Pathway Proteins
Kareem N. Mohni, Alexander R. Dee, Samantha Smith, April J. Schumacher, Sandra K. Weller
Department of Molecular, Microbial and Structural Biology and the Molecular Biology and Biochemistry Graduate Program, University of Connecticut Health Center, Farmington, Connecticut, USAa
Herpes simplex virus 1 (HSV-1) is a double-stranded DNA virus that replicates in the nucleus of the host cell and is known to
interact with several components of the cellular DNA-damage-signaling machinery. We have previously reported that the DNA
damage response kinase, ATR, is specifically inactivated in HSV-1-infected cells. On the other hand, we have also shown that
ATR and its scaffolding protein, ATRIP, are recruited to viral replication compartments, where they play beneficial roles during
HSV-1 replication. In order to better understand this apparent discrepancy, we tested the hypothesis that some of the
compo-nents of the ATR pathway may exert an antiviral effect on infection. In fact, we learned that all 10 of the canonical ATR pathway
proteins are stable in HSV-infected cells and are recruited to viral replication compartments; furthermore, short hairpin RNA
(shRNA) knockdown shows that several, including ATRIP, RPA70, TopBP1, Claspin, and CINP, are required for efficient HSV-1
replication. We also determined that activation of the ATR kinase prior to infection did not affect virus yield but did result in
reduced levels of recombination between coinfecting viruses. Together, these data suggest that ATR pathway proteins are not
antiviral
per se
but that activation of ATR signaling may have negative consequences during viral replication, such as inhibiting
recombination.
H
erpes simplex virus 1 (HSV-1) is a large double-stranded
DNA (dsDNA) virus that replicates in the nucleus of the host
cell. After entry into the nucleus, viral DNA is recognized by
cel-lular homeostatic mechanisms, including the ND10 components
PML, sp100, and hDaxx (
1
,
2
), as well as cellular double-strand
break (DSB) repair proteins (
3
,
4
). It is becoming increasingly
clear that the ND10 and DSB repair pathways represent intrinsic
cellular antiviral defense strategies, and both are counteracted by
the viral E3 ubiquitin ligase ICP0 (
1
,
3
). Viral DNA replication
itself also elicits a cellular DNA damage response and results in the
specific recruitment of cellular DNA repair proteins to sites of
viral DNA replication (
4
–
9
).
The cellular DNA damage response is orchestrated by three
phosphoinositide 3-kinase-related kinases (PIKKs): DNA-PK
(DNA-dependent protein kinase), ATM (ataxia telangiectasia
mutated), and ATR (ATM and Rad3 related) (
10
–
12
). DNA-PK
and ATM respond primarily to double-strand breaks, and ATR
responds to stalled replication forks and stretches of
single-stranded DNA (ssDNA). DNA-PK stimulates nonhomologous
end joining (NHEJ), and ATM is primarily thought to stimulate
repair via homologous recombination (HR) (
11
). During HSV-1
infection, DNA-PK is degraded by ICP0 in some cell types, and
this might be expected to inactivate the NHEJ pathway (
13
–
15
).
ATM is activated following the onset of DNA replication, and
several ATM pathway proteins play a positive role during
infec-tion (
5
,
7
,
9
). Consistent with ATM activation, high rates of
re-combination have also been observed between coinfecting HSV-1
viruses (
16
,
17
).
Following a DSB, ATM is activated, and dsDNA ends are
re-sected, generating long stretches of ssDNA adjacent to dsDNA.
Resected DNA provides the primary molecular trigger for ATR
activation, leading to the phosphorylation of the ATR substrates
Chk1 (checkpoint kinase 1) and RPA (replication protein A) (
12
).
ATR signaling requires the precise recruitment of cellular sensors
and effectors to stretches of ssDNA adjacent to dsDNA at sites of
DNA damage. The cellular ssDNA binding protein, RPA, coats the
ssDNA and recruits ATR through a direct interaction with ATRIP
(ATR-interacting protein) (
18
). In a second independent
recruit-ment event, the PCNA-like damage-specific clamp 9-1-1 (for
Rad9, Rad1, and Hus1) is loaded at the dsDNA junction, followed
by the recruitment of the ATR activator TopBP1, resulting in the
activation of ATR (
11
,
12
). We have previously reported that ATR
is specifically inactivated in HSV-1-infected cells (
6
,
19
). Although
we initially reported that ATR and ATRIP were redistributed to
different cellular compartments (
19
), Mohni et al., using more
specific antibodies, showed that not only are ATR and ATRIP both
recruited to replication compartments, they play beneficial roles
during HSV-1 replication (
6
).
In this study, we set out to test the hypothesis that ATR
path-way proteins themselves or activation of the ATR pathpath-way exerts a
cellular antiviral effect on infection. Using short hairpin RNA
(shRNA) knockdown, we report that none of the ATR pathway
proteins are antiviral, and many of them actually play beneficial
roles during HSV-1 infection. Furthermore, activation of the ATR
pathway had no effect on total virus yields but did result in a
reduction in recombination between two coinfecting viruses.
Thus, HSV-1 may have evolved to disable ATR signaling to
pro-mote recombination during infection.
MATERIALS AND METHODS
Cells and reagents.HeLa, HFF-1, U2OS, Vero, and pEAK (293T deriva-tive) cells were obtained from the American Type Culture Collection
Received13 September 2012Accepted15 October 2012
Published ahead of print24 October 2012
Address correspondence to Sandra K. Weller, [email protected].
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02504-12
on November 7, 2019 by guest
http://jvi.asm.org/
(ATCC). GP2-293 cells were purchased from Clontech. All cells were maintained in Dulbecco’s modified Eagle medium with 10% fetal bovine serum, except Vero cells, which were maintained in 5% fetal bovine se-rum. MG132 was purchased from Sigma and used at a final concentration of 10M. Calf intestinal phosphatase was purchased from New England BioLabs and used as suggested by the manufacturer.
Viruses.The KOS strain was used as wild-type HSV-1, and all mutant viruses used in this study were derived from KOS and were described previously:⌬ICP0 (0) (20),⌬ICP4 (d120) (21),⌬ICP22 (d22lacZ) (22), ⌬ICP27 (d27-1) (23), and⌬ICP8 (HD2) (24). Virus tsK13, also called ts276, was derived from KOS and contains a temperature-sensitive muta-tion in UL5 (P236L) (25). Virus tsR was derived from strain 17⫹and contains a temperature-sensitive mutation in UL9 (V220M) (26,27). Temperature-sensitive mutants were grown in Vero cells at 34°C and titrated at both 34°C and 39.5°C to check for reversion. Both mutants have a very tight temperature-sensitive phenotype at 39.5°C. Virus in1863 is derived from strain 17⫹and was obtained from Chris Preston (MRC Virology Unit, Glasgow, Scotland). The virus contains thelacZgene under the control of the human cytomegalovirus (HCMV) promoter/enhancer inserted in thetkgene (1,6).
DNA constructs. pEGFP was purchased from Clontech. pEGFP-TopBP1-978-1286 wild type (also referred to as GFP-TopBP1-AAD be-low), pEGFP-TopBP1-978-1286 W1145R, Myc-TopBP1, and HA-CINP were provided by David Cortez (Vanderbilt University School of Medi-cine, Nashville, TN) (28,29). Flag-Hus1 and Flag-Rad17 were purchased from Origene. GFP-Rad1 was provided by Veronique Smits (30). Flag-Chk1 was purchased from Addgene (Addgene plasmid 22894) (31).
IF analysis.Immunofluorescence (IF) analysis was performed as de-scribed previously (4,6,32). Briefly, cells adhering to glass coverslips were washed with phosphate-buffered saline (PBS), fixed with 4% paraformal-dehyde, and permeabilized with 1% Triton X-100. The cells were blocked in 3% normal goat serum and reacted with antibodies as indicated. Pri-mary antibodies include polyclonal rabbit anti-ATRIP (rATRIP Upstate) (Upstate; 1:200), monoclonal mouse anti-ICP8 (1:200; Abcam), poly-clonal rabbit anti-ICP8 367 (1:400) (33), monoclonal rat anti-Hsc70 (1: 200; Stressgen), polyclonal rabbit anti-hemagglutinin (HA) (1:200; Clon-tech), monoclonal rat HA (1:200; Roche), monoclonal mouse anti-FLAG M2 (1:200; Sigma), monoclonal mouse anti-RPA32 9H8 (1:200; GeneTex), monoclonal mouse anti-Myc 9B11 (1:200; Cell Signaling), and monoclonal mouse anti-␥H2AX (1:200; Upstate). Alexa Fluor secondary antibodies (1:200; Molecular Probes) were used with fluorophores excitable at a wavelength of 488, 594, or 647. Images were captured using a Zeiss LSM 510 confocal NLO microscope equipped with argon and HeNe lasers and a Zeiss 63⫻objective lens (numerical aperture, 1.4). Images were processed and arranged using Adobe Photoshop CS3 and Illustrator CS3.
Western blot analysis.Cells in 35-mm dishes were lysed in 2⫻SDS sample buffer (4% SDS, 20% glycerol, 100 mM Tris, pH 6.8, 100 mM dithiothreitol [DTT], 10%-mercaptoethanol, 1 mM sodium orthovana-date, 10 mM NaF, 1⫻protease inhibitor cocktail [Roche], and 0.1% bro-mophenol blue) and boiled for 5 min. Proteins were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked for 1 h in 5% nonfat dry milk or 2% bovine serum albumin (BSA) dissolved in Tris-buffered saline–Tween (TBST). Primary antibodies were diluted in blocking solution and incubated over-night at 4°C. The primary antibodies used included polyclonal rabbit anti-ATRIP (rATRIP Upstate) (1:3,000; Upstate), monoclonal mouse anti-ICP8 (1:10,000; Abcam), monoclonal mouse anti-ICP4 (1:10,000; US Biologics), monoclonal mouse anti--actin (1:15,000; Sigma), mono-clonal mouse anti-HA (F7) (1:3,000; Santa Cruz), monomono-clonal mouse anti-Myc (1:5,000; Cell Signaling), polyclonal rabbit anti-GFP (green flu-orescent protein) (1:1,000; Santa Cruz), polyclonal goat anti-ATR (N19) (1:1,000; Santa Cruz), polyclonal rabbit anti-phospho-ATR S428 (1: 1,000; Santa Cruz), monoclonal mouse anti-Chk1 (1:1,000; Santa Cruz), monoclonal rabbit anti-phospho-Chk1 S345 (1:5,000; Cell Signaling), polyclonal goat anti-PML (N19) (1:1,000; Santa Cruz), mouse
monoclo-nal anti-RPA32 (9H8) (1:1,000; GeneTex), polyclomonoclo-nal rabbit anti-phos-pho-RPA S33 (1:3,000; Bethyl), polyclonal rabbit anti-phosanti-phos-pho-RPA S4/S8 (1:3,000; Bethyl), polyclonal goat anti-RPA70 (C-21) (1:1,000; Santa Cruz), polyclonal goat anti-Rad1 (N-18) (1:1,000; Santa Cruz), polyclonal rabbit anti-Hus1 (M-281) (1:1,000; Santa Cruz), polyclonal rabbit anti-Rad9 (1:1,000; GeneTex), polyclonal rabbit anti-phospho-Rad9 S387 and S272 (1:1,000; Abgent), polyclonal goat anti-Rad17 (1: 5,000; Bethyl), polyclonal rabbit anti-Claspin (1:10,000; Bethyl), mono-clonal mouse anti-TopBP1 (1:1,000; BD Bioscience), and polymono-clonal rabbit anti-Ku86 (1:1,000; Santa Cruz). Polyclonal rabbit anti-ATRIP 403 (rATRIP 403) (1:3,000) (34), anti-CINP (29), and anti-phospho-ATRIP S224 (1:3,000) (35) were previously described and provided by David Cortez (Vanderbilt University School of Medicine, Nashville, TN). Rabbit antiserum to HCLK2 was previously described and provided by Siegfried Hekimi (McGill University, Montreal, Quebec) (36).
Lentivirus generation and use.The pLKO.1 system was used to pack-age lentiviruses and to deliver shRNA into target cells as previously de-scribed (4,6). Lentiviruses expressing short hairpin RNA targeting GFP (shGFP), shATR, shATRIP-1, and shATRIP-2 were described previously (6). The following shRNA target sequences were cloned into the pLKO.1-TRC cloning vector (Addgene plasmid 10878) according to the manufac-turer’s suggestions: shRPA70-1, 5=-GGAAUUAUGUCGUAAGUCA (37); shRPA70-2, 5=-AACACUCUAUCCUCUUUCAUG (38); shRPA32, 5=-CCUAGUUUCACAAUCUGUU (39); shRad9, 5=-AAGUCUUUCCU GUCUGUCUUC (40–42); shRad17, 5=-CAGACUGGGUUGACCCAUC (41,43); shTopBP1, 5=-GUGGUUGUAACAGCGCAUC (44); shClaspin, 5=-GGAAAGAAAGGCAGCCAGA (45); shChk1, 5=-GCGUGCCGUAG
ACUGUCCA (45); shCINP, 5=-AAACCUGUCUUAUCUGUCAUU
(29); and shHCLK2-1, 5=-GCGGUAUCUCGGUGAGAUGUU (46). Retrovirus generation and use.The pLPCX retrovirus system based on murine leukemia virus was purchased from Clontech. To package retrovirus particles, pLPCX was transfected with a vesicular stomatitis virus G protein (VSV-G)-expressing plasmid (pVSV-G; Clontech) at a ratio of 2:1 into GP2-293 packaging cells, which supply thegagandpol genes. pLPCX-HA-Rad9 (38), pLPCX-HA-Rad9-CRD (38), and pLPCX-HA-ATRIP (47) were provided by David Cortez (Vanderbilt University School of Medicine, Nashville, TN). pLPCX-HA-ATRIP-CRD was de-scribed previously (6). pLPCX-Flag-ATR was made by subcloning the BamHI fragment containing Flag-ATR from pBJF-ATR-wt, provided by Karlene Cimprich (Stanford University School of Medicine, Stanford, CA) (48,49), into pLPCX. pLPC-MYC-hTel2 was purchased (Addgene plasmid 22802) (50) and is referred to as Myc-HCLK2 in the text. Target cells were infected with retroviruses to generate cell lines. The medium was changed the following day, and selection with puromycin was started at 72 h postinfection. Vero cells were selected with 10g/ml Puromycin, and HeLa and HFF-1 cells were selected with 2g/ml puromycin. Cell lines were maintained in half the concentration of puromycin used for selection.
Growth curves and yields.All growth curves and yield experiments were performed in cells infected with in1863 at a multiplicity of infection (MOI) of 0.1. Virus was collected at the indicated times postinfection, and titers were determined on Vero cells by staining for -galactosidase-pos-itive plaques, as previously described (6). Where indicated, HeLa and HFF-1 cells were infected with lentiviruses, selected with 2g/ml puro-mycin, and infected with HSV-1 at 72 h post-lentiviral infection. In some experiments, HeLa cells were transfected with the indicated plasmids 18 h prior to infection with HSV-1.
Virus recombination assay.HeLa cells were infected with tsK13 alone, tsR alone, or both tsK13 and tsR (tsK13-tsR) at an MOI of 1 PFU/cell for each virus and incubated at 34°C. Progeny virus was collected at 24 h postinfection and titrated on Vero cells at 34°C and 39.5°C. The percentage of recombinants was calculated as follows: 2⫻[(tsK13-tsR39.5°C⫺tsK1339.5°C⫺tsR39.5°C)/
(tsK13-tsR34°C)]⫻100, where the subscript numbers refer to the titer of the
virus at the indicated temperature. The number was multiplied by 2 to
on November 7, 2019 by guest
http://jvi.asm.org/
count for the equal possibility of generating a recombinant virus with both temperature-sensitive mutations.
RESULTS
All ATR pathway proteins are stable during infection and are
recruited to replication compartments.
We have reported that
ATR signaling is disabled in HSV-1-infected cells between 1 and 5
h postinfection (
6
,
7
). To test whether this inactivation is due to
the destabilization of one or more of the essential ATR pathway
proteins, HeLa cells were infected with HSV-1 and subjected to
Western blot analysis. Uninfected cells were also treated with
hy-droxyurea (HU) or UV light as positive controls for ATR
activa-tion.
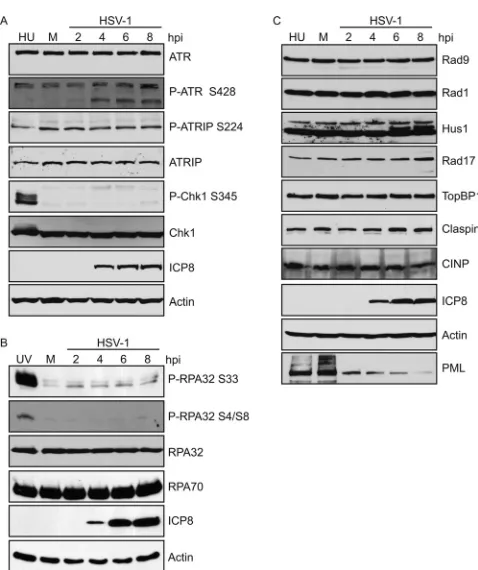
Figure 1A
shows that ATR, ATRIP, and Chk1 are all stable
during infection. The phosphorylation marks, S428 on ATR and
S224 on ATRIP, are required for ATR activity but are not
phos-phorylated by ATR (
35
,
51
). Both of these marks were observed in
mock-infected cells, and the levels of phosphorylation did not
change during infection or in the presence of HU (
Fig. 1A
). On the
other hand, ATR-specific phosphorylation of Chk1 on S345 is
detected in response to HU, but not during HSV-1 infection,
con-sistent with previous reports (
6
,
7
,
52
).
In response to DNA-damaging agents, the N terminus of
RPA32 is phosphorylated on S33 by ATR and on S4 and S8 by
DNA-PK (
53
,
54
). HeLa cells were infected with HSV-1 or
dam-aged with UV as a positive control for ATR and DNA-PK
activa-tion.
Figure 1B
indicates that RPA32 levels were stable during
infection; however, RPA32 was robustly phosphorylated on S33
and S4/S8 only in the UV-treated sample and not in the
HSV-1-infected samples. The lack of phosphorylation on RPA32 S4/S8 is
consistent with the previously reported degradation of DNA-PK
in HSV-1-infected HeLa cells (
14
,
15
) and suggests that the
DNA-PK/NHEJ pathway is inactivated in infected cells.
As described in the introduction, several additional essential
ATR pathway proteins are required for ATR activation.
Figure 1C
shows that all of the essential pathway proteins, including
compo-nents of the 9-1-1 clamp (Rad9, Rad1, and Hus1), Rad17,
TopBP1, Claspin, and CINP, are stable during HSV-1 infection in
HeLa cells, while the known ICP0 target, PML, was efficiently
degraded (
Fig. 1C
). Thus, ATR inactivation is not due to
degrada-tion of any of the known components of the pathway.
Another method of preventing ATR activation would be to
exclude an essential ATR pathway protein from HSV-1 replication
compartments, the sites of viral DNA synthesis. To test this
pos-sibility, Vero cells were infected with HSV-1 and analyzed for the
recruitment of ATR pathway proteins using tagged expression
constructs (
Fig. 2
). Vero cells were chosen for their flat
morphol-ogy and well-defined replication compartments. Consistent with
previous reports, ATRIP was recruited to replication
compart-ments (
6
,
52
), and Hus1, Rad1, Rad17, TopBP1, CINP, and Chk1
were also recruited to replication compartments.
The study of Rad9 has been hampered by the lack of available
high-quality antibodies that detect endogenous Rad9 (
30
,
38
,
55
).
To study the role of Rad9 during HSV-1 infection, we generated
Vero cell lines stably expressing either an empty vector (Vero
empty vector) or HA-Rad9 (Vero HA-Rad9) by retroviral
infec-tion, as described in Materials and Methods. Two independently
derived cell lines expressing HA-Rad9 (numbers 1 and 2) behaved
identically in that Rad9 was efficiently recruited to replication
compartments after HSV-1 infection (
Fig. 2
).
Regulation of the ATR pathway requires the
cofactor/chaper-one HCLK2, which functions to stabilize both ATR and Chk1 (
46
)
and is required for ATR activation (
44
).
Figure 3A
indicates that
HCLK2 is stable in both Vero and HeLa cells after HSV-1
infec-tion, although much lower levels of HCLK2 are detected in Vero
cells. We suspected that the HCLK2 antibody does not efficiently
recognize monkey HCLK2, and we therefore generated a Vero cell
line stably expressing Myc-HCLK2.
Figure 3B
and
C
indicate that
Myc-HCLK2 was stable following infection and was recruited
both to replication compartments and to virus-induced
chaper-one-enriched (VICE) domains following infection. VICE
do-mains have been shown to contain components of the nuclear
protein quality control machinery and putative misfolded
pro-teins (
56
), and it is possible that a population of the overexpressed
HCLK2 is misfolded or recruited there to chaperone misfolded
proteins. Virus growth was identical on Vero Myc-HCLK2 cells
and cells expressing an empty vector (
Fig. 3D
). Together, the data
presented in
Fig. 1
to
3
support the conclusion that ATR pathway
proteins and regulators are stable during infection and are
re-cruited to replication compartments. Thus, the lack of ATR
sig-naling during HSV-1 infection is not due to degradation or
mis-localization of ATR pathway proteins.
Rad9 is posttranslationally modified during HSV-1
infec-tion.
Another possible explanation for the lack of ATR signaling is
FIG 1All ATR pathway proteins are stable during HSV-1 infection. HeLa cells were either mock infected or infected with HSV-1 (KOS) at an MOI of 10 PFU/cell, and cell lysates were collected at the indicated time points and ana-lyzed by Western blotting. Alternatively, cells were treated with either 3 mM HU (A and C) for 2 h or 50 J/m2UV (B) and allowed to recover for 1 h as a
positive control for DNA damage-induced changes in mobility and phosphor-ylation. Panels A and C represent the same samples run on separate gels. Western blotting was performed as indicated for the ATR pathway proteins, and actin was used as a loading control. In panel A, the faster-migrating band (approximately 150 kDa) of ATR S428 likely represents a cleavage product. hpi, hours postinfection.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.43.282.60.345.2]that one or more of the ATR components are regulated by
post-translational modification. Both Hus1 and Rad9 have been
re-ported to be modified by ubiquitination (
55
). Interestingly a
slow-er-migrating band of Hus1 was observed at 6 and 8 h post-HSV-1
infection (
Fig. 1C
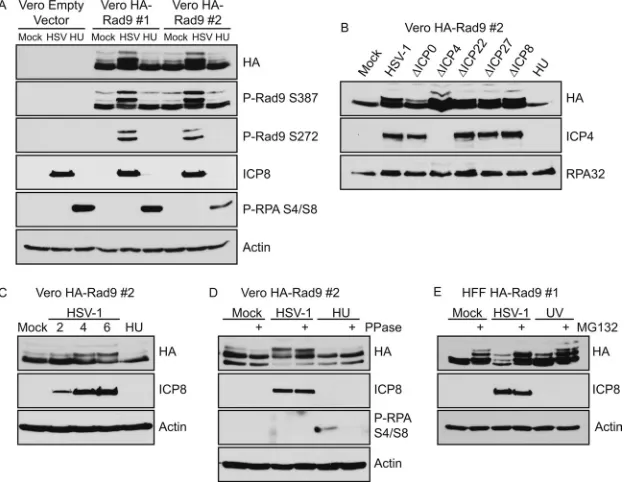
). Vero cells stably expressing HA-Rad 9 were
either mock infected, infected with HSV-1, or treated with HU
and analyzed by Western blotting (
Fig. 4A
). Two
slower-migrat-ing forms of Rad9 were observed in the HSV-1-infected samples
that were absent, or in much reduced amounts, in the mock- and
HU-treated controls. Phosphospecific antibodies were used to
show that these slower-migrating bands were phosphorylated on
S387, a constitutively phosphorylated site on Rad9 (
57
), and S272,
an ATM-mediated DNA damage-inducible site on Rad9 (
58
). The
S272 phosphorylation mark was present only on the two
slower-migrating forms in infected cells (
Fig. 4A
). The slower-migrating
forms of HA-Rad9 were detected in cells infected with mutants in
individual immediate-early genes, as well as in cells infected with
an ICP8-null virus (
Fig. 4B
), and appeared as early as 4 h
postin-fection (
Fig. 4C
). Therefore, while the timing of the shift in
HA-Rad9 may correlate with the onset of viral DNA replication, DNA
replication
per se
and early-gene expression are not required. The
only viral mutant that resulted in reduced levels of the
slower-migrating form was the ICP0-null virus, suggesting that the
pres-ence of ICP0 may help to stabilize the slower-migrating form.
The slower-migrating form of Rad9 exhibited increased
mo-bility after phosphatase treatment; however, this treatment still
resulted in a band that migrated more slowly than in
mock-in-fected controls (
Fig. 4D
). Thus, the phosphorylation does not
ac-count for the entire mobility shift in Rad9. HA-Rad9 also
exhib-ited a mobility shift after HSV-1 infection in HFF-1 cells that
stably express HA-Rad9 (
Fig. 4E
). The addition of proteasome
inhibitors increased the amount of the slower-migrating forms of
FIG 2All ATR pathway proteins are recruited to HSV-1 replication compart-ments. Vero cells or Vero cells stably expressing HA-Rad9 were infected with HSV-1 (KOS) at an MOI of 10 PFU/cell and fixed at 6 h postinfection. For all other samples, Vero cells were transiently transfected with the indicated tagged expression construct for 18 h prior to infection. Immunofluorescence assays were performed as described in Materials and Methods. Cells transfected with GFP-Rad1 exhibit Rad1 in replication compartments and a subpopulation of GFP-Rad1 in VICE domains. The population of Rad1 in VICE domains may be a result of overexpression of one component of the 9-1-1 clamp.
FIG 3ATR/ATRIP chaperone HCLK2 is stable during HSV-1 infection. (A) Vero and HeLa cells were either mock infected or infected with HSV-1 (KOS) at an MOI of 10 PFU/cell, and the cell lysates were analyzed by Western blot-ting at 6 h postinfection using antibodies directed against endogenous HCLK2. (B) Vero cells stably expressing Myc-HCLK2 were generated by retroviral in-fection and continuous selection in puromycin. Cells were treated as for panel A, and 2 mM HU treatment was included as indicated. Cell lysates were ana-lyzed by Western blotting using antibodies directed against the tagged overex-pressed proteins. (C) Vero-Myc-HCLK2 cells were either mock infected or infected with HSV-1 (KOS) at an MOI of 10 PFU/cell and fixed at 6 h postin-fection. Immunofluorescence assays were done with antibodies directed against Myc, ICP8, and Hsc70 as described in Materials and Methods. (D) Vero empty vector and Vero-Myc-HCLK2 were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell. Progeny virus was collected at 24 h postinfection, and viral titers were determined on Vero cells. The values represent the aver-ages of three independent experiments, and the error bars represent the stan-dard errors of the means.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.299.543.63.285.2] [image:4.585.78.250.64.522.2]HA-Rad9 in both the HSV-1- and UV-treated samples. We also
observed the stabilization of slower-migrating forms in
mock-infected cells in the presence of the proteasome inhibitors. Thus,
the slower-migrating form of Rad9 may be targeted for
degrada-tion via the proteasome.
In an attempt to identify the nature of the posttranslational
modifications on Rad9, we purified Rad9 from infected cells and
subjected it to mass spectrometry analysis. The only
posttransla-tional modification detected in the mass spectrometry analysis
was phosphorylation (
Table 1
). We also analyzed proteins that
copurified with Rad9 in mock-infected, HSV-1-infected, and
HU-treated cells (
Table 2
). In HSV-1-infected cells, the E3 ubiquitin
ligase UBR4 was identified as a Rad9-interacting protein (
Table
2
); however, we were unable to detect ubiquitinated species of
Rad9 during HSV-1 infection by coimmunoprecipitation and
Western blotting (data not shown). Interestingly, UBR4 was also
reported to copurify with Rad9 in the presence of overexpressed
ubiquitin (
55
). Further experiments will be required to determine
the nature of the posttranslational modifications on Hus1 and
Rad9 in HSV-infected cells and to determine if these
modifica-tions occur on endogenously expressed protein in addition to the
overexpressed forms. We are also intrigued by recent reports that
Rad9 may be modified by SUMO addition (
59
) and arginine
methylation (
60
). It is possible that ATR activation is regulated by
posttranslational modification.
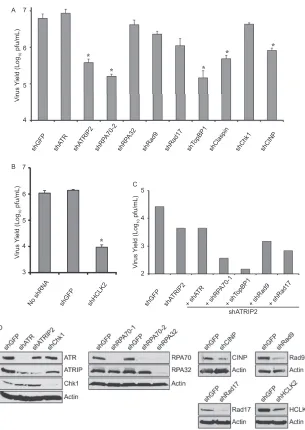
Some ATR pathway proteins are required for efficient HSV-1
replication.
If one or more of the ATR pathway components were
antiviral, we would predict that depletion using shRNA would
allow HSV-1 to replicate to higher titers. We have previously
re-ported that in the absence of ATRIP, HSV-1 replication is reduced
by 1 log unit, suggesting that ATRIP plays a positive role during
HSV-1 replication (
6
). HFF-1 cells were infected with lentiviruses
expressing shRNA to individual ATR pathway proteins to deplete
endogenous proteins. All of the shRNA sequences used in this
study have been validated for specific knockdown and an absence
FIG 4Rad9 is posttranslationally modified during HSV-1 infection. Vero cells stably expressing HA-Rad9 (Vero HA-Rad9) or an empty vector (Vero Empty Vector) were generated by retroviral infection and continuous selection in puromycin. (A) Two independently derived cell lines of Vero HA-Rad9 (numbers 1 and 2) were infected with HSV-1 (KOS) at an MOI of 10 PFU/cell or treated with 3 mM HU, and cell lysates were collected at 6 h posttreatment. (B) Vero HA-Rad9 number 2 cells were infected with HSV-1 or the indicated mutant viruses at an MOI of 5 PFU/cell and collected at 6 h postinfection and post-HU treatment. (C) Vero HA-Rad9 number 2 cells were treated as for panel A, and HSV-1-infected samples were collected at 2, 4, and 6 h postinfection and 6 h post-HU treatment. (D) Vero HA-Rad9 number 2 cells were treated as for panel A, and cell lysates were treated with calf intestinal phosphatase for 30 min at 37°C. (E) HFF-1 cells stably expressing HA-Rad9 were generated in the same manner as Vero HA-Rad9 cells and infected with HSV-1 at an MOI of 10 PFU/cell or treated with 50 J/m2UV. Where indicated, MG132 was added at a final concentration of 10M at 3 h post-HSV-1 infection or 1 h post-UV treatment and
[image:5.585.137.451.68.309.2]maintained for an additional 3 h prior to collection. All samples were analyzed by Western blotting with the indicated antibodies.
TABLE 1Phosphorylated residues on Rad9
Residue
Phosphorylationa
Mock HSV-1 HU
S21 MS
S86 MS
S100 MS
S106 MS
S272 WB WB
S277 MS MS MS
S324 MS
S328 MS MS
S336 MS
S341 MS MS MS
T351 MS MS
T355 MS MS MS
S387 MS/WB MS/WB MS/WB
aMock, uninfected cells; HSV-1, cells that were infected with KOS at an MOI of 10 for
6 h; HU, cells that were treated with 3 mM HU for 6 h; MS, identified by mass spectrometry; WB, identified by Western blotting.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.298.544.533.697.2]of off-target effects (
6
,
37
–
42
,
44
–
47
). Furthermore, all
knock-down cells generated in these experiments behaved similarly to
control cells in terms of growth and doubling times during the
course of the experiment, except for cells expressing shRNA
tar-geting Chk1, which exhibited rounded edges and longer doubling
times than control cells. To avoid problems associated with
long-term knockdown of DNA repair proteins, we generated a new
batch of lentiviral knockdown cells for each repeat of every
exper-iment and used them at 72 h post-lentiviral infection.
Consistent with our published observations, HFF-1 cells
de-pleted of ATRIP resulted in a greater than 1-log-unit reduction in
virus yield (
Fig. 5A
) (
6
), and cells depleted of ATR exhibited no
reduction in virus yield. We also observed this dependence on
ATRIP, but not ATR, in HeLa cells (data not shown). Knockdown
of ATR still leaves residual ATRIP, which may be sufficient to
support HSV-1 growth (
Fig. 5D
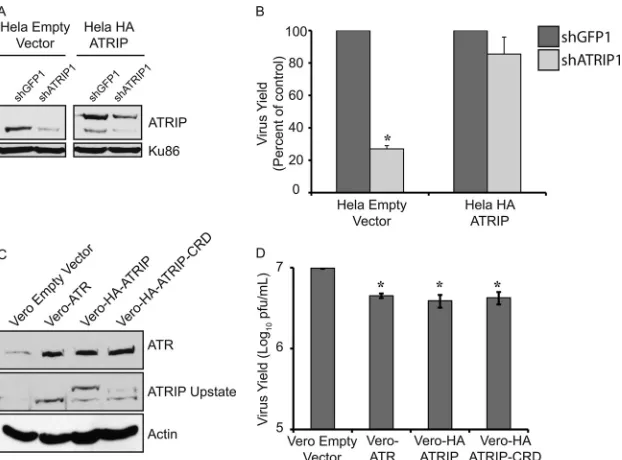
). HeLa cells stably expressing an
shRNA-resistant ATRIP were able to rescue the defect in HSV-1
growth seen in the presence of shATRIP, while cells expressing an
empty vector could not (
Fig. 6A
and
B
). Thus, the reduction in
HSV-1 yield seen with shATRIP was specific to ATRIP.
HFF-1 cells depleted of CINP and Claspin also exhibited a
1-log-unit reduction in virus yield, while depletion of RPA70,
TopBP1, and HCLK2 reduced the virus yield by almost 2 log units
(
Fig. 5A
and
B
). We did not observe a reduction in virus yield with
shRPA32, suggesting that RPA70 may play a unique role during
HSV-1 replication. Depletion of Rad9, Rad17, or Chk1 caused
little or no reduction in virus yield in HFF-1 cells, suggesting that
these proteins are dispensable for HSV-1 growth. Depletion of
Chk1 had the most significant effect on cell morphology and
dou-bling time (data not shown) but had little effect on virus yield,
suggesting that reductions in virus yield are not caused by toxicity
after knockdown. Cells expressing the shGFP control behaved
identically to uninfected controls, suggesting that lentivirus
infec-tion and puromycin selecinfec-tion did not adversely affect the ability of
the cells to support HSV-1 growth (
Fig. 5B
).
In addition to their known roles in the ATR-mediated DNA
damage response, RPA70 and TopBP1 also play essential roles in
DNA replication (
18
,
37
,
61
–
63
). The observation that
knock-down of RPA70 and TopBP1 resulted in greater reductions in
virus yield than knockdown of ATRIP prompted us to ask whether
these proteins were playing an additional role. If so, knockdown of
either protein in combination with ATRIP would be expected to
result in a larger decrease in virus yield.
Figure 5C
shows that this
is indeed the case, suggesting that RPA70 and TopBP1 play roles
outside of ATR signaling during infection, perhaps in DNA
repli-cation.
Our observation that ATR components play positive roles in
infection led us to examine the mechanism of recruitment to
rep-lication compartments. ATRIP is recruited to sites of DNA
dam-age through a direct interaction with RPA-ssDNA; however, we
previously reported that ATRIP is recruited to replication
com-partments independently of RPA, suggesting a possible
interac-tion with a viral protein (
6
). In uninfected cells, an acidic region in
the C-terminal tail of Rad9 called the checkpoint recruitment
do-main (CRD) binds a basic cleft on the N terminus of RPA70 and is
required to localize Rad9 at sites of DNA damage even in the
presence of endogenous protein (
38
). To determine if RPA
bind-ing was required for Rad9 localization to viral replication
com-partments and for the posttranslational modification of Rad9, we
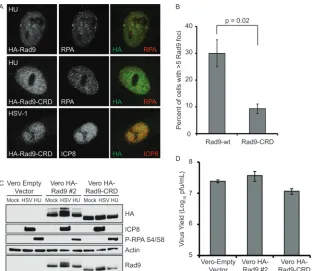
generated a Vero cell line stably expressing HA-Rad9-CRD, a
mu-tant that fails to bind RPA (
38
). HA-Rad9-CRD was not recruited
to HU-induced DNA damage foci (
Fig. 7A
and
B
), consistent with
previous reports (
38
); however, HA-Rad9-CRD was recruited to
replication compartments (
Fig. 7A
). The slower-migrating forms
of HA-Rad9 seen in
Fig. 7C
following infection were not seen in
cell lines expressing HA-Rad9-CRD, suggesting that the
post-translational modification reported above requires RPA
interac-tion. HA-Rad9-CRD migrates faster than wild-type HA-Rad9 due
to replacement of acidic residues with basic residues (
38
), and we
[image:6.585.41.286.79.580.2]cannot rule out the possibility that the increased mobility of
HA-Rad9-CRD could mask the presence of potential posttranslational
TABLE 2Proteins that copurify with Rad9
Gene name/function % Coverage
Mock
Eukaryotic translation elongation factor 1 alpha 1-like 14 19.6
hnRNP K 13.1
Eukaryotic translation elongation factor 1 alpha 1 variant 11.5
Hsc70 8
Eukaryotic initiation factor 4A-I isoform 1 6.5
Chaperonin (Hsp60) 6.3
Hsp70 4.7
E3 ubiquitin-protein ligase NEDD4 isoform 2 3.9
HSV-1
Hsc70 18.9
Proteasome (prosome, macropain) 26S subunit, non-ATPase, 3
15.2
DNA-binding protein B 15.1
hnRNP K 9.1
hnRNP H 9.1
Histone H1.3 9
RNase-like protein 12 precursor 7.5
Hsp70 6.7
Paraspeckle component 1 5.4
TATA-binding protein-associated factor 2N isoform 2 5.4
Chaperonin (Hsp60) 3.7
Transcription factor SOX-10 4.7
Histone-binding protein RBBP4 isoform a 3.5
E3 ubiquitin-protein ligase UBR4 3.4
Probable ATP-dependent RNA helicase DDX17 isoform 1 3.4 Eukaryotic translation elongation factor 1 alpha 1 variant 3.2
HDAC2 protein 3.2
Eukaryotic initiation factor 4A-I isoform 1 3
HU
Hsc70 32.4
hnRNP K 26.9
Eukaryotic initiation factor 4A-I isoform 1 22.9
Histone H1.3 20.8
Eukaryotic translation elongation factor 1 alpha 1 variant 19
Chaperonin (Hsp60) 15.9
Elongation factor 1-alpha 2 13.2
DNA-binding protein B 10.4
hnRNP H 8.9
Hsp70 7.4
Poly[ADP-ribose] polymerase 8 isoform 1 6
TATA-binding protein-associated factor 2N isoform 2 5.4
E3 ubiquitin-protein ligase NEDD4 5
DNA-binding protein A 4.1
HDAC2 protein 3.2
Nucleosome assembly protein 1-like 1 2.6
on November 7, 2019 by guest
http://jvi.asm.org/
modifications. Indeed, the band corresponding to HA-Rad9-CRD
in
Fig. 7C
is somewhat diffuse. Finally, virus growth rates were
similar in Vero cells expressing HA-Rad9, HA-Rad9-CRD, or an
empty vector (
Fig. 7D
). The observation that the recruitment of
Rad9 to replication compartments is independent of RPA may
indicate that Rad9 is recruited by a viral protein or by interaction
with one of its other cellular interacting partners, such as MLH1
(
64
), which is also in replication compartments (
4
).
Overexpression of ATR/ATRIP reduces viral yields.
To test
the effect of ATR/ATRIP overexpression on virus yield, we
gener-ated Vero cells stably overexpressing either Flag-ATR,
HA-ATRIP, or HA-ATRIP-CRD (which is unable to bind RPA or
lo-calize to sites of DNA damage [
6
]). These cell lines expressed
higher levels of ATR and ATRIP than cells expressing an empty
vector (
Fig. 6C
), and HSV-1 growth was reduced by 2-fold in all
three of the overexpression cell lines (
Fig. 6D
). This decrease in
virus yield was small but reproducible; on the other hand, no
decrease in virus yield was observed in cells constitutively
overex-pressing HA-Rad9 or HCLK2 (
Fig. 3
and
4
), suggesting that the
decrease was specific to ATR and ATRIP.
ATR activation prior to infection reduces viral
recombina-tion.
We were surprised to observe that even though ATR
signal-ing is not active dursignal-ing infection, many of the ATR pathway
pro-teins appear to play a positive role during infection, suggesting
FIG 5Several ATR pathway proteins are required for efficient HSV-1 replication. (A and B) HFF-1 cells were infected with lentiviruses expressing the indicated shRNA and selected with puromycin. After selection, the cells were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell. Progeny virus was collected at 24 h postinfection, and titers were determined on Vero cells. The values represent the averages of three independent experiments, and the error bars represent the standard errors of the mean. *,P⬍0.05. (C) HFF-1 cells were infected with shGFP or shATRIP and selected with puromycin. After selection, the cells were infected a second time with either shGFP, shATRIP, or the indicated shRNA. After an additional 48 h, the cells were infected with HSV-1 (in1863) at an MOI of 0.01 PFU/cell. Progeny virus was collected at 24 h postinfection, and titers were determined on Vero cells. The data from a representative experiment are shown. Rad9 and Rad17 served as controls for known ATR pathway proteins. (D) Western blot analysis of cells treated in parallel with the growth yields presented in panels A and B.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.139.444.61.491.2]that none of the proteins themselves are inherently antiviral. We
next posed the question of whether ATR activation itself was
an-tiviral. ATR activation is known to inhibit cellular-gene
expres-sion, and activation prior to infection might be expected to reduce
viral-gene expression and subsequently reduce viral replication.
We monitored the effects of UV and HU pretreatment on the
expression of immediate-early gene products and on virus yield.
UV pretreatment slightly reduced the amount of ICP4 at 6 h
postinfection, and HU had no noticeable effect on ICP4
expres-sion (
Fig. 8A
). We did not observe a significant decrease in virus
yield in response to UV or HU pretreatment (
Fig. 8B
). These data
suggest that ATR activation is not antiviral.
Another way to induce ATR activation without causing the
redistribution of the ATR pathway proteins is to express a
previ-ously described fragment of TopBP1 (amino acids 978 to 1286)
that is able to activate ATR signaling in the absence of DNA
dam-age. Transfection with this TopBP1 fragment (AAD) results in
activation of ATR substrates, including
␥
H2AX, RPA-S33, and
Chk1-S345 (
28
,
65
,
66
). Transfection of HeLa cells with a plasmid
expressing GFP-TopBP1-AAD resulted in the phosphorylation of
␥
H2AX (
Fig. 9A
), while transfection with GFP alone or
GFP-TopBP1-AAD W1145R, an inactivating mutation that does not
result in ATR activation, did not. Expression was confirmed by
Western blotting using antibodies raised against GFP (
Fig. 9B
).
Growth curves on transfected cells did not exhibit any differences
in the growth of HSV-1 (
Fig. 9C
), suggesting that activation of
ATR in the absence of DNA damage has no effect on virus yield, at
least at these multiplicities of infection.
ATR activation is also known to play a role in HR (
12
). To ask
whether prior activation of ATR affected HR, we measured
re-combination frequencies between two temperature-sensitive
HSV-1 mutants in cells expressing TopBP1-AAD or the negative
controls. Cells were coinfected with temperature-sensitive
mu-tants in UL5 and UL9 as described in Materials and Methods. In
cells overexpressing GFP alone or GFP-TopBP1-AAD W1145R, a
recombination frequency of 12% was observed (
Fig. 9D
), whereas
in cells overexpressing TopBP1-AAD, this percentage was reduced
to 7%. These data suggest that ATR activation may limit
recom-bination between coinfecting viruses without affecting overall
vi-ral yields.
DISCUSSION
We have previously reported that ATR is not activated in
HSV-1-infected cells (
6
), and this study was initiated to investigate the
status of the essential proteins involved in ATR signaling. Several
observations were made. (i) All ATR pathway proteins tested were
stable during HSV-1 infection and were recruited to viral
replica-tion compartments. (ii) No ATR-induced phosphorylareplica-tion events
were detectable on Chk1 or RPA during HSV-1 infection. (iii)
Posttranslational modifications were identified on Hus1 and
Rad9, and Rad 9 was shown to be phosphorylated. (iv) A Rad9
mutant that cannot bind RPA and cannot localize to sites of DNA
damage is recruited to HSV-1 replication compartments in an
RPA-independent fashion. (v) The ATR pathway proteins ATRIP,
RPA70, TopBP1, CINP, and Claspin are required for efficient
HSV-1 replication. (vi) ATRIP and RPA70/TopBP1 participate in
separate pathways during HSV-1 replication. (vii) Overexpression
of ATR or ATRIP reduced HSV-1 replication by 2-fold. (viii)
Ac-FIG 6Overexpression of ATR or ATRIP slightly reduces HSV-1 replication. HeLa cells stably expressing either an empty vector or shRNA-resistant HA-ATRIP were generated by retroviral infection and continuous selection in puromycin. HeLa empty vector and HeLa-HA-ATRIP were infected with lentiviruses expressing either shGFP or shATRIP. (A) ATRIP levels were monitored by Western blotting using an antibody that recognizes both endogenous ATRIP (bottom) and HA-ATRIP (top). (B) Cells were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell. (C and D) Vero cells stably expressing either an empty vector, Flag-ATR, HA-ATRIP, or HA-ATRIP-CRD were generated by retroviral infection and continuous selection in puromycin. (C) Overexpression of ATR and ATRIP was confirmed by Western blotting using antibodies raised against endogenous proteins that also cross-react with the overexpressed forms. The slower-migrating forms of ATRIP in the HA-ATRIP and HA-ATRIP-CRD lanes correspond to the slower-migrating HA fusion proteins. (D) Cells were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell. (B and D) Progeny virus was collected at 24 h postinfection, and viral titers were determined on Vero cells. The values represent the averages of three independent experiments, and the error bars represent the standard errors of the means. *,P⬍0.05.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.138.449.65.295.2]tivation of ATR signaling prior to infection had no effect on virus
yield but resulted in reduced frequency of recombination between
coinfecting HSV-1 viruses. Taken together, these data indicate
that the ATR pathway proteins themselves play beneficial roles
during HSV-1 replication but that activation of ATR signaling
may have negative consequences for the virus, perhaps by
decreas-ing recombination frequencies.
All ATR pathway proteins are stable during HSV-1 infection.
Adenoviruses 5 and 12 have been shown to disable ATR signaling
by specifically disabling or degrading MRN or TopBP1,
respec-tively (
67
,
68
). The observation that all ATR pathway proteins
tested are stable during HSV-1 infection suggests that ATR is
dis-abled by a mechanism distinct from that used by adenoviruses.
The observation that two components of the 9-1-1 clamp are
modified during HSV-1 infection provides a possible mechanism
by which ATR signaling could be disabled. Alternatively, Rad9
could be recruited to DNA substrates that arise during HSV
rep-lication and therefore not be available to interact with ATR
path-way proteins. For example, Rad9 is known to localize to sites of
double-strand breaks following ionizing radiation in an
Mre11-and CtIP-dependent mechanism (
69
–
71
), suggesting that
resec-tion from a DSB is capable of producing a suitable substrate for
loading Rad9. Interestingly, the virus-encoded exonuclease UL12
has recently been shown to interact with cellular repair machinery
and to stimulate recombination by a single-strand annealing
mechanism (
72
,
73
). If UL12 resects either viral or cellular DNA in
infected cells, the resected DNA would be expected to recruit
Rad9. We suggest that during viral infection, ATR signaling could
be prevented either by posttranslational modification of Rad 9 or
by the recruitment of Rad9 to substrates or complexes that
pre-vent it from participating in ATR signaling.
ATR pathway proteins are required for efficient HSV-1
rep-lication.
ATRIP, CINP, Claspin, RPA70, TopBP1, and HCLK2
were shown to be necessary for efficient HSV-1 replication, as
knockdown resulted in a 1- to 2-log-unit decrease in virus yield.
Knockdown of RPA70 and TopBP1 in combination with ATRIP
exhibited even more severe decreases in virus yield, suggesting
that these proteins may act outside the ATR signaling pathway.
RPA and TopBP1 have well-documented roles in DNA replication
(
18
,
37
,
61
–
63
), and it is possible that RPA and TopBP1 participate
directly in viral DNA replication. Consistent with this suggestion,
RPA and ATRIP are present in the earliest detectable
prereplica-tive structures (
6
).
Another reported role of RPA in uninfected cells is to regulate
homologous recombination. RPA70 can interact with Mre11 (
38
)
and may participate in the high rates of recombination observed in
infected cells. A class of mutants in the N terminus of RPA70 has been
described in budding yeast (rfa-t11) (
74
–
77
) and in mammalian cells
(RPA70, R41E, and Y42F) (
37
), which separate DNA repair functions
from DNA replication. It will be of interest to determine whether
FIG 7A Rad9 mutant that cannot bind RPA is recruited to HSV-1 replication compartments. Vero cells stably expressing HA-Rad9-CRD were generated in parallel with Vero HA-Rad9 number 2. (A) Vero HA-Rad9 number 2 and Vero HA-Rad9-CRD cells were treated with 2 mM HU for 6 h or infected with HSV-1 (KOS) at an MOI of 10 PFU/cell and fixed at 6 h postinfection. Immunofluorescence assays were done with antibodies directed against HA and either RPA or ICP8 as described in Materials and Methods. (B) Cells were treated as for panel A, except that Vero cells were transiently transfected with the same expression constructs used to generate HA-Rad9 and HA-Rad9-CRD cell lines 24 h prior to HU treatment. The cells were processed for immunofluorescence and scored by eye for the number of DNA damage foci induced by HU, and at least 100 cells were counted for each replicate of the experiment. (C) The Vero empty vector, Vero HA-Rad9 number 2, or Vero HA-Rad9-CRD cell line was treated as for panel A, and cell lysates were analyzed by Western blotting at 6 h posttreatment. (D) Cells were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell. Progeny virus was collected at 24 h postinfection, and viral titers were determined on Vero cells. (B and D) The values represent the averages of three independent experiments, and the error bars represent the standard errors of the means.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.137.454.64.335.2]similar separation-of-function mutants can be used to distinguish
between various possible roles of RPA during HSV infection.
ATR also plays negative roles during HSV-1 replication.
Overexpression of ATR/ATRIP resulted in a reproducible 2-fold
reduction in virus yield. When ATR was activated prior to
infec-tion, virus yields were unaffected; however, a 2-fold reduction in
recombination frequencies between coinfecting HSV-1 viruses
was observed. Although these are relatively small effects, they are
reproducible, and we suggest that HSV has evolved to closely
reg-ulate ATR to achieve efficient infection.
ATR is known to stabilize stalled forks and prevent their
col-lapse into double-strand breaks; furthermore, ATR has been
shown to restrict homologous recombination after replication
fork collapse (
78
). Inhibition of ATR signaling also reduces
ho-mologous recombination at DSBs in uninfected cells (
79
,
80
), as
does overexpression of a dominant-negative kinase-dead allele of
ATR (
81
). The viral genome is known to contain nicks and gaps
(
82
), and replication through a nick can cause replication fork
stalling, potentially resulting in the generation of a DSB. DSBs are
known to be recombinogenic, and we and others have suggested
that HSV-1 DNA replication proceeds through a
recombination-dependent mechanism (
83
). It is thus possible that during viral
DNA replication, overexpression of ATR or prior activation of
ATR activity could stabilize collapsing replication forks, thus
de-creasing the probability of fork collapse and DSB formation,
events that may be beneficial to the virus in order to promote
homologous recombination. HSV-1 may have evolved to utilize
the beneficial aspects of ATR components to promote DNA
rep-lication without the negative consequences of ATR activation that
FIG 8DNA damage-induced ATR activation slightly reduces HSV-1 replica-tion. (A) Vero cells were left undamaged or treated with UV or HU and then infected with HSV-1 (KOS) at an MOI of 10 PFU/cell. Cell lysates were col-lected at the indicated times and analyzed by Western blotting. (B) Vero cells were pretreated with HU and UV, and then cells were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell. Progeny virus was collected at 24 h postinfection, and viral titers were determined on Vero cells. The values represent the averages of three independent experiments, and the error bars represent the standard errors of the mean. In all cases, ATR activation was induced with 10 J/m2UV, and cells were allowed to recover for 1 h
prior to infection, or the cells were treated with 3 mM HU for 24 h and used immediately following HU wash out.
FIG 9ATR activation prior to infection does not reduce HSV-1 replication but inhibits viral recombination. (A to C) HeLa cells were transfected with either GFP, GFP-TopBP1 978-1286, or GFP-TopBP1 978-1286 W1145R and used at 18 h posttransfection. The cells were processed for immunofluores-cence and stained for␥H2AX (A) or were analyzed for GFP expression by Western blotting (B), and parallel samples were infected with HSV-1 (in1863) at an MOI of 0.1 PFU/cell (C). Progeny virus was collected at the indicated time points, and viral titers were determined on Vero cells. The values sent the averages of three independent experiments, and the error bars repre-sent the standard errors of the mean. For all samples used in viral growth curves, GFP expression was confirmed in live cells using an inverted micro-scope equipped with a UV laser at the start of the experiment prior to infection. (D) Cells were infected with HSV-1 temperature-sensitive mutants tsK13 (UL5) and tsR (UL9) at an MOI of 1 PFU/cell for each virus. Progeny virus was collected at 24 h postinfection, and viral titers were determined at both the permissive and nonpermissive temperatures. The recombination frequency represents the percentage of wild-type recombinants that are able to grow at the nonpermissive temperature. The values represent the averages of three independent experiments, and the error bars represent the standard errors of the mean.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.79.249.64.267.2] [image:10.585.335.506.70.527.2]could stabilize forks and decrease homologous recombination.
This is the first report of a cellular protein that appears to restrict
recombination. Although overexpression and prior activation of
ATR had relatively modest effects on viral yields at the MOIs
tested in this study, it is possible that natural infections in which
much smaller amounts of virus are expected to be present would
be more reliant on recombination.
ACKNOWLEDGMENTS
We thank members of the Weller laboratory for helpful comments and discussions.
This work was supported by Public Health Service grants AI21747 and AI069136.
REFERENCES
1.Everett RD, Parada C, Gripon P, Sirma H, Orr A.2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol.82:2661–2672.
2.Lukashchuk V, Everett RD.2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol.84:4026 – 4040.
3.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD.
2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog.7:e1002084. doi:10.1371/journal.ppat.1002084.
4.Mohni KN, Mastrocola AS, Bai P, Weller SK, Heinen CD.2011. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J. Virol.85:12241–12253.
5.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD.2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A.102:5844 –5849.
6.Mohni KN, Livingston CM, Cortez D, Weller SK.2010. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J. Virol.84:12152–12164. 7.Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T,
Sugaya Y, Isomura H, Ishizaki K, Tsurumi T.2005. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elic-ited by herpes simplex virus infection. J. Biol. Chem.280:30336 –30341. 8.Taylor TJ, Knipe DM.2004. Proteomics of herpes simplex virus
replica-tion compartments: associareplica-tion of cellular DNA replicareplica-tion, repair, re-combination, and chromatin remodeling proteins with ICP8. J. Virol.
78:5856 –5866.
9.Wilkinson DE, Weller SK.2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol.78:4783– 4796.
10. Abraham RT.2004. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst.)3:883– 887.
11. Ciccia A, Elledge SJ.2010. The DNA damage response: making it safe to play with knives. Mol. Cell40:179 –204.
12. Cimprich KA, Cortez D.2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol.9:616 – 627.
13. Jackson SA, DeLuca NA. 2003. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. U. S. A.100:7871–7876.
14. Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA.
1996. Attenuation of DNA-dependent protein kinase activity and its cat-alytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol.70:7471–7477.
15. Parkinson J, Lees-Miller SP, Everett RD.1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol.73:650 – 657.
16. Brown SM, Ritchie DA, Subak-Sharpe JH.1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mu-tants, their arrangement into complementation groups and recombina-tion analysis leading to a linkage map. J. Gen. Virol.18:329 –346. 17. Schaffer PA, Tevethia MJ, Benyesh-Melnick M.1974. Recombination
between temperature-sensitive mutants of herpes simplex virus type 1. Virology58:219 –228.
18. Wold MS.1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem.66:61–92.
19. Wilkinson DE, Weller SK.2006. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci.
119:2695–2703.
20. Samaniego LA, Wu N, DeLuca NA. 1997. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol.71: 4614 – 4625.
21. DeLuca NA, McCarthy AM, Schaffer PA.1985. Isolation and character-ization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol.56:558 –570. 22. Long MC, Leong V, Schaffer PA, Spencer CA, Rice SA.1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol.
73:5593–5604.
23. Rice SA, Knipe DM.1990. Genetic evidence for two distinct transactiva-tion functransactiva-tions of the herpes simplex virus alpha protein ICP27. J. Virol.
64:1704 –1715.
24. Gao M, Knipe DM.1989. Genetic evidence for multiple nuclear functions of the herpes simplex virus ICP8 DNA-binding protein. J. Virol.63:5258 – 5267.
25. Zhu L, Weller SK.1988. UL5, a protein required for HSV DNA synthesis: genetic analysis, overexpression in Escherichia coli, and generation of polyclonal antibodies. Virology166:366 –378.
26. Blumel J, Matz B.1995. Thermosensitive UL9 gene function is required for early stages of herpes simplex virus type 1 DNA synthesis. J. Gen. Virol.
76:3119 –3124.
27. Matz B, Subak-Sharpe JH, Preston VG. 1983. Physical mapping of temperature-sensitive mutations of herpes simplex virus type 1 using cloned restriction endonuclease fragments. J. Gen. Virol.64:2261–2270. 28. Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D.
2007. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol.27:3367–3377.
29. Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao R, Ye F, Sirbu BM, Titus LC, Shyr Y, Cortez D.2009. Functional genomic screens identify CINP as a genome maintenance protein. Proc. Natl. Acad. Sci. U. S. A.
106:19304 –19309.
30. Warmerdam DO, Kanaar R, Smits VA.2010. Differential dynamics of ATR-mediated checkpoint regulators. J. Nucleic Acids2010:319142. doi: 10.4061/2010/319142.
31. Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK.2007. The human Tim/Tipin complex coor-dinates an intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol.27:3131–3142.
32. Livingston CM, DeLuca NA, Wilkinson DE, Weller SK.2008. Oligomer-ization of ICP4 and rearrangement of heat shock proteins may be impor-tant for herpes simplex virus type 1 prereplicative site formation. J. Virol.
82:6324 – 6336.
33. Shelton LS, Albright AG, Ruyechan WT, Jenkins FJ.1994. Retention of the herpes simplex virus type 1 (HSV-1) UL37 protein on single-stranded DNA columns requires the HSV-1 ICP8 protein. J. Virol.68:521–525. 34. Cortez D, Guntuku S, Qin J, Elledge SJ.2001. ATR and ATRIP: partners
in checkpoint signaling. Science294:1713–1716.
35. Myers JS, Zhao R, Xu X, Ham AJ, Cortez D.2007. Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M check-point response to DNA damage. Cancer Res.67:6685– 6690.
36. Jiang N, Benard CY, Kebir H, Shoubridge EA, Hekimi S.2003. Human CLK2 links cell cycle progression, apoptosis, and telomere length regula-tion. J. Biol. Chem.278:21678 –21684.
37. Haring SJ, Mason AC, Binz SK, Wold MS.2008. Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and check-points. J. Biol. Chem.283:19095–19111.
38. Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D.
2008. The basic cleft of RPA70N binds multiple checkpoint proteins, in-cluding RAD9, to regulate ATR signaling. Mol. Cell. Biol.28:7345–7353. 39. Vassin VM, Wold MS, Borowiec JA.2004. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell. Biol.24:1930 –1943.
40. Hirai I, Wang HG.2002. A role of the C-terminal region of human Rad9 (hRad9) in nuclear transport of the hRad9 checkpoint complex. J. Biol. Chem.277:25722–25727.
on November 7, 2019 by guest
http://jvi.asm.org/
41. Medhurst AL, Warmerdam DO, Akerman I, Verwayen EH, Kanaar R, Smits VA, Lakin ND.2008. ATR and Rad17 collaborate in modulating Rad9 localisation at sites of DNA damage. J. Cell Sci.121:3933–3940. 42. Sorensen CS, Syljuasen RG, Lukas J, Bartek J.2004. ATR, Claspin and
the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle3:941–945.
43. Zou L, Cortez D, Elledge SJ.2002. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev.16:198 –208.
44. Rendtlew Danielsen JM, Larsen DH, Schou KB, Freire R, Falck J, Bartek J, Lukas J.2009. HCLK2 is required for activity of the DNA damage response kinase ATR. J. Biol. Chem.284:4140 – 4147.
45. Yang XH, Shiotani B, Classon M, Zou L. 2008. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev.22:1147–1152.
46. Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ.2007. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat. Cell Biol.9:391– 401.
47. Ball HL, Myers JS, Cortez D.2005. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dis-pensable for Chk1 phosphorylation. Mol. Biol. Cell16:2372–2381. 48. Cimprich KA, Shin TB, Keith CT, Schreiber SL.1996. cDNA cloning and
gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl. Acad. Sci. U. S. A.93:2850 –2855.
49. Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH.1998. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle check-points. EMBO J.17:159 –169.
50. Takai H, Wang RC, Takai KK, Yang H, de Lange T.2007. Tel2 regulates the stability of PI3K-related protein kinases. Cell131:1248 –1259. 51. Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB, Cortez D.
2011. Thr-1989 phosphorylation is a marker of active ataxia telangiecta-sia-mutated and Rad3-related (ATR) kinase. J. Biol. Chem.286:28707– 28714.
52. Vogel R, Seyffert M, Strasser R, de Oliveira AP, Dresch C, Glauser DL, Jolinon N, Salvetti A, Weitzman MD, Ackermann M, Fraefel C.2012. Adeno-associated virus type 2 modulates the host DNA damage response induced by herpes simplex virus 1 during coinfection. J. Virol.86:143– 155.
53. Anantha RW, Vassin VM, Borowiec JA.2007. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem.282:35910 –35923.
54. Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA.2009. Human RPA phosphorylation by ATR stimulates DNA synthesis and pre-vents ssDNA accumulation during DNA-replication stress. J. Cell Sci.122: 4070 – 4080.
55. Warmerdam DO.2010. Biological and functional analysis of the ATR checkpoint pathway. Ph.D. thesis. Erasmus University Rotterdam, Rotter-dam, The Netherlands.
56. Livingston CM, Ifrim MF, Cowan AE, Weller SK.2009. Virus-induced chaperone-enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog.5:e1000619. doi: 10.1371/journal.ppat.1000619.
57. Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM.
2007. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev.21:1472–1477.
58. Chen MJ, Lin YT, Lieberman HB, Chen G, Lee EY. 2001. ATM-dependent phosphorylation of human Rad9 is required for ionizing radi-ation-induced checkpoint activation. J. Biol. Chem.276:16580 –16586. 59. Eichinger CS, Jentsch S.2011. 9-1-1: PCNA’s specialized cousin. Trends
Biochem. Sci.36:563–568.
60. He W, Ma X, Yang X, Zhao Y, Qiu J, Hang H.2011. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res.39:4719 – 4727.
61. Araki H, Leem SH, Phongdara A, Sugino A.1995. Dpb11, which inter-acts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc. Natl. Acad. Sci. U. S. A.92:11791–11795.
62. Fanning E, Klimovich V, Nager AR.2006. A dynamic model for repli-cation protein A (RPA) function in DNA processing pathways. Nucleic Acids Res.34:4126 – 4137.
63. Masumoto H, Sugino A, Araki H.2000. Dpb11 controls the association between DNA polymerases alpha and epsilon and the autonomously rep-licating sequence region of budding yeast. Mol. Cell. Biol.20:2809 –2817. 64. He W, Zhao Y, Zhang C, An L, Hu Z, Liu Y, Han L, Bi L, Xie Z, Xue P, Yang F, Hang H.2008. Rad9 plays an important role in DNA mismatch repair through physical interaction with MLH1. Nucleic Acids Res.36: 6406 – 6417.
65. Kumagai A, Lee J, Yoo HY, Dunphy WG.2006. TopBP1 activates the ATR-ATRIP complex. Cell124:943–955.
66. Mordes DA, Glick GG, Zhao R, Cortez D.2008. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev.22:1478 – 1489.
67. Blackford AN, Patel RN, Forrester NA, Theil K, Groitl P, Stewart GS, Taylor AM, Morgan IM, Dobner T, Grand RJ, Turnell AS. 2010. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc. Natl. Acad. Sci. U. S. A.107:12251–12256.
68. Carson CT, Orazio NI, Lee DV, Suh J, Bekker-Jensen S, Araujo FD, Lakdawala SS, Lilley CE, Bartek J, Lukas J, Weitzman MD. 2009. Mislocalization of the MRN complex prevents ATR signaling during ad-enovirus infection. EMBO J.28:652– 662.
69. Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP.
2006. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol.8:37– 45.
70. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP.2007. Human CtIP promotes DNA end resection. Nature
450:509 –514.
71. Warmerdam DO, Freire R, Kanaar R, Smits VA. 2009. Cell cycle-dependent processing of DNA lesions controls localization of Rad9 to sites of genotoxic stress. Cell Cycle8:1765–1774.
72. Balasubramanian N, Bai P, Buchek G, Korza G, Weller SK. 2010. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol.
84:12504 –12514.
73. Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK.2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog.8:e1002862. doi: 10.1371/journal.ppat.1002862.
74. Barlow JH, Lisby M, Rothstein R.2008. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol. Cell30:73– 85. 75. Kanoh Y, Tamai K, Shirahige K.2006. Different requirements for the association of ATR-ATRIP and 9-1-1 to the stalled replication forks. Gene
377:88 –95.
76. Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE.1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell94:399 – 409.
77. Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD.1998. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics148:989 –1005.
78. Sirbu BM, Lachmayer SJ, Wulfing V, Marten LM, Clarkson KE, Lee LW, Gheorghiu L, Zou L, Powell SN, Dahm-Daphi J, Willers H.2011. ATR-p53 restricts homologous recombination in response to replicative stress but does not limit DNA interstrand crosslink repair in lung cancer cells. PLoS One6:e23053. doi:10.1371/journal.pone.0023053.
79. Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, Cliby WA, Sarkaria J, Beale G, Edmondson RJ, Curtin NJ.2011. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer105:372–381.
80. Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB.2012. The novel ATR inhibitor VE-821 in-creases sensitivity of pancreatic cancer cells to radiation and chemother-apy. Cancer Biol. Ther.13:1072–1081.
81. Wang H, Wang H, Powell SN, Iliakis G, Wang Y.2004. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res.64:7139 – 7143.
82. Roizman B.1979. The structure and isomerization of herpes simplex virus genomes. Cell16:481– 494.
83. Wilkinson DE, Weller SK.2003. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life55:451– 458.
on November 7, 2019 by guest
http://jvi.asm.org/