There was little to no detectable precipitation of any of the mutant F proteins when the anti-HN antibody was used (Fig. 7A, lanes 9, 13, 17, 23, and 26), although the wild-type F protein was efficiently precipitated with HN protein anti-body (Fig. 7A, lanes 5 and 20) in parallel transfections. In addition, in the reciprocal coimmunoprecipitation experiment illustrated in Fig. 7B, anti-F protein antibody precipitated only trace amounts of HN protein coexpressed with the F-tmMV21 or F-tmSV21 mutant (Fig. 7B, lanes 10 and 14), although anti-F protein antibody efficiently precipitated HN protein co-expressed with either the cleaved (Fig. 7B, lane 2) or uncleaved (Fig. 7B, lane 6) wild-type F protein, as previously reported (35). These results show that the NDV HN protein does not form significant levels of a complex containing the mutant F proteins under conditions that can detect significant levels of virus-specific wild-type F and HN protein-containing com-plexes. These results are consistent with the conclusion that these mutant F proteins are defective in fusion activation. The
trace amounts of HN protein coimmunoprecipitated with the F-tmMV21, F-tmSV21, and F-tmMV mutants may account for the low levels of pore formation detected (Fig. 5) after the 40 h of incubation of effector and target cells.
Conformation of the ectodomains of mutant F proteins.The
fusion defects of the mutant proteins coupled with the defec-tive complex formation of these mutant proteins with NDV HN protein suggested that the conformation of the NDV F protein ectodomain may be altered in these mutant proteins. We previously characterized two antibodies that can detect conformational differences in the NDV F protein, a monoclo-nal antibody, Fu1a, and a polyclomonoclo-nal antibody raised against the HR1 domain of the F protein (anti-HR1) (21, 22, 34). To assess the conformations of the mutant proteins, we asked if the reactivities of mutant proteins to these antibodies were altered.
[image:7.585.111.473.69.266.2]The monoclonal antibody anti-Fu1a binds to NDV F protein expressed alone. It also binds to F protein coexpressed with the NDV HN protein if the attachment of HN protein is blocked by the incubation of cells in neuraminidase (21). However, the binding of this antibody to cells actively undergoing cell-cell fusion is significantly reduced (21). These results suggest that this antibody binds preferentially to a prefusion form of the protein. The reactivities of the mutant proteins expressed in the absence of HN protein to anti-Fu1a were visualized by immunofluorescence, as shown in the upper panel of Fig. 8A, and these results were quantified as shown in the lower panel of Fig. 8A. The results show that substitution of the TM do-main significantly increased the signal obtained with anti-Fu1a, indicating that the binding site for this antibody was affected by changes in the TM domain sequence of the protein and that the binding site for this antibody was even more accessible in FIG. 7. Coimmunoprecipitation of wild-type HN protein and mutant F proteins. Plasmids used for transfections are indicated at the bottoms of the lanes (all cDNAs were inserted into pSVL), and the antibodies used to precipitate proteins (IP) are indicated at the top of each lane. The anti-F protein antibody used for IP was anti-Fu1a. The anti-HN protein antibody used for IP was a mixture of anti-HN protein monoclonal antibodies. T is the total extract and represents 20% of the amount used in immunoprecipitations. Results shown are representative of at least two separate determinations for each mutant protein. (A) The F protein, detected in the Western blot (WB) using anti-F protein antibody (anti-HR2), was present in each precipitate and was electrophoresed in the absence of reducing agent. Results with HN and F-tmNDV controls were always accomplished in parallel and are shown. (B) HN protein present in each sample electrophoresed in the presence of reducing agent and detected in the Western blot using anti-HN (anti-HN-AS) protein antibody. Results with HN and F-tmNDV controls were always accomplished in parallel and are shown.
on November 7, 2019 by guest
http://jvi.asm.org/
FIG. 8. Binding of conformation-sensitive antibodies to surface-expressed mutant F proteins. (A) Binding of anti-Fu1a to cell surfaces. (Top) Representative immunofluorescence detected after binding anti-Fu1a to COS-7 cells expressing wild-type or mutant F proteins or empty vector (in the absence of HN protein expression). (Bottom) Graphs showing the quantification of these results and the results of identical experiments, accomplished as described in Materials and Methods. All images used for each data set were acquired using the same exposures and were processed through Adobe Photoshop using identical settings for all images. Error bars represent standard deviations. (B) Binding of anti-HR1 to cell surfaces expressing mutant proteins. (Top) Representative immunofluorescence detected after binding anti-HR1 antibody to COS-7 cells expressing wild-type or mutant F proteins or empty vector (in the absence of HN protein expression). (Bottom) Graphs showing the quantification of these results as well as those from identical experiments, accomplished as described in Materials and Methods. All images for each data set were acquired using the same exposures and were processed through Adobe Photoshop using identical settings for all images. Error bars represent standard deviations. The binding of anti-NDV antibody is quantified in Table 1.
on November 7, 2019 by guest
http://jvi.asm.org/
mutant F proteins than in the prefusion form of the wild-type F protein.
In contrast, another antibody, a polyclonal antibody raised against a peptide corresponding to the HR1 domain of the protein (anti-HR1), does not bind to F protein expressed alone (34), nor does it bind to F protein coexpressed with HN protein under conditions in which HN protein receptor binding is inhibited (21) or under conditions in which fusion is inhibited (21, 22). Nor does the antibody bind to the NDV F protein coexpressed with the SV HN protein or the MV H protein (34). However, it does bind to cells undergoing cell-to-cell fusion (21, 22, 34). These combined results suggest that the anti-HR1 antibody binds to a postactivation form of the F protein.
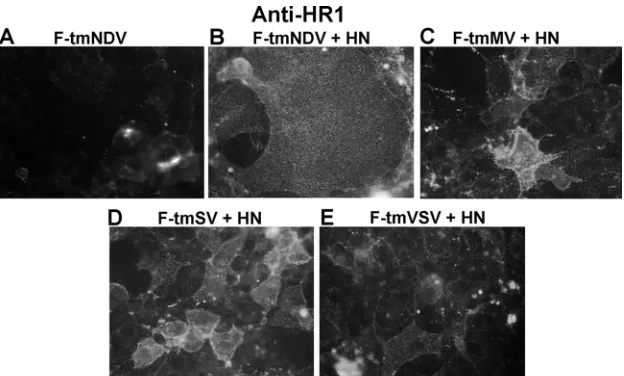
Figure 8B shows that while anti-HR1 antibody minimally bound to the wild-type F protein expressed alone, as previously reported (34), it did bind to NDV F proteins with foreign TM domains, indicating that the HR1 domains of these mutant proteins were more accessible to anti-HR1 antibodies than the HR1 domain in the wild-type protein. Indeed, anti-HR1 bind-ing to mutant proteins was characteristic of a postactivation form of the wild-type F protein. Not surprisingly, since we found no evidence of an HN protein-mutant F protein com-plex, the results were very similar if the mutant F proteins were coexpressed with HN protein (Fig. 9).
Thus, binding of both the monoclonal anti-Fu1a antibody and the HR1 peptide antibody was significantly altered in the mutant proteins, indicating that the foreign transmembrane domain sequences affect the conformation of the ectodomain of the F protein.
DISCUSSION
The role of the transmembrane domains of paramyxovirus fusion proteins has not been extensively investigated. In a recent study, Bissonnette et al. (6) reported the results of a mutational analysis of the TM domain of the parainfluenza virus 5 (PIV5) F protein TM domain. Alanine-scanning
mu-tagenesis, which substituted three alanines sequentially for au-thentic residues across the domain, resulted in mutant proteins whose fusion activities were not drastically affected by these substitutions. Most mutant proteins directed at least 50% of wild-type levels of hemifusion, and most directed syncytia with sizes similar to that of the wild type. There were, however, two residues near the amino-terminal end of the domain that were important for the fusion activity of the protein. The authors concluded that specific resides in the TM domain, particularly those at the amino-terminal end, are important for conforma-tional intermediates of the F protein formed after fusion acti-vation and en route to the final postfusion conformation. The effects of the mutations on the preactivation form of the pro-tein were not specifically addressed in this study.
To explore the role of specific sequences in the structure and function of the NDV fusion protein, we chose to replace the entire TM domain of the NDV F protein with the TM domain of two other paramyxovirus F proteins. It seemed possible that paramyxovirus F protein TM domains might be interchange-able, since these domains have all evolved to support the con-formation and activities of proteins that are likely very similar in structure and function. However, if the specific sequence were important for the structure and activity of each F protein, then replacement of one paramyxovirus F protein TM domain with another would result in a nonfunctional protein.
[image:9.585.136.448.68.256.2]The results presented here indicate that the specific se-quence of the NDV F protein TM domain is important for its structure and function. While replacement of the NDV F pro-tein TM domain with that of the MV F propro-tein or the SV F protein resulted in mutant proteins that were expressed and cleaved at or near wild-type levels, the mutant proteins were quite defective in membrane fusion. None of the mutant pro-teins formed significant levels of complexes with the NDV HN protein, and the conformation of the ectodomain of the pre-activation form of the mutant proteins was different from that of the wild-type protein, as determined by binding of confor-mation-sensitive antibodies.
FIG. 9. Effect of HN protein expression on anti-HR1 binding to surface-expressed mutant F proteins. The panels show representative immunofluorescence detected after binding anti-HR1 antibody to COS-7 cells coexpressing wild-type or mutant F proteins and the HN protein. All images, acquired using the same exposures, were processed through Adobe Photoshop using identical settings for all images.
on November 7, 2019 by guest
http://jvi.asm.org/
TM domain mutant proteins and membrane fusion. Mem-brane fusion is classically divided into sequential stages (8, 13). The first step, hemifusion, is the fusion of the outer leaflets of the target and effector membranes and is monitored by the transfer of a fluorescent lipid between target and effector membranes (22, 24, 27). Pore formation follows hemifusion and involves the fusion of both outer and inner leaflets of the two membranes, resulting in a pore through which molecules may pass between target and effector membranes, and is mea-sured by reporter assays that depend upon this content mixing (for examples, see reference 33). The final stage of paramyxo-virus fusion is pore expansion, which is measured by syncytium formation. The NDV F TM mutant proteins were defective in all stages of fusion. They all showed virtually no hemifu-sion. However, three of the mutant proteins, the F-tmMV, F-tmMV21, and F-tmSV21 mutants, could direct low levels of pore formation, up to 30% of that of the wild-type con-trol. The minimal levels of hemifusion and low levels of pore formation detected with these mutants likely indicate that these mutants can direct membrane fusion with very slow kinetics, since the hemifusion assay was monitored after a short-term (1-h) incubation of target and effector membranes, while content mix-ing was measured after 40 h. All the mutant proteins were incapable of directing significant levels of syncytium formation, measured after 72 h, indicating major defects in pore expan-sion. The low levels of syncytium formation may relate to slow fusion kinetics or failure of these mutant proteins to support pore expansion.
Since the mutant F proteins were defective in the earliest stages of membrane fusion, it is possible that these mutant proteins were defective in fusion activation. Proteolytic cleav-age of paramyxovirus F0 to the disulfide-linked F1⫹F2
het-erodimer is required for fusion activation (reviewed in refer-ence 29). Indeed, the F proteins containing the VSV G protein TM domain were defective in cleavage, which could account for their failure to direct any detectable fusion. However, the mutant proteins with the SV or MV TM domains were pro-teolytically cleaved at levels similar to that of the wild type; thus, cleavage defects are not likely the primary explanation for the defective fusion activities of these proteins.
Paramyxovirus F protein activation is also linked to virus-specific interactions of the F protein with the attachment pro-tein, the HN, H, or G protein (reviewed in reference 58). All available evidence indicates that in most paramyxovirus sys-tems, this interaction allows attachment protein receptor bind-ing to stimulate the conformational changes in F protein re-quired to mediate membrane fusion (reviewed in references 29 and 58). Thus, one possible explanation for the fusion defects of these mutant proteins is a failure to form a complex con-taining the NDV HN protein, thus blocking any transmission of a signal between the HN and F proteins upon HN protein receptor binding. We and others (1, 2, 11, 32, 35, 46, 59, 71) have demonstrated a complex containing the HN and F pro-teins but most often under conditions that are also used to isolate detergent-resistant membranes. To explore the pos-sibility that the complexes detected under these conditions reflect only colocalization in a detergent-resistant membrane fragment rather than direct protein-protein interactions, we first asked if HN-F protein-containing complexes were virus specific. A hallmark of the detected attachment protein-F
pro-tein complex was its virus specificity, which correlates with the virus specificity of the promotion of membrane fusion by the attachment protein (18, 71). This specificity has been inter-preted as evidence of a direct attachment protein-F protein interaction. Indeed, our results indicated that these complexes are virus specific. We showed that coexpression of the SV F protein with the NDV HN protein did not result in coimmu-noprecipitation of the two proteins. Detection of HN and F protein-containing complexes under these conditions certainly does not prove a direct interaction between the two proteins. However, the virus specificity of these complexes correlates with the established virus-specific fusion promotion activity of HN proteins. We also showed, in results consistent with our previous observations (35), that more of the complex is de-tected between a fusion-inactive, uncleaved NDV F protein and an HN protein than between the two proteins precipitated from monolayers undergoing extensive cell-cell fusion. Thus, failure to detect an NDV HN-SV F protein complex cannot be due to the absence of cleavage of the SV F protein.
Using these conditions, the mutant F proteins with foreign TM domains were defective in forming complexes with the NDV HN protein. Thus, failure to activate the fusion activity of these mutant proteins is one possible explanation for their fusion defects. It is difficult to assess the reasons for failure to detect complexes of the mutant F proteins and the HN protein since the domain of the F protein that binds to the HN protein has not been clearly defined. Mutant F proteins would fail to complex with the HN protein if the transmembrane domain of the NDV F protein were part of its HN protein-interacting domain. The one study of the fusion activities of different chime-ric F proteins with sequences from two different paramyxovirus F proteins did not clearly indicate if the F protein TM domain has a role in the type-specific interactions with an attachment protein (64). Indeed, the authors note that they cannot exclude the possibility that the TM domain has a role in attachment protein interactions. Alternatively, and perhaps more likely, the foreign TM domains could alter the conformation of the NDV F protein ectodomain, resulting in loss or alteration of the HN protein binding site on the F protein ectodomain.
TM domain mutant proteins and the conformation of the
ectodomain.That a foreign TM domain can significantly affect
the conformation of the NDV F protein ectodomain is clearly shown by the failure of the F protein with the VSV G protein TM domain to be cleaved efficiently. Altered migration of the proteins in polyacrylamide gels also suggests an altered con-formation. To assess the conformation of the ectodomain of NDV F protein with TM domains of other paramyxoviruses, we utilized two different antibodies, antibodies that we have previously shown to be dependent upon the conformation of the F protein. One antibody, anti-Fu1a, binds a prefusion form of F protein preferentially, while another, anti-HR1, binds only a postfusion activation form of the protein. Using these two antibodies, we found that the mutant F proteins, expressed in the absence of HN protein, bound both antibodies at levels significantly higher than that of the wild-type protein. Most significantly, the mutant proteins bound anti-HR1 in the ab-sence of the HN protein. These results may indicate that these mutant proteins are in an aberrant conformation that does not resemble either the pre- or postfusion form of the protein but is rather more open and in which both the anti-Fu1a epitope
on November 7, 2019 by guest
http://jvi.asm.org/
and the HR1 domain are readily accessible. Alternatively, these mutant proteins, in the absence of the HN protein, may be in a very early postactivation conformation in which the anti-Fu1a epitope has not been lost but the HR1 domain is accessible. In either case, replacement of the NDV F protein TM domain with another paramyxovirus F protein TM domain has significant effects on the conformation of the ectodomain of the NDV F protein, effects that may account for the failure of the proteins to complex with HN protein and to direct membrane fusion.
In summary, replacement of the NDV F protein TM domain with comparable domains from closely related paramyxovirus F proteins resulted in proteins with defective fusion activities. The most likely explanation for defective fusion activity is the abnormal conformation of the mutant F protein ectodomains. Altered conformation may affect proper folding of a metasta-ble form of the protein or subsequent conformational inter-mediates important for membrane merger, although our re-sults are more consistent with the former possibility. That alterations in the TM domain of a viral glycoprotein can affect the conformational intermediates has recently been proposed to account for defective membrane fusion by the HIV Env glycoprotein with a TM domain from either the VSV G protein or the glycophorin A glycoprotein (25). Shang and Hunter (54) have reported that alteration of specific residues in the HIV Env TM domain correlated with conformational changes in specific regions of gp120 and gp41 and slower and less efficient fusion.
ACKNOWLEDGMENTS
This work was supported by grant AI30572 from the National Insti-tutes of Health.
We thank M. Peeples and R. Iorio for anti-F protein (Fu1a) and anti-HN protein monoclonal antibodies, respectively, and M. B. A. Oldstone, D. Nayak, and M. Schmidt for MV F, SV F, and VSV G protein cDNAs, respectively. We thank Reid Gilmore for dog pancreas membranes and the peptide NYT.
REFERENCES
1.Aguilar, H. C., et al.2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signal-ing. J. Virol.81:4520–4532.
2.Aguilar, H. C., et al.2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and virus entry. J. Virol. 80:4878–4889.
3.Ali, A., and N. P. Nayak.2000. Assembly of Sendai virus: M protein interacts with F and HN proteins and with the cytoplasmic tail and transmembrane domain of F protein. Virology276:289–303.
4.Armstrong, R. T., A. S. Kushnir, and J. M. White.2000. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol.151:425–438. 5.Bagai, S., and R. A. Lamb.1996. Truncation of the COOH-terminal region
of paramyxovirus SV5 fusion protein leads to hemifusion but not complete fusion. J. Cell Biol.135:73–84.
6.Bissonnette, M. L. Z., J. E. Donald, W. F. DeGrado, T. S. Jardetzky, and R. A. Lamb.2009. Functional analysis of the transmembrane domain in paramyxovirus F protein-mediated membrane fusion. J. Mol. Biol.386: 14–36.
7.Chen, L., et al.2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure9:255–266.
8.Chernomordik, L. V., and M. M. Kozlov.2008. Mechanics of membrane fusion. Nat. Struct. Mol. Biol.15:675–683.
9.Cleverley, D. Z., and J. Lenard.1998. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. U. S. A.95:3425–3430.
10.Colman, P. M., and M. C. Lawrence.2003. The structural biology of type 1 viral membrane fusion. Nat. Rev. Mol. Cell Biol.4:309–316.
11.Deng, R., et al.1999. Mutations in the Newcastle disease virus
hemaggluti-nin-neuraminidase protein that interfere with its ability to interact with homologous F protein in the promotion of fusion. Virology253:43–54. 12.Dolganiuc, V., L. McGinnes, E. J. Luna, and T. G. Morrison.2003. Role of
the cytoplasmic domain of the Newcastle disease virus fusion protein in association with lipid rafts. J. Virol.77:12968–12979.
13.Earp, L. J., S. E. Delos, H. E. Park, and J. M. White.2005. The many mechanisms of viral membrane fusion. Curr. Top. Microbiol. Immunol. 285:25–66.
14.Harder, T., P. Scheiffele, P. Verkade, and K. Simons.1998. Lipid domain structure of the plasma membrane revealed by patching of membrane com-ponents. J. Cell Biol.141:929–942.
15.Heinz, F. X., and S. L. Allison.2001. The machinery for flavivirus fusion with host cell membranes. Curr. Opin. Microbiol.4:450–455.
16.Hernandez, L. D., L. R. Hoffman, T. G. Wolfsberg, and J. M. White.1996. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol.12:627–661. 17.Hernandez, L. D., et al.1997. Activation of a retroviral membrane fusion
protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol.139:1455–1464.
18.Hu, X., R. Ray, and R. W. Compans.1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainflu-enza viruses. J. Virol.66:1528–1534.
19.Iorio, R. M., R. L. Glickman, A. M. Riel, J. P. Sheehan, and M. A. Bratt. 1989. Functional and neutralization profile of seven overlapping antigenic sites on the HN glycoprotein of Newcastle disease virus: monoclonal anti-bodies to some sites prevent viral attachment. Virus Res.13:245–262. 20.Iorio, R. M., et al.1991. Neutralization map of the
hemagglutinin-neuramin-idase glycoprotein of Newcastle disease virus: domains recognized by mono-clonal antibodies that prevent receptor recognition. J. Virol.65:4999–5006. 21.Jain, S., L. W. McGinnes, and T. G. Morrison.2008. Overexpression of thiol/disulfide isomerases enhances membrane fusion directed by Newcastle disease virus fusion protein. J. Virol.82:12039–12048.
22.Jain, S., L. W. McGinnes, and T. G. Morrison.2007. Thiol/disulfide ex-change is required for membrane fusion directed by the Newcastle disease virus fusion protein. J. Virol.81:2328–2339.
23.Jardetzky, T. S., and R. A. Lamb.2004. A class act. Nature427:307–308. 24.Kemble, G. W., T. Danieli, and J. W. White.1994. Lipid-anchored influenza
hemagglutinin promotes hemifusion, not complete fusion. Cell76:383–391. 25.Kondo, N., K. Miyauchi, F. Meng, A. Iwamoto, and Z. Matsuda.2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem.285:14681–14688.
26.Laliberte, J. P., L. W. McGinnes, M. E. Peeples, and T. G. Morrison.2006. Integrity of membrane lipid rafts is necessary for the ordered assembly and release of infectious Newcastle disease virus particles. J. Virol.80:10652– 10662.
27.Laliberte, J. P., L. W. McGinnes, and T. G. Morrison.2007. Incorporation of functional HN-F glycoprotein-containing complexes into Newcastle disease virus is dependent on cholesterol and membrane lipid raft integrity. J. Virol. 81:10636–10648.
28.Lamb, R. A.1993. Paramyxovirus fusion: a hypothesis of changes. Virology 197:1–11.
29.Lamb, R. A., and G. D. Parks.2007. Paramyxoviridae: the viruses and their replication, p. 1450–1496.InD. M. Knipe et al. (ed.), Fields virology, 5th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, PA.
30.Lamb, R. A., R. G. Paterson, and T. S. Jardetzky.2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37.
31.MacCallum, J. L., W. F. D. Bennett, and D. P. Tieleman.2007. Partitioning of amino acid side chains into lipid bilayers: results from computer simula-tions and comparison to experiments. J. Gen. Physiol.129:371–377. 32.Malvoisin, E., and T. F. Wild.1993. Measles virus glycoproteins: studies on
the structure and interaction of the haemagglutinin and fusion proteins. J. Gen. Virol.74:2365–2372.
33.McGinnes, L., T. Sergel, J. Reitter, and T. Morrison.2001. Carbohydrate modifications of the NDV fusion protein heptad repeat domains influence maturation and fusion activity. Virology283:332–342.
34.McGinnes, L. W., K. Gravel, and T. G. Morrison.2002. Newcastle disease virus HN protein alters the conformation of the F protein at cell surfaces. J. Virol.76:12622–12633.
35.McGinnes, L. W., and T. G. Morrison.2006. Inhibition of receptor binding stabilizes Newcastle disease virus HN and F protein-containing complexes. J. Virol.80:2894–2903.
36.McGinnes, L. W., H. Pantua, J. Reitter, and T. G. Morrison.1 June 2006, posting date. Newcastle disease virus: propagation, quantification, and storage. Curr. Protoc. Microbiol. 1:15F2.1-15F.2.18. doi:10.1002/ 9780471729259.mc15f02s01.
37.McGinnes, L. W., J. N. Reitter, K. Gravel, and T. G. Morrison.2003. Evidence for mixed membrane topology of the Newcastle disease virus fusion protein. J. Virol.77:1951–1963.
38.McGinnes, L. W., et al.2001. Mutational analysis of the membrane proximal heptad repeat of Newcastle disease virus fusion protein. Virology289:343– 352.
on November 7, 2019 by guest
http://jvi.asm.org/
39.Melikyan, G. B., S. Lin, M. G. Roth, and F. S. Cohen.1999. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell 10:1821–1836.
40.Melikyan, G. B., et al.2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol.151:413–423.
41.Melikyan, G. B., R. M. Markosyan, M. G. Roth, and F. S. Cohen.2000. A point mutation in the transmembrane domain of the hemagglutinin of in-fluenza virus stabilizes a hemifusion intermediate that can transit to fusion. Mol. Biol. Cell11:3765–3775.
42.Morrison, T. G., M. E. Peeples, and L. W. McGinnes.1987. Conformational change in a viral glycoprotein during maturation due to disulfide bond disruption. Proc. Natl. Acad. Sci. U. S. A.84:1020–1029.
43.Niwa, H., K. Yamamura, and J. Miyazaki.1991. Efficient selection for high expression transfectants with a novel eukaryotic vector. Gene108:193–199. 44.Odell, D., E. Wanas, J. Yan, and H. P. Ghosh.1997. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of the vesicular stomatitis virus glycoprotein G. J. Virol.71:7996–8000.
45.Owens, R. J., C. Burke, and J. K. Rose.1994. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycopro-tein that affect fusion activity. J. Virol.68:570–574.
46.Plemper, R. K., A. L. Hammond, D. Gerlier, A. K. Fielding, and R. Cattaneo. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol.76:5051–5061.
46a.Promega.2009. Promega protocols and application guide. Promega, Madi-son, WI.
47.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin.2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191.
48.Russell, C. J., T. S. Jardetzky, and R. A. Lamb.2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034.
49.Schroth-Diez, B., E. Ponimaskin, H. Reverey, M. G. Schmidt, and A. Herrmann.1998. Fusion activity of transmembrane and cytoplasmic domain chimeras of the influenza virus glycoprotein hemagglutinin. J. Virol.72:133– 141.
50.Sergel, T., L. McGinnes, and T. Morrison.2001. Mutations in the fusion peptide and adjacent heptad repeat inhibit folding or activity of the New-castle disease virus fusion protein. J. Virol.75:7934–7943.
51.Sergel, T., L. W. McGinnes, and T. G. Morrison.1993. The fusion promotion activity of the NDV HN protein does not correlate with neuraminidase activity. Virology196:831–834.
52.Sergel, T., and T. G. Morrison.1995. Mutations in the cytoplasmic domain of the fusion glycoprotein of Newcastle disease virus depress syncytia for-mation. Virology210:264–272.
53.Sergel-Germano, T., C. McQuain, and T. Morrison.1994. Mutations in the fusion peptide and heptad repeat regions of the Newcastle disease virus fusion protein block fusion. J. Virol.68:7654–7658.
54.Shang, L., and E. Hunter.2010. Residues in the membrane-spanning domain core modulate conformation and fusogenicity of the HIV-1 envelope glyco-protein. Virology404:158–167.
55.Simons, K., and E. Ikonen.1997. Functional rafts in cell membranes. Nature 387:569–572.
56.Simons, K., and D. Toomre.2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol.1:31–39.
57.Skehel, J. J., and D. C. Wiley.2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem.69:531– 569.
58.Smith, E. C., A. Popa, A. Chang, C. Masante, and R. E. Dutch.2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227.
59.Stone-Hulslander, J., and T. G. Morrison.1997. Detection of an interaction between the HN and F proteins in Newcastle disease virus-infected cells. J. Virol.71:6287–6295.
60.Swanson, K., et al.2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology402:372–379.
61.Takizawa, N., et al.2006. Supervillin modulation of focal adhesions involving TRIP6/ZRP-1. J. Cell Biol.174:447–458.
62.Taylor, G. M., and D. A. Sanders.1999. The role of the membrane-spanning domain sequence in glycoprotein-mediated membrane fusion. Mol. Biol. Cell10:2803–2815.
63.Tong, S., et al.2002. Regulation of fusion activity by the cytoplasmic domain of a paramyxovirus F protein. Virology301:322–333.
64.Tsurudome, M., et al.1998. Identification of regions on the fusion protein of human parainfluenza virus type 2 which are required for haemagglutinin-neuraminidase proteins to promote cell fusion. J. Gen. Virol.79:279–289. 65.von Heijne, G.2007. Formation of transmembrane helices in vivo—is
hydro-phobicity all that matters? J. Gen. Physiol.129:353–356.
66.Waning, D. L., C. J. Russell, T. S. Jardetzky, and R. A. Lamb.2004. Acti-vation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A.101:9217–9222. 67.Weissenhorn, W., et al.1999. Structural basis for membrane fusion by
en-veloped viruses. Mol. Membr. Biol.16:3–9.
68.White, J. M., S. E. Delos, M. Brecher, and K. Schornberg.2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol.43:189–219.
69.Wilk, T., T. Pfeiffer, A. Bukovsky, G. Moldenhauer, and V. Bosch.1996. Glycoprotein incorporation and HIV-1 infectivity despite exchange of the gp160 membrane-spanning domain. Virology218:269–274.
70.Wolfenden, R.2007. Experimental measures of amino acid hydrophobicity and the topology of transmembrane and globular proteins. J. Gen. Physiol. 129:357–362.
71.Yao, Q., X. Hu, and R. Compans.1997. Association of the parainfluenza virus fusion and hemagglutinin-neuraminidase glycoproteins on cell surfaces. J. Virol.71:650–656.
72.Yin, H.-S., R. G. Paterson, X. Wen, R. A. Lamb, and T. S. Jardetzky.2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A.102:9288–9293.
73.Yin, H.-S., X. Wen, R. G. Paterson, R. A. Lamb, and T. S. Jardetzky.2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature439:38–44.