Copyright © 2001, American Society for Microbiology. All Rights Reserved.
NOTES
Enhancer and Long-Term Expression Functions of Herpes Simplex
Virus Type 1 Latency-Associated Promoter Are both
Located in the Same Region
HERVE BERTHOMME,1,2JOE¨LLE THOMAS,1PASCALE TEXIER,1ALBERTO EPSTEIN,1
ANDLAWRENCE T. FELDMAN2*
Centre de Ge´ne´tique Mole´culaire et Cellulaire, UMR5534 CNRS, Universite´ Claude Bernard Lyon I, Villeurbanne,
France,1and Department of Microbiology and Immunology, UCLA School of Medicine,
Los Angeles, California 900952
Received 29 November 2000/Accepted 6 February 2001
During herpes simplex virus type 1 (HSV-1) latent infection in vivo, the latency-associated promoter (LAP) is the only promoter to remain highly active long term. In a previous attempt to characterize LAP activity in vitro and in a mouse model, we showed that a 1.5-kb fragment called the long-term expression element (LTE), located immediately downstream from the transcriptional start site of LAP, was able to (i) increase gene expression in an orientation-independent manner, regardless of the cell type or the promoter used in vitro (enhancer activity) and (ii) keep LAP active during latency in vivo (long-term expression activity) (H. Berthomme, J. Lokensgard, L. Yang, T. Margolis, and L. T. Feldman, J. Virol. 74:3613–3622, 2000). To determine if these two functions could be separated genetically, we conducted a mutational analysis on the LTE and analyzed the effect on the LAP-LTE properties in both transient expression in cell culture and mouse dorsal root ganglia lytic and latent infection. In this report, we show that the first half of the LTE sequence, corresponding to the region previously described as LAP2 or exon1, encodes the enhancer function. This same region is also required to keep the LAP active during latency. These results exclude the intron region as containing any significant enhancer activity or any ability to keep the LAP active during latency. The results also show that these two functions have not been separated, leaving open the possibility that there is no long-term expression function per se but that the enhancer itself may function to keep the LAP active during latency by raising the level of expression to a detectable one. Further mutational analysis will be required to determine if these two potential functions continue to cosegregate.
The common and most intriguing property among herpesvi-ruses is the ability to establish a latent infection in their hosts. This strategy of infection allows the virus to persist in the cell until it reactivates and produces infectious particles, making the host a perpetual reservoir for virus transmission. Following this strategy, herpes simplex virus type 1 (HSV-1), the proto-type virus of theAlphaherpesviridaesubfamily, is able to estab-lish latency primarily in trigeminal ganglia in humans (36). HSV-1 maintains latency throughout the life of the host but can periodically reactivate under conditions such as stress, fever, and UV light. To study HSV-1 latency and reactivation, several animal models have been developed, mainly in mice and rabbits. These models have proven useful for better un-derstanding the mechanisms of establishment, maintenance of latency, and reactivation in vivo. An interesting aspect of HSV-1 latency is that only one transcription unit is abundantly expressed (7, 8, 23, 34, 41, 44, 45), whereas expression of all the lytic genes is not detected. This transcription unit leads to the synthesis of latency-associated Transcripts (LATs)
correspond-ing to the minor LATs and the major LATs (29, 48, 50). The minor LATs represent a less abundant polyadenylated RNA species which is about 8.3 kb in length and is dependent on the latency-associated promoter (LAP) (2, 12, 53, 55). The major LATs are highly abundant RNAs which are thought to be spliced from the primary minor LATs into stable introns of 1.5 and 2.0 kb (9, 13, 24, 35, 42, 49, 52).
The LAT RNAs were first described as not essential for establishment of latency (20, 21, 25, 39, 43). However, it was shown more recently that LAT promoter mutants not able to produce any of the LAT RNAs were in fact impaired for efficient establishment of latency (32, 38, 47). A possible ex-planation for this effect could derive from a LAT antisense mechanism, as suggested previously (45), since the LAT tran-scripts are complementary in part to the ICP0 mRNA (13, 23, 34, 51). This hypothesis is corroborated by other studies show-ing that LAT mutant viruses were able to produce more pro-ductive-cycle gene expression during acute infection in sensory neurons in vivo (5, 15). Thus, the role of the LAT RNAs would be to reduce immediate-early gene expression during the lytic process of in vivo infection, therefore enhancing establishment of latency. Another function mapped to the LAT region was an increased ability to reactivate from latency. In fact, several lines of evidences have shown that LAT promoter and/or LAT * Corresponding author. Mailing address: Department of
Microbi-ology and ImmunMicrobi-ology, 43-169CHS, UCLA School of Medicine, Los Angeles, CA 90095-1405. Phone: (310) 206-1014. Fax: (310) 206-3865. E-mail: [email protected].
4386
on November 9, 2019 by guest
http://jvi.asm.org/
transcript deletion mutants were altered for reactivation from the latent state (4, 17–19, 25, 30). One hypothesis for this activity is that a LAT RNA-associated function would preserve the integrity of the neurons until all the essential steps of reactivation are accomplished. This function could be due to an antiapoptotic effect of the LAT in reactivating infected cells (31). Another possibility concerns a putative viral protein en-coded by the LAT RNAs, which was described as a virulence factor capable of complementing growth deficiencies of imme-diate-early gene expression in ICP0 and vmw65 mutants (46). As a consequence, this factor could promote reactivation and help it proceed.
Regarding the role that the LAT RNAs play during estab-lishment of latency and/or reactivation, we are interested in understanding why LAT RNAs are highly expressed during latency, whereas all the other genes of the genome are not. This led us to study the structure of the LAT promoter respon-sible for the LAT RNAs’ transcription during latency. The LAT promoter has been very well characterized, and several motifs have been identified in the proximal region upstream of the transcriptional start site (1, 2, 10, 14, 26, 40, 54). More recently, we identified another region which is required for the LAT promoter to remain active long term in dorsal root gan-glia (DRG) latently infected neurons (27). This sequence, the long-term expression (LTE) element, corresponds to a 1.5-kbp fragment located immediately downstream of the LAT tran-script cap site. We further showed that the LTE element con-tained several functions, including (i) an enhancer activity in-dependent of the cell type or the promoter used in vitro, (ii) a promoter activity in productively infected cells in vitro, and (iii) a long-term expression activity in sensory neurons in vivo (3). To further define the structure of the LAP and to deter-mine if in fact these three functions colocalized to the same region or originated from different segments of the LTE se-quence, we conducted a mutational analysis on the LTE ele-ment. Using transient-expression experiments in cells cultured in vitro as well as recombinant viruses in a mouse model, we were able to show that a 0.6-kb fragment, corresponding to the first half of the LTE sequence, was sufficient when associated with the LAT promoter for both enhancer and long-term ex-pression activities. However, this same region did not allow the previously described promoter-like activity (3), suggesting that other elements were required for this function.
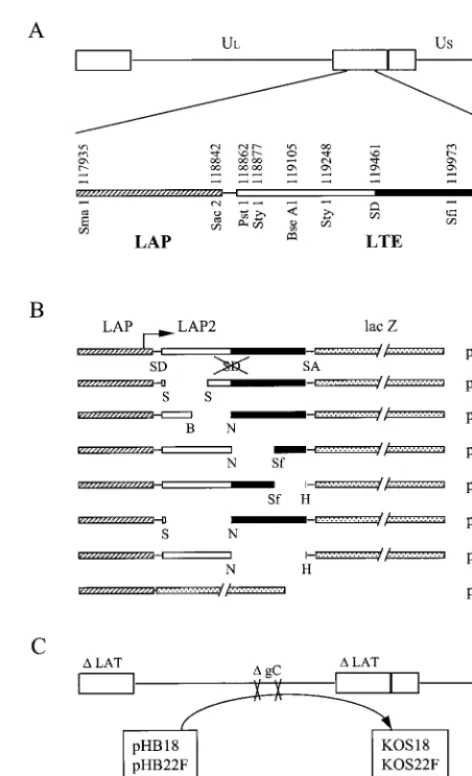
[image:2.612.314.550.88.476.2]In a previous report, we showed that the 1.5-kb LTE se-quence which is located immediately downstream from the transcriptional start site of LAP was sufficient to increase gene expression in an orientation-independent manner, regardless of the cell type or the promoter used in vitro (3). The increase in LAT promoter expression was measured in transient-expres-sion assays and in viral infections by inserting the entire LTE DNA fragment into an intron cassette located between the LAT promoter and thelacZreporter gene. These data sup-ported the idea that the LTE region contained an enhancer activity. As shown in Fig. 1A, the LTE sequence encompasses the previously termed LAP2 promoter extending from aPstI site (position 118862) to the 5⬘end of the 2.0-kb LAT (position 119461), as well as the first 837 bp of the 2.0-kb LATs (posi-tions 119462 to 120298). Thus, we questioned whether the enhancer activity originated from LAP2, part of the LAT in-tron, or another smaller but unidentified region inside the LTE
FIG. 1. Graphic map of the DNA structures of plasmids and vi-ruses. (A) The HSV-1 genome is classically represented in a linear form, including the two unique regions ULand US, flanked by the inverted repeats (open rectangle). In the expanded view of the LAT promoter (LAP) LTE sequence from one of the two copies present in the genome, the open bar represents the region of exon 1 or the leader RNA 5⬘of the intron. SD, splice donor site of the LAT intron; note that in all LAT promoter-LTE-lacZconstructs, this splice donor site is mutated, as indicated by the crossed-out SD in the first line of panel B. This site is mutated so as not to interfere with splicing of DNA within the intron cassette. The beginning of the LAT intron is indicated by a solid bar. The LTE sequence overlaps the LAP2 promoter (16), as well as the first 837 bp of the 2.0-kb LAT. (B) The overall structure of the plasmid set used in this study is composed of the LAT promoter (hatched bar), thelacZreporter gene (stippled bar), and a synthetic intron (SD and SA sequences) with the LTE region more or less deleted. The deletions are indicated by the flanking sites of the en-zymes used. B, BseAI; H,HpaI; N, NheI; Sf, SfiI; S, StyI. PHB18 contains only a LAT promoter attached to the lacZgene, with no splicing cassette in between. (C) The four recombinant viruses KOS18, KOS22F, KOS37, and KOS39 were constructed by insertion of plasmid pHB18, pHB22F, pHB37, and pHB39, respectively, at the gC locus of the parental virus KOSdl1.8 (25), which is deleted essentially from the
PstI site to just past theHpaI site shown in panel A. Therefore, the genotype of these viruses is LAT⫺and gC⫺.
VOL. 75, 2001 NOTES 4387
on November 9, 2019 by guest
http://jvi.asm.org/
element. Despite its potential proximity to LAP2, it is impor-tant to note that the enhancer functions in the inverted direc-tion. This excludes the possibility that the enhancer and LAP2 have identical functions.
To more closely map the enhancer, we first performed a mutational analysis of the LTE element and checked the ef-fects of these mutations on enhancer activity. We therefore constructed the plasmid set described in Fig. 1B and analyzed
lacZreporter gene expression after transfection of these plas-mids into BHK (fibroblast) and ND7 (neuron) cells in culture. We constructed four internal deletions spanning the LTE el-ement, pHB7.1, pHB8.1, pHB9.1, and pHB14.1. The activity of these plasmids in transient-transfection assays was compared to that of the full-length plasmid (pHB22F), to a plasmid containing only the LAT promoter-lacZreporter without an
LTE element insertion (pHB18), and to a plasmid containing a LAT promoter-lacZreporter with cellular DNA inserted into the intron cassette (pHB23).
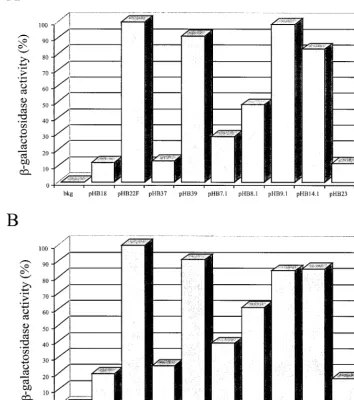
[image:3.612.127.481.89.489.2]The data in Fig. 2 showed that thelacZexpression originat-ing from plasmid pHB22F was 8 and 5 times higher in BHK and ND7 cells, respectively, than the expression observed with pHB18, as expected (3). A-galactosidase activity similar to that with pHB22F was obtained for plasmids pHB9.1 and pHB14.1. These plasmids had deletions in sequences within the LAT intron part of the LTE DNA fragment. Deletion of theStyI-StyI (pHB7.1) or theBseAI-NheI fragment (pHB8.1) resulted in a significant decrease inlacZgene expression by 60 to 70% (pHB7.1) or by 40 to 50% (pHB8.1), depending on the cells used for transfection. These deletions were located in the part of the LTE element upstream of the LAT intron. This FIG. 2. Histogram representing the-galactosidase activity originating from transfections with plasmids. Six-well plates containing 5⫻105 BHK (A) or ND7 (B) cells per well were transfected with 1g of DNA from plasmids pHB18, pHB22F, pHB37, pHB39F, pHB7.1, pHB8.1, pHB9.1, pHB14.1, and pHB23. At 2 days posttransfection, cells were harvested and lysed, and-galactosidase activity was determined in vitro using chlorophenol red--D-galactopyranoside as the substrate. Values are average means from experiments conducted in triplicate and are expressed
as the percentage of activity with plasmid pHB22F. bkg, background.
on November 9, 2019 by guest
http://jvi.asm.org/
would seem to place the enhancer activity within the region upstream of the LAT intron. To verify this conclusion, two additional plasmids were constructed. pHB37 had a deletion encompassing the deletions in pHB7.1 and pHB8.1, spanning pHB7.1 and pHB8.1, and pHB39 carried the combined dele-tions of pHB9.1 and pHB14.1 Removing the entire 5⬘ end of the LTE sequence (pHB37) led to a background activity which was not significantly different from that of either of the control plasmids, pHB18 (Pⱖ0.15), containing no insert, or pHB23 (Pⱖ0.12), containing a control, nonviral DNA insert. Thus, the enhancer activity found to be associated with the LTE fragment seemed to be reduced only partially when part of the 5⬘ end of the LTE element was deleted, suggesting that the entire 5⬘half of the LTE element is required for full enhancer activity. Moreover, theStyI-NheI fragment of the LTE element seemed to be sufficient by itself to obtain the maximal level of enhancer activity (compare pHB22F and pHB39). Also, these results were obtained in a neuronal cell line (ND7 cells) as well as in fibroblasts (BHK cells), indicating that the enhancer activity associated with that region is not specific to a cell line. We also demonstrated previously that the LTE sequence was required for long-term expression in vivo (3). In this work, we discussed the possible meaning of having two functions in the same region. One possibility is that the enhancer and long-term functions are different. In that case, they should be genetically separable. The alternative possibility is that the functions are actually the same. We measured long-term ex-pression by determining if a certain promoter construct can function well into latency to provide a detectable level of -ga-lactosidase activity from alacZreporter gene. All viral pro-moters, including LAP, show a decrease in activity after the first week of infection, and LAP by itself falls to a level of activity that is no longer detectable in our reporter assay sys-tem. It is conceivable that the LTE element merely acts as an enhancer to raise the baseline of LAP expression so that when it falls, it still remains functioning at a detectable level. This would mean that the LTE element and the enhancer have the same function and so could not be separated into two distinct functions by mutational analysis.
In order to determine if the 5⬘ end of the LTE element, which is necessary and sufficient for the enhancer activity, also contained the LTE function, we constructed a set of four recombinant viruses. Plasmids pHB18, pHB22F, pHB37, and pHB39 were inserted into the virus at gC by cotransfection with virion DNA. These viruses were screened for insertion of thelacZgene and plaque purified a total of five times. Their DNA structure was verified by Southern blot hybridization (data not shown). The four viruses, hereafter designated KOS18, KOS22F, KOS37, and KOS39 contained the expres-sion units of plasmids pHB18, pHB22F, pHB37, and pHB39, respectively, inserted at the gC locus. These viruses were then used to infect BALB/c mice through the footpad of the rear limbs in order to establish a latent infection in the DRG. At 4 days postinfection (corresponding to the acute phase of infec-tion) or at 28 days postinfection (corresponding to the latent phase of infection), mouse DRG L3 to L6 were stained with X-Gal (5-bromo-4-chloro-3-indolyl--D-galactopyranoside) in
situ in order to detect the level of -galactosidase activity produced from the recombinant viruses.
As shown in Fig. 3, the lacZ expression observed with
KOS22F and KOS39 was similar and much higher at 4 days postinfection than the expression detected with viruses KOS18 and KOS37 (compare DRG in Fig. 3A and E with that in C and G). Most importantly, during latency we were able to detect blue neurons in DRG infected with KOS22F and KOS39 (Fig. 3D and H), but no activity was observed in DRG infected by KOS18 or KOS37 (Fig. 3B and F). As shown in Table 1, titers of KOS18, KOS22F, KOS37, and KOS39 ranged from 1⫻104
to 2.9 ⫻ 104 at 4 days postinfection, and as expected, no
infectious virus could be recovered at 28 days postinfection. We showed previously that KOS22F does not further decrease in activity throughout latency, so that its activity at 28 days is similar to its activity at 42 and 60 days and even at 6 months postinfection. Thus, the differences in expression observed on days 4 and 28 are actual differences in levels of expression at early and at latent times. Furthermore, these differences were not due to the ability of the recombinant viruses to grow differently in vivo. Therefore, the tremendous decrease inlacZ
expression from KOS37 to a level similar to that in KOS18 during acute and latent infection seemed to be due to deletion of the 5⬘ half of the LTE element. Also, the presence of the
StyI-NheI fragment associated with the LAT promoter (KOS39) was sufficient to restore a level of expression similar to that obtained with the entire LTE fragment (KOS22F). These data suggested that theStyI-NheI fragment contained all the elements required for both the enhancer activity observed at 4 days postinfection in neurons and the long-term expression activity detected during latency. The data also indicate that the long-term expression and enhancer functions have not been separated.
As we reported previously, the third function that is associ-ated with the LTE region is a lytic cycle promoter function that operates only when the LTE is placed in the forward or natural direction (3). In order to determine if theStyI-NheI fragment located within the 5⬘half of the LTE region also contained a promoter activity (besides the enhancer and long-term expres-sion activities), we performed productive infection of cells cul-tured in vitro using the four recombinant viruses KOS18, KOS22F, KOS37, and KOS39. As can be seen in Fig. 4, the level oflacZexpression obtained from KOS22F is 2.5 and 4 times higher than that obtained with KOS18 in the BHK and ND7 cell lines, respectively. These increases were both highly significant (Pⱕ0.03). However, the viruses with deletions of the LTE element expressed similar levels oflacZas in control KOS18-infected cells. For instance, the level oflacZexpression in ND7 cells infected with KOS18 was not significantly differ-ent from that with KOS37 (P ⫽0.11) or KOS39 (P⫽0.44). These results suggested that each fragment, corresponding ei-ther to the 5⬘end (LAP2 region) or the 3⬘end (part of the LAT intron) of the LTE, did not contain all the elements required for the promoter activity that is detected with the entire LTE sequence.
HSV-1 is able to establish a latent infection in humans and in a variety of animal models. During the latent state, the LAT promoter is the only promoter that remains active, whereas all the other viral promoters are repressed. In an attempt to un-derstand the molecular mechanisms leading to the LAT pro-moter’s constitutive activity, we demonstrated in previous work that a region located immediately downstream from the LAT was necessary for that function (27) and was therefore termed
VOL. 75, 2001 NOTES 4389
on November 9, 2019 by guest
http://jvi.asm.org/
the LTE sequence. Further study revealed that the LTE se-quence contained three different functions, including long-term expression, enhancer, and promoter activities (3). As stated before, the LTE region is about 1.5 kb in length and
[image:5.612.98.509.67.634.2]corresponds to a segment of the LAT locus from positions 118862 to 120298 on the HSV-1 genome. This segment over-laps the LAP2 promoter (positions 118862 to 119461) and part of the 2.0-kb LAT intron (positions 119462 to 120298). FIG. 3.-Galactosidase activity in whole-mount infected DRG by histochemical staining. BALB/c mice were inoculated with 107PFU per footpad with either KOS18, KOS22F, KOS37, or KOS39 virus. At the indicated times postinfection, DRG were removed and stained as indicated in Materials and Methods. The L4 and L5 pairs of DRG infected with (A and B) KOS18, (C and D) KOS22F, (E and F) KOS37, (G and H) KOS39 are shown at 4 and 28 days postinfection, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
The aim of this present work was to more precisely map the different functions associated with the LTE region and to de-termine if the long-term expression and enhancer functions could be separated. We first used transient expression in cell culture and showed that deletion of the 5⬘ end of the LTE sequence led to complete loss of the enhancer function. Other, smaller deletions within this segment resulted in only a partial decrease in enhancer function, suggesting that several ele-ments spanning the entireStyI-NheI fragment are required for full enhancer activity. These results were further confirmed, since the StyI-NheI fragment associated with the LAT pro-moter was sufficient alone to restore the maximal level of gene expression. Therefore, our data strongly suggest that the en-hancer activity is located over the entire LAP2 region and is not associated with the 2.0-kb LAT intron part.
Previous studies have assigned different functions to the LAP2 region. First, the LAP2 sequence has been described as a promoter that is highly active during the productive infection, but its intrinsic contribution to the expression of the LATs during latency is not significant (6). Second, deletion of a segment within the LAP2 promoter, extending from positions 119007 to 119355, led to a decreased ability of the mutant virus (termed 17⌬348) to reactivate following epinephrine-induced iontophoresis into the cornea in a rabbit model (4). Interest-ingly, in that study, three smaller and nonoverlapping deletions inside this 348-bp region were not sufficient alone to reduce the epinephrine-induced reactivation. Instead, all recombinant vi-ruses were able to reactivate with wild-type efficiency, suggest-ing that the smaller deletions within the 348-bp region did not individually remove the segment required for the reactivation process. With respect to our work, theStyI-NheI deletion re-moved the entire 348-bp region described by Bloom and col-leagues, whereas theStyI-StyI and BseAI-NheI deletions re-moved only 243 and 250 bp, respectively. Therefore, there is a strong correlation between the epinephrine-induced reactiva-tion process, which depends on the entire 348-bp region, and the enhancer activity, which required theStyI-NheI fragment. One hypothesis to correlate these two activities is that the enhancer function leads to increased expression of the LAT intron during the few days following primary infection, as de-scribed previously (3). This transient increase in LATs could favor latency establishment by reducing synthesis of the imme-diate-early gene ICP0 (15), thereby limiting the productive infection. As a consequence, the reactivation potential would be augmented, since it has been shown that there is a strong correlation between the number of latent sites and reactivation (37).
In order to determine if the region required for full en-hancer activity (i.e., the LAP2 region) was also sufficient for
[image:6.612.315.555.80.469.2]the long-term expression function associated with the whole LTE sequence, we performed in vivo experiments in a mouse model using recombinant viruses. In these experiments, we showed that the LAP2 portion of the LTE region, associated with the LAT promoter, was very capable of constitutive ex-pression during latency. Therefore, both the enhancer and the long-term expression activities colocalized to the LAP2 se-quence. In a previous report, we could not determine if the enhancer and long-term expression functions were identical or separate entities. The fact that the two activities colocalize to the LAP2 region suggests that they may be the same function, FIG. 4. Histogram representing the-galactosidase activity origi-nating from infections of cells in culture. The four viruses KOS18, KOS22F, KOS37, and KOS39 were used to independently infect BHK (A) and ND7 (B) cells grown to near confluence in 60-mm dishes at a multiplicity of infection of 10 PFU per cell. Once total cytopathic effect was achieved (2 to 3 days), cells were harvested and lysed, and -ga-lactosidase activity was determined in the cellular extracts using the in vitro chlorophenol red--D-galactopyranoside assay. Three
indepen-dent experiments were performed, and the average means of these values are indicated as a percentage of the activity in KOS22-infected cells.
TABLE 1. Virus titers in infected DRG in vivo
Virus Titer (PFU/ganglion) at time postinfection: 4 days 28 days
None 0 0
KOS18 1.8⫻104 0
KOS22F 1.0⫻104 0
KOS37 2.9⫻104 0
KOS39 1.9⫻104 0
VOL. 75, 2001 NOTES 4391
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.53.292.83.165.2]but more mutations must be analyzed before a firm conclusion can be reached.
Finally, we also tried to map the lytic cycle promoter that lies within the LTE region when the LTE element is placed in the forward or natural direction. We first hypothesized that this function could originate from the LAP2 sequence, since it has already been described as a promoter. Alternatively, a TATA box consensus sequence located within the 2.0-kb intron could also promote transcription, especially during productive infec-tion, when viral transactivation occurred. We showed that re-moval of the 5⬘end or the 3⬘end of the LTE element com-pletely abolished the promoter activity. These results indicated that each half of the LTE sequence is not sufficient to promote gene expression during productive infection in vitro. There-fore, it is likely that several elements are required, which are present on each half of the LTE. One possibility is that these elements span the entire LTE region, which is doubtful, as we would probably obtain an intermediate level of transcription with at least one end, 5⬘ or 3⬘of the LTE element. Alterna-tively, if the LAP2 sequence were to carry the promoter activity linked to the LTE sequence, our results would suggest that an essential part of the LAP2 promoter lies downstream of the
NheIsite of the LTE element. In fact, the study performed to characterize the LAP2 promoter in vitro used plasmids con-taining deletions from the 5⬘ end of or within LAP2 (16). However, the segment downstream from the initiation site of transcription was not studied, although the region used in these constructs ended at aPpuMIsite 42 bp downstream from the⫹1 site. Thus, if this sequence is crucial for the initiation of transcription from the LAP2 promoter, ourNheI-HpaI dele-tion, which removed this small part (leaving 4 bp after the⫹1 site intact), would therefore abrogate any activity from the LAP2 promoter.
Altogether, our data showed that the 5⬘ end of the LTE sequence, corresponding to practically the entire LAP2 region, seemed to contain multiple functions which can function alter-natively during acute or latent infection. We demonstrated that the enhancer activity required the entire 5⬘end segment of the LTE element and colocalized with the long-term expression function. Further work based on mutagenesis of the different motifs that we have identified in this region will be necessary to define more precisely which of these motifs are required for these different activities.
This work was supported by Public Health Service grant AI28338 to L.T.F.
REFERENCES
1.Batchelor, A. H., and P. O’Hare.1992. Localization ofcis-acting sequence requirements in the promoter of the latency-associated transcript of herpes simplex virus type 1 required for cell type-specific activity. J. Virol.66:3573– 3582.
2.Batchelor, A. H., and P. O’Hare.1990. Regulation and cell type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J. Virol.64:3269–3279.
3.Berthomme, H., J. Lokensgard, L. Yang, T. Margolis, and L. T. Feldman.
2000. Evidence for a bidirectional element located downstream from the herpes simplex virus type 1 latency-associated promoter that increases its activity during latency. J. Virol.74:3613–3622.
4.Bloom, D. C., J. M. Hill, G. Devi-Rao, E. K. Wagner, L. T. Feldman, and J. G. Stevens.1996. A 348-base-pair region in the latency-associated transcript facilitates herpes simplex virus type 1 reactivation. J. Virol.70:2449–2459. 5.Chen, S. H., M. F. Kramer, P. A. Schaffer, and D. M. Coen.1997. A viral
function represses accumulation of transcripts from productive- cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol.71:
5878–5884.
6.Chen, X., M. C. Schmidt, W. F. Goins, and J. C. Glorioso.1995. Two herpes
simplex virus type 1 latency-active promoters differ in their contributions to latency-associated transcript expression during lytic and latent infections. J. Virol.69:7899–7908.
7.Croen, K. D., J. M. Ostrove, L. J. Dragovic, J. E. Smialek, and S. E. Straus.
1987. Latent herpes simplex virus in human trigeminal ganglia: detection of an immediate early gene “anti-sense” transcript by in situ hybridization. N. Engl. J. Med.317:1427–1432.
8.Deatly, A. M., J. G. Spivack, E. Lavi, and N. W. Fraser.1987. RNA from an immediate early region of the type 1 herpes simplex virus genome is present in the trigeminal ganglia of latently infected mice. Proc. Natl. Acad. Sci. USA
84:3204–3208.
9.Devi-Rao, G. B., S. A. Goodart, L. M. Hecht, R. Rochford, M. K. Rice, and E. K. Wagner.1991. Relationship between polyadenylated and nonpolyade-nylated herpes simplex virus type 1 latency-associated transcripts. J. Virol.
65:2179–2190.
10. Dobson, A. T., T. P. Margolis, W. A. Gomes, and L. T. Feldman.1995. In vivo deletion analysis of the herpes simplex virus type 1 latency-associated tran-script promoter. J. Virol.69:2264–2270.
11. Dobson, A. T., T. P. Margolis, F. Sedarati, J. G. Stevens, and L. T. Feldman.
1990. A latent, nonpathogenic HSV-1-derived vector stably expresses beta-galactosidase in mouse neurons. Neuron5:353–360.
12. Dobson, A. T., F. Sederati, G. Devi-Rao, W. M. Flanagan, M. J. Farrell, J. G. Stevens, E. K. Wagner, and L. T. Feldman.1989. Identification of the latency-associated transcript promoter by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J. Virol.63:3844–3851.
13. Farrell, M. J., A. T. Dobson, and L. T. Feldman.1991. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA
88:790–794.
14. Frazier, D. P., D. Cox, E. M. Godshalk, and P. A. Schaffer.1996. Identifi-cation ofcis-acting sequences in the promoter of the herpes simplex virus type 1 latency-associated transcripts required for activation by nerve growth factor and sodium butyrate in PC12 cells. J. Virol.70:7433–7444. 15. Garber, D. A., P. A. Schaffer, and D. M. Knipe.1997. A LAT-associated
function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol.71:5885– 5893.
16. Goins, W. F., L. R. Sternberg, K. D. Croen, P. R. Krause, R. L. Hendricks, D. J. Fink, S. E. Straus, M. Levine, and J. C. Glorioso.1994. A novel latency-active promoter is contained within the herpes simplex virus type 1 UL flanking repeats. J. Virol.68:2239–2252.
17. Hill, J. M., H. H. Garza, Jr., Y. H. Su, R. Meegalla, L. A. Hanna, J. M. Loutsch, H. W. Thompson, E. D. Varnell, D. C. Bloom, and T. M. Block.
1997. A 437-base-pair deletion at the beginning of the latency-associated transcript promoter significantly reduced adrenergically induced herpes sim-plex virus type 1 ocular reactivation in latently infected rabbits. J. Virol.
71:6555–6559.
18. Hill, J. M., J. B. Maggioncalda, H. H. Garza, Jr., Y. H. Su, N. W. Fraser, and T. M. Block.1996. In vivo epinephrine reactivation of ocular herpes simplex virus type 1 in the rabbit is correlated to a 370-base-pair region located between the promoter and the 5⬘end of the 2.0-kilobase latency-associated transcript. J. Virol.70:7270–7274.
19. Hill, J. M., F. Sedarati, R. T. Javier, E. K. Wagner, and J. G. Stevens.1990. Herpes simplex virus latent phase transcription facilitates in vivo reactiva-tion. Virology174:117–125.
20. Ho, D. Y., and E. S. Mocarski.1989. Herpes simplex virus latent RNA (LAT) is not required for latent infection in the mouse. Proc. Natl. Acad. Sci. USA
86:7596–7600.
21. Javier, R. T., J. G. Stevens, V. B. Dissette, and E. K. Wagner.1988. A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology166:254–257.
22. Krause, P. R., K. D. Croen, J. M. Ostrove, and S. E. Straus.1990. Structural and kinetic analyses of herpes simplex virus type 1 latency-associated tran-scripts in human trigeminal ganglia and in cell culture. J. Clin. Investig.
86:235–241.
23. Krause, P. R., K. D. Croen, S. E. Straus, and J. M. Ostrove.1988. Detection and preliminary characterization of herpes simplex virus type 1 transcripts in latently infected human trigeminal ganglia. J. Virol.62:4819–4823. 24. Krummenacher, C., J. M. Zabolotny, and N. W. Fraser.1997. Selection of a
nonconsensus branch point is influenced by an RNA stem-loop structure and is important to confer stability to the herpes simplex virus 2-kilobase latency-associated transcript. J. Virol.71:5849–5860.
25. Leib, D. A., C. L. Bogard, M. Kosz-Vnenchak, K. A. Hicks, D. M. Coen, D. M. Knipe, and P. A. Schaffer.1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol.63:2893–2900.
26. Leib, D. A., K. C. Nadeau, S. A. Rundle, and P. A. Schaffer.1991. The promoter of the latency-associated transcripts of herpes simplex virus type 1 contains a functional cAMP-response element: role of the latency-associated transcripts and cAMP in reactivation of viral latency. Proc. Natl. Acad. Sci. USA88:48–52.
27. Lokensgard, J. R., H. Berthomme, and L. T. Feldman.1997. The
on November 9, 2019 by guest
http://jvi.asm.org/
associated promoter of herpes simplex virus type 1 requires a region down-stream of the transcription start site for long-term expression during latency. J. Virol.71:6714–6719.
28. Margolis, T. P., F. Sedarati, A. T. Dobson, L. T. Feldman, and J. G. Stevens.
1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology189:150–160.
29. Mitchell, W. J., R. P. Lirette, and N. W. Fraser.1990. Mapping of low abundance latency-associated RNA in the trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J. Gen. Virol.71:125–132. 30. Perng, G. C., E. C. Dunkel, P. A. Geary, S. M. Slanina, H. Ghiasi, R. Kaiwar,
A. B. Nesburn, and S. L. Wechsler.1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol.68:8045–8055. 31. Perng, G. C., C. Jones, J. Ciacci-Zanella, M. Stone, G. Henderson, A. Yukht,
S. M. Slanina, F. M. Hofman, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.
2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science287:1500–1503.
32. Perng, G. C., S. M. Slanina, A. Yukht, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.2000. The latency-associated transcript gene enhances establish-ment of herpes simplex virus type 1 latency in rabbits. J. Virol.74:1885–1891. 33. Perry, L. J., and D. J. McGeoch.1988. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol.69:2831–2846.
34. Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L. Lewis, J. R. Lokensgard, and S. L. Wechsler.1987. Detection of latency-related viral RNAs in tri-geminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol.61:3820–3826.
35. Rodahl, E., and L. Haarr.1997. Analysis of the 2-kilobase latency-associated transcript expressed in PC12 cells productively infected with herpes simplex virus type 1: evidence for a stable, nonlinear structure. J. Virol.71:1703–1707. 36. Roizman, B., and A. E. Sears.1987. An inquiry into the mechanisms of
herpes simplex virus latency. Annu. Rev. Microbiol.41:543–571. 37. Sawtell, N. M.1998. The probability of in vivo reactivation of herpes simplex
virus type 1 increases with the number of latently infected neurons in the ganglia. J. Virol.72:6888–6892.
38. Sawtell, N. M., and R. L. Thompson.1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol.66:2157–2169. 39. Sedarati, F., K. M. Izumi, E. K. Wagner, and J. G. Stevens.1989. Herpes
simplex virus type 1 latency-associated transcription plays no role in estab-lishment or maintenance of a latent infection in murine sensory neurons. J. Virol.63:4455–4458.
40. Soares, K., D. Y. Hwang, R. Ramakrishnan, M. C. Schmidt, D. J. Fink, and J. C. Glorioso.1996.cis-Acting elements involved in transcriptional regula-tion of the herpes simplex virus type 1 latency-associated promoter 1 (LAP1) in vitro and in vivo. J. Virol.70:5384–5394.
41. Spivack, J. G., and N. W. Fraser.1987. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol.61:3841–3847. 42. Spivack, J. G., G. M. Woods, and N. W. Fraser.1991. Identification of a
novel latency-specific splice donor signal within the herpes simplex virus type 1 2.0-kilobase latency-associated transcript (LAT): translation inhibition of
LAT open reading frames by the intron within the 2.0-kilobase LAT. J. Virol.
65:6800–6810.
43. Steiner, I., J. G. Spivack, R. P. Lirette, S. M. Brown, A. R. MacLean, J. H. Subak-Sharpe, and N. W. Fraser.1989. Herpes simplex virus type 1 latency-associated transcripts are evidently not essential for latent infection. EMBO. J.8:505–511.
44. Steiner, I., J. G. Spivack, D. R. O’Boyle 3rd, E. Lavi, and N. W. Fraser.1988. Latent herpes simplex virus type 1 transcription in human trigeminal ganglia. J. Virol.62:3493–3496.
45. Stevens, J. G., E. K. Wagner, G. B. Devi-Rao, M. L. Cook, and L. T. Feldman.
1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science235:1056–1059.
46. Thomas, S. K., G. Gough, D. S. Latchman, and R. S. Coffin.1999. Herpes simplex virus latency-associated transcript encodes a protein which greatly enhances virus growth, can compensate for deficiencies in immediate-early gene expression, and is likely to function during reactivation from virus latency. J. Virol.73:6618–6625.
47. Thompson, R. L., and N. M. Sawtell.1997. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J. Virol.71:5432–5440.
48. Wagner, E. K., G. Devi-Rao, L. T. Feldman, A. T. Dobson, Y. F. Zhang, W. M. Flanagan, and J. G. Stevens.1988. Physical characterization of the herpes simplex virus latency-associated transcript in neurons. J. Virol.62:
1194–1202.
49. Wagner, E. K., W. M. Flanagan, G. Devi-Rao, Y. F. Zhang, J. M. Hill, K. P. Anderson, and J. G. Stevens.1988. The herpes simplex virus latency-associ-ated transcript is spliced during the latent phase of infection. J. Virol.
62:4577–4585.
50. Wechsler, S. L., A. B. Nesburn, R. Watson, S. Slanina, and H. Ghiasi.1988. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J. Gen. Virol.69:3101–3106.
51. Wechsler, S. L., A. B. Nesburn, R. Watson, S. M. Slanina, and H. Ghiasi.
1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J. Virol.62:4051–4058.
52. Zabolotny, J. M., C. Krummenacher, and N. W. Fraser.1997. The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J. Virol.71:4199–4208.
53. Zwaagstra, J., H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.1989. In vitro promoter activity associated with the latency-associated transcript gene of herpes simplex virus type 1. J. Gen. Virol.70:2163–2169.
54. Zwaagstra, J. C., H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.1991. Identification of a major regulatory sequence in the latency associated tran-script (LAT) promoter of herpes simplex virus type 1 (HSV-1). Virology
182:287–297.
55. Zwaagstra, J. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, S. C. Wheatley, K. Lillycrop, J. Wood, D. S. Latchman, K. Patel, and S. L. Wechsler.1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J. Virol.64:5019–5028.
VOL. 75, 2001 NOTES 4393