0022-538X/89/073072-08$02.00/0
Copyright © 1989,American Society for Microbiology
Chromatin
Structure of Recombinant Moloney Murine Leukemia
Virus
Proviral DNAs That Contain
tax-Responsive
Sequences from
Human
T-Cell
Lymphotropic
Virus
Type
IIin
the
Presence
and
Absence of
tax
HARUO KITADO ANDHUNG FAN*
Department of Molecular Biology andBiochemistry andCancer ResearchInstitute, University of California,
Irvine,
California
92717Received7 September1988/Accepted 24 March 1989
Human T-cell lymphotropic virus types I and II (HTLV-I and HTLV-II) are replication-competent
retroviruses which containtwo additional regulatory proteins, taxandrex. taxis atranscriptional
transacti-vator of the HTLV-I orHTLV-II long terminal repeat (LTR) and also ofsomeheterologous promoters. To
investigate the mechanism of tax transactivation, we used chimeric Moloney murine leukemia viruses (M-MuLVs) with LTRs containing tax-responsive sequences from the HTLV-II LTR (nucleotides -273 to -32). Mo+HTLV-II+M-MuLV contained the HTLVIIsequencesinsertedintothewild-typeM-MuLV LTR
atnucleotide -150, whereas deltaMo+HTLV-II+ M-MuLV contained thesamesequences inserted intoan
M-MuLV LTR lacking its own enhancer region. HTLV-II tax(tax II)-positive mouse cells (15S-5a)infected with Mo+HTLV-II+ M-MuLVordeltaMo+HTLV-II+ M-MuLV showedhigherrates of viraltranscription
innuclearrun-onassaysthan did infectedtax-negative NIH 3T3 cells. The chromatinstructureofthese viruses
wasinvestigated by high-resolution mapping ofDNaseI-hypersensitive (HS) sites.Three prominentHS sites
wereassociated with HTLV-II sequences inproviral chromatin both intax-positiveand intax-negativecells. Thespacing resembled that of the 21-base-pair (bp) repeats, but the HSsitesweredisplaced approximately 50
bp upstream of the21-bp repeats.Thissuggested that cellularproteins boundtothe HTLV-IIsequencesinthe
presence orabsence oftax.Nodirecteffectoftaxonchromatinstructurewasfound. Theseinvivo resultswere
consistent with results ofin vitroDNasefootprinting studies performed byother investigators.
HumanT-cell lymphotropicvirus typesI and II (HTLV-I
and HTLV-II) are two retroviruses associated with T-cell disorders in humans (16, 20, 26). Togetherwith the closely related bovine leukemia virus,these virusesformasubclass
ofretroviruseswhichencodetworegulatory proteinsknown
astaxandrex(19, 28, 30). taxisa nuclearproteinthat is a
transcriptional transactivator oftarget promoters (12, 26). These target promoters include the HTLV-I or HTLV-II long terminal repeats (LTRs) themselves (5, 9, 32, 37), as
wellasheterologouspromotersfor the cellularinterleukin-2 and the interleukin-2 receptorgenes and the adenovirus E3 gene(6, 13, 18, 35).In the HTLV-Iand HTLV-IILTRs,the
sequences necessaryfortaxresponsehavebeenmapped(4,
11, 22, 24, 27). In particular, HTLV-I and HTLV-II both contain 21-base-pair (bp) sequences which are repeated
threetimes withintheU3 regions of each virus(31,33). The 21-bp repeat sequences themselves can confer tax
respon-siveness toheterologous promoters and arebothnecessary
andsufficient fortransactivation (4, 11, 34).
The mechanism of tax transactivation is currently not
known. The simplest model is that tax protein interacts directly with the HTLV 21-bprepeats to effect transactiva-tion. However, other tax-responsive promoters have little homology with the HTLV 21-bp repeats. Moreover,
at-tempts to demonstrate direct binding oftax to the HTLV 21-bprepeatshave beenunsuccessful. Thissuggeststhattax
protein mightinteract with other cellularfactors, which, in turn, interact with the 21-bp repeats. In vitro DNase foot-printing experiments have detected cellular proteins that
* Corresponding author.
bind withthe 21-bprepeats, supportingthe latter model (1, 17).
In vivoexperimentsontaxtransactivationwouldprovide important information in addition to the in vitro
experi-ments. Unfortunately, HTLV-I andHTLV-II arerelatively
difficulttogrowinculture,andinfection is achievedonly by
cocultivation of infected cells with susceptible uninfected cells (7, 43). We previously developed chimeric Moloney murine leukemia viruses (M-MuLVs) which contain U3
sequences from the HTLV-II LTR (21). These viruses are
replication competent and are responsive to HTLV-II tax
(tax II)ininfectivityassays.LikemostMuLVs, it ispossible
toinfect stocks of thesevirusesathigh multiplicityinto cells in the presenceor absence oftax II. Thus, it ispossible to
introduce a tax II-responsive retrovirus into cells and
ob-servetheeffects oftaxII. In the experimentswhose results
are reportedhere, the effect oftaxon thechromatin struc-ture of these chimeric HTLV-II-containing M-MuLVs was
studied.
MATERIALS AND METHODS
Cells. Allcellsweregrown astissue culturemonolayersin Dulbecco modified Eagle medium supplemented with 10%
calf serum (Irvine Scientific, Santa Ana, Calif.). NIH 3T3
cells havebeen describedpreviously (40). 15S-5acellsare a
line of NIH 3T3 cells thatexpress tax11(21).
Generation of celllines thatproduce infectiousM-MuLVs. Cell lines thatproduceinfectiouswild-typeand Mo+HTLV-II+ M-MuLV were obtained by transfection of plasmid
DNAscontaining corresponding provirus organizationsonto NIH 3T3 cells as described previously (21). In the case of
3072
on November 10, 2019 by guest
http://jvi.asm.org/
delta
Mo+HTLV-II+
M-MuLV, our previous producer cell line was generated by transfection on NIH 3T3 cells that contained a retrovirus vector expressing tax II(15S-1cells). It was theoretically possible that the delta Mo+HTLV-II+M-MuLV virus stocks produced from this cell line also contained the genome for the tax II expression vector, which might complicate the interpretation of the experiments. Therefore, we constructed another delta Mo+HTLV-II+
M-MuLV producer cell line. The plasmid containing the delta
Mo+HTLV-II+
M-MuLV provirus organization was cotransfected onto NIH 3T3 cells together with a tax II expression plasmid driven by the simian virus 40 promoter and with a neomycin resistance gene expression plasmid in which the neomycin resistance gene was driven by the deltaMo+HTLV-II+
LTR. Cotransfected cells were grown in thepresence of G418. Since the deltaMo+HTLV-II+LTR does not function well in the absence of tax11 (21), growth in G418 selected for cells producing significant amounts of tax II,
which, in turn, would support the efficient replication and production of delta Mo+HTLV-II+ M-MuLV. G418-resis-tant cell clones were screened for expression of virus by the XC plaque assay (29), and one such producer clone was used for the experiments. Southern blot hybridizations confirmed that the cells produced the desired delta Mo+HTLV-II+
M-MuLV (results not shown).
Virus infections. For nuclear run-on transcription experi-ments,
106
NIH 3T3 or 15S-5a cells were seeded per 9-cm dish 5 h prior to infection. Following treatment with 4 ml of Polybrene (20,ug/ml)
in Tris-buffered saline for 1 h at room temperature, cells were adsorbed with 1 ml of wild-type M-MuLV,Mo+HTLV-II+
M-MuLV, or deltaMo+HTLV-II+ M-MuLV stock for 1 h at
37°C
and then fed with the medium. The wild-type stock contained 106 XC PFU of the virus, and equivalent numbers of virus particles compared with the wild-type stock (as measured by reverse tran-scriptase assays) were used forMo+HTLV-II+
M-MuLV and deltaMo+HTLV-II+
M-MuLV infections. The infected cells were passaged once so that two 15-cm dishes were seeded from each 9-cm dish. While the cells were still subconfluent, nuclei were harvested and frozen for run-on transcription reactions as described previously (39a). South-ern blots confirmed that at this time point the majority of the viral DNA was integrated (data not shown). Similar infection procedures were used for DNase I hypersensitive (HS) site-mapping experiments. In these cases, undiluted tissue culture supernatants from producer cell lines were used as virus stocks. The infected cells were passaged once to seed five 15-cm dishes, and nuclei were harvested while the cells were subconfluent. The nuclei were immediately digested with DNaseI.
Cell fractionation and DNase I digestion of nuclei. Fraction-ation and DNase I digestion were performed essentially as described by Weintraub and Groudine (41) with modifica-tions previously reported (39). Briefly, nuclei were isolated from trypsinized cells by lysis in 1% Nonidet P-40, harvested by centrifugation, and suspended in reticulocyte standard buffer. DNase I (RQ1 DNase; Promega Biotec, Madison, Wis.) was added to various concentrations, and digestions were carried out for 20 min at
20°C.
Reactions were termi-nated by the addition of EDTA, and high-molecular-weight DNA was purified as previously described (15).Restriction enzyme digestion, gel electrophoresis, and prep-aration of labeled DNA probes. Restriction enzymes were obtained commercially, and digestions were performed in recommended buffers. Agarose gel electrophoresis was
per-formed as previously described (2). Labeled DNA probes were prepared by the randomprimerextension method (8). Southern blot hybridization. DNA was transferred by capillary blotting, essentiallyasdescribed by Southern (38), onto nylon membranes (GeneScreen Plus; Dupont, NEN Research Products, Boston, Mass.). Hybridization and washing of nylon membraneswere done as specified by the manufacturer. Autoradiographic exposures were performed at -80°Conpreflashed X-rayfilm (XAR-5; Eastman Kodak Co., Rochester, N.Y.) with two intensifying screens.
Nuclear run-on transcription. Nuclei were isolated as described above, except that following the last wash, the nuclear pellet was suspended in an equal volume of nuclear storage buffer (20 mM Tris hydrochloride [pH 7.9], 75 mM NaCl, 0.5 mM EDTA, 0.85 mM dithiothreitol, 0.125 mM phenylmethylsulfonyl fluoride, 50% glycerol), and nuclei were counted and stored at -80°C. In vitro elongation
reactions were performed as described by Firzlaff and Diggelmann (10) with modifications by Thompson and Fan (39a). Briefly, reactions took place in a mixture
containing
100 mM Tris hydrochloride (pH 7.9), 0.4 mM EDTA, 10%glycerol, 8 mM dithiothreitol, 1 mM MnSO4, 60 mM
(NH4)2SO4,
0.6 mM each ATP, GTP, and CTP, 50RCi
of[ot-32P]UTP
(400 Ci/mM; Amersham Corp.,Arlington
Heights, Ill.), and approximately 2 x 106 nuclei in nuclear storage buffer, in a total volume of 100 ,ul. Reactions were incubated at25°C for 40 min. After incubation, the reaction mixtures were adjusted to 10 mM MgCl2, and 2 ,ul ofRQ1 DNase Iwas added. Following incubation for 5 min at
26°C,
samples were treated with 100RI
ofproteinase K per ml in the presence of0.5% sodium dodecyl sulfate for 30 min at42°C. RNA was then purified by phenol-chloroform extrac-tion and ethanol precipitation. Transcripts were further purified bypassage through a 1-mlcolumnofSephadexG-50 and reprecipitated with ethanol in 2 M ammonium acetate. Pellets were again washed and suspended in TE buffer (0.1
M Tris [pH 7.5], 1 mM EDTA).
Hybridization of in vitro-elongated transcripts to slot-blot filters. Nylonmembrane filters (GeneScreen Plus)
containing
1 ,ug each ofthree DNA fragments were prepared asrecom-mended by the manufacturer. The three DNA
fragments
were an 8.3-kilobase (kb) HindlIl fragmentcontaining
M-MuLV sequences (representing the entire viralgenome)
(3)
at the top, a 1.6-kbBamHI fragmentcontaining
the human gamma actin gene(providedbyL. Kedes)atthebottom,
and an unrelated plasmid vector fragment of 4.4 kb in between. Prehybridization was carried out for 3to 4h at45°C in1.5 ml of hybridization buffer containing 50 mMpiperazine-N,N'-bis(2-ethanesulfonic acid) (PIPES; pH 7.0), 50% deionized formamide, 0.5 MNaCl,0.8%sodium
dodecyl
sulfate,
2 mM EDTA, and 200 ,ug of denatured salmon sperm DNA per ml. Following prehybridization, 2 x 105 cpm of run-ontran-scripts was added to the above mixture, and the
hybridiza-tion was continued for 2 days at 45°C. The filters werewashed briefly with 2x SSC (lx SSC is 0.15 M NaCl
plus
0.015 Msodium citrate) and then washed for30 min at
65°C
in 2x SSC-1% sodium dodecyl sulfate and for 30 min at
roomtemperature in 0.5x SSC. Filters were thenincubated for 30 min at 37°C in a solution
containing
10 mM Tris hydrochloride (pH 7.5), 0.3M NaCl,2 mMEDTA,
100,ugof bovine serum albumin perml, 10,ugofRNase Aperml,
and 200 U ofRNase T1 perml. After RNase treatment, thetwoprevious washes wererepeated and the filters were
exposed
to preflashed XAR-5 film with two
intensifying
screens at-800C.
on November 10, 2019 by guest
http://jvi.asm.org/
a.
M-MuLVLTR
b.
Xbal-150
-450 -340 -180 80-30 +145
a I . _
CAT TATA
U3 RU
+1 +70
MoENHANCERS
iRI
Mo+HTLV 11 M-MuLV LTR
XbAI Xb
Mo ENHANCERS
AMo+HTLV 11M-MuLVLTR
(Mo enhancersdeleted)
XbaI
C.
HTLVII
Sma-Bstprobe
XbPl M.
|Enh
-t
SPst-Xhoprobe
I-MuLV provirus
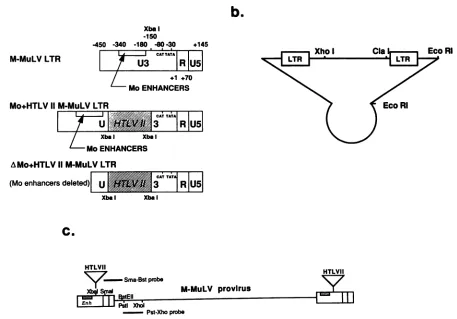
FIG. 1. (a)Schematicdiagrams of wild-type,Mo+HTLV-II+,anddeltaMo+HTLV-I1 LTRs.Thelocations ofM-MuLVpromoter(CAT and TATA)andenhancerelements as well asXbaIrestriction sites are shown. Numbersarerelativetothe capsiteas +1. Symbol: , insertedHTLVIIsequences. (b) Schematicdiagramofproviral organizationsthatwereusedtogenerate infectious MuLVs. Boxes labeled LTR represent any oneofthe three LTRsdescribedinpanela, andthecircularportionrepresents thepBR322plasmidvector. Fordetailed constructionsofequivalentplasmids,seereference23.(c)Restriction sites usedtogenerateMuLV-specific probesinrelationtotheproviral structure. Positions ofthe twoprobesareindicatedbytheheavylines above andbelowthemap. TheBstEII andXhoI siteswerealsoused asreference restriction sitesin the DNase IHS site mapping.
RESULTS
Diagrams ofthewild-type and chimeric LTRs driving the
infectiousM-MuLVs usedinthis study are shown in Fig. la. Twochimeric LTRs were used in these studies, Mo+HTLV-II+ and delta Mo+HTLV-II+ (+ indicates that the HTLV sequences were inserted in the same transcriptional direc-tion as the M-MuLV LTR). Sequences from the HTLV-II LTR U3 region (nucleotides -273 to -32) wereinserted at nucleotide -150 into the wild-type M-MuLV LTR to give the Mo+HTLV-II+ LTR orinto an M-MuLV LTRlacking the enhancerregion (nucleotides -357 to -150) to give the deltaMo+HTLV-II+LTR.Thesechimeric LTRs were used to construct plasmids containing M-MuLV proviral organi-zations with the altered LTRs at either end (Fig. lb), and cells producing the corresponding infectious M-MuLVs wereobtainedbytransfection of NIH 3T3 cells as described in Materials and Methods.
For the experiments, cells were freshly infected at high multiplicity with the various M-MuLVs and pooled and harvested after only one passage. We previously showed that thiswasadvantageous, since the infected cells had large numbers of proviral copies per cell (ca. 10 to 20) (39a). The
large proviral copy number made it easier to detect M-MuLVDNA or RNA in the nuclearrun-on and DNAse I HS
site-mapping
experiments. Moreover, the fact that thecul-tures were not clonal meant that they contained a large
numberofproviruses integrated at multiple sites. Analyses of such cultures minimize effects from particular adjacent cellsequences on viral expression.
Run-ontranscription.As described in theintroduction, the M-MuLVs containing HTLV-II sequences appeared to be good models for investigating the mechanisms ofHTLV-II taxprotein action. Itwasfirst necessary toconfirm thatthe increased infectivities of these chimeric M-MuLVs in tax-expressing cells reflected increased viral transcription. Therefore, viral RNA synthesis rates in the presence or absence of tax were measured by nuclearrun-on transcrip-tion assays (10). Wild-type and chimeric viruses were in-fected into NIH 3T3 cells or 15S-Sa cells, NIH 3T3 cells expressingtaxII.Equal numbers of viruses asmeasuredby reverse transcriptase activities were used in the infections. Nuclei were harvested from the infected cells 4 to 6 days after infection, and in vitro transcription reactions were carriedoutin thepresence of[ot-32P]UTP. Equalamounts of labeledRNA from the different reactions were then hybrid-ized with filterscontaining one M-MuLV-specificDNAband aswell astwocontrol DNA bands. One control DNA band wasspecific forthehumangamma-actin gene (14) and served asapositive hybridization control. The other control DNA band containedonly bacterial plasmid sequences andserved as anegative control fornonspecific binding.
Figure 2shows the results ofoneexperiment. Very little
CA;TAiTA
3R US
CA; T;TA _
R US
Xba I
HTLVII
T
111
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.94.551.78.397.2]A
a
b
4,
c
d
M
A
e
f
gh
."x_a4
B a
C,
d
am M
A
e
'
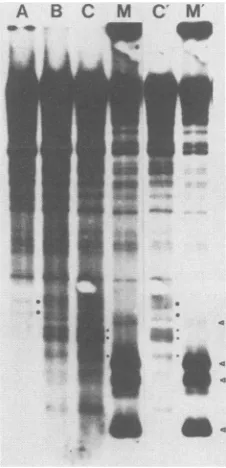
h'FIG. 2. Run-ontranscription experiments. NIH 3T3 cells(lanes
ato d and a' tod')and15S-5a cells(laneseto h and e' toh')were
infected with wild-type M-MuLV (lanes b, b', f, and f'), Mo+HTLV-II+ M-MuLV (lanes c, c', g, and g'), or delta
Mo+HTLV-11'M-MuLV(lanes d, d', h,andh')asdescribed in the text. Lanes a,a',e,ande'wereuninfected controls. Invitro-labeled transcriptswereprepared,andequalamountsofradioactivitywere
hybridizedtoslot-blot filters. Each filter had three DNAfragments:
an8.3-kbfragment containingthe entire M-MuLV genome at thetop (marked M), a 1.6-kbfragment containingthe human gamma actin gene at the bottom(marked A),andanunrelated vectorfragmentof 4.4 kb in between. Two different exposures of thesamefiltersare
shown: 5 days (A)and 1day (B).
hybridization to the plasmid DNA bands was observed,
confirmingthe specificityof the hybridizationconditions. In vitro-labeled transcripts from uninfected NIH 3T3 cells showed almost no hybridization to the M-MuLV-specific
band and detectable (although low) hybridization to the
gamma-actin band, asexpected (lanes a). Uninfected15S-5a
cells showedlow-levelhybridizationto the
M-MuLV-specif-ic band, as well as hybridization to the gamma-actin band
(lanes e). The 15S-5a cells were originally infected with
amphotropicMuLV(21),andradioactivityin the
M-MuLV-specificbandpresumably represented cross-hybridizationof
amphotropicMuLVtranscripts. Transcripts fromwild-type
M-MuLV-infected NIH 3T3 and 15S-5acells showed
equiv-alent amounts of M-MuLV-specific transcripts, indicating
that M-MuLV-transcription is not responsive to HTLV-II
tax, as expected (lanes b and f). In contrast, infection of
Mo+HTLV-II+ M-MuLV or delta Mo+HTLV-II+ M-MuLV resulted in higher viral transcription in the
tax-positive 15S-5acells (Fig. 2A, lane g or h, compared with
lane c or d). In
Mo+HTLV-II+,
this difference is morereadilyseeninalighterexposureof thesamefilters shown in
Fig. 2B (lane g' compared with lane c'). These results confirmed that the chimeric HTLV-II-containing M-MuLV
proviruses were transcriptionally responsive to HTLV-IT tax protein.
One alternative explanation of the results in Fig. 2 was that theamountofproviralDNA in infected 15S-Sacells was larger than in infected NIH 3T3 cells. However, Southern blothybridizationsindicated that the amount of Mo+HTLV-1 proviralDNA in the infected 15S-5a cellswasthe same as that in NIH 3T3 cells (results not shown). For delta Mo+HTLV-II+-infected cultures, the difference was at most fivefold, which was clearly less than the difference in
transcription. Thus, for both Mo+HTLV-II+ and delta
Mo+HTLV-II+, the results in Figure 2 reflected transcrip-tional activation bytaxII.
It should be noted that the infectivities of these viruses showastrongerdependenceon tax II(e.g., 1.5to2 ordersof magnitude for Mo+HTLV-II+) than is evident from the run-ontranscriptions.This mayreflectthe fact that infectiv-ity assaysmeasuretheoverall efficienciesof infection, which may not be linearly proportional to the transcription rate. Wehave also observedasimilarphenomenonwith hormone-responsive M-MuLVs carrying sequences from mouse mam-mary tumorvirus (MMTV) (23, 39a).
DNase I HS sitemapping of wild-typeandchimericLTRs. Since the nuclearrun-on assays indicated that the HTLV-II-containing M-MuLV proviruses were transcriptionally activated by HTLV-II tax protein, we wished to know whether thechromatinstructuresof the transcriptional con-trolregions (LTRs) for these viruseswerealtered in vivo by the presence oftax. Regions containingthe HTLV-II 21-bp repeats wereofparticular interest.Thechromatin structures were investigated by mapping DNase I HS sites by the
indirectend-labeling method (39, 42). Nuclei from infected cellswereincubated with differentconcentrations of DNase I, and then high-molecular-weight DNA was extracted and
digestedtocompletionwith areference restriction enzyme. Southern blots of the restriction digests were then hybrid-ized with a radioactively labeled M-MuLV-specific DNA probe.
Figures 3a and b, lanes A to C, show an analysis of
wild-type MuLVproviral chromatinin the absence or pres-enceoftaxII.Nuclei from NIH 3T3or15S-Sa cells infected with wild-typeM-MuLVwereincubated with DNase I, and DNAswerethen digested with XhoI. Southern blot hybrid-ization with an M-MuLV-specific probe from nucleotides
563to1560(Fig. lc,Pst-Xhoprobe)is shown. Aspreviously reported (39a), M-MuLV proviruses in NIH 3T3 cells
showed four HS sites in the enhancerregion.Twoprominent
siteswereassociated with the75-bptandem repeats, and the other two were located immediately upstream and down-stream. Inaddition, another HS site mapped tothe TATA
box, 30 bp upstream from the start site for transcription. M-MuLV proviruses in tax II-positive 15S-5a cells showed the same pattern and location of HS sites asthose in NIH 3T3cells.Thus, taxIIproteindidnotaffect the distribution of HS sites within the
wild-type
M-MuLVLTR,asexpected.For amap of the location of HS sites within the
wild-type
M-MuLV LTR, seeFig. 6.Next, we examined the HS sites within the HTLV-II sequencesof the chimeric M-MuLVs.
Initially,
Mo+HTLV-11 M-MuLV-infected cellswerestudied, because this virus has ahigher infectivitythan deltaMo+HTLV-II+ M-MuLV (21). As a result, morecopies
ofintegrated
proviral DNA would be expected for freshly infected cultures of Mo+HTLV-II+M-MuLV,leading
tostrongerhybridization
signals. HS
mapping
of Mo+HTLV-II+ M-MuLV provi-rusesinNIH3T3and15S-5a cells with XhoIasthereferenceon November 10, 2019 by guest
http://jvi.asm.org/
a8 A B C D E F M
.
0 -mo_ *E0
b. A' B' C' D' E/ F M'
_1w
If
lw
itA Aa
h
IA B C M C' M'
__
-~
_WR.
..i.s.
.$: Z ,
.:..
_ ___~~~~~~~~~~~~~~~~~~~~~~~4w:C
_ VIIF _ ~~~~~4
j;E . _
FIG. 3. (a) DNase IHS sitemappingof wild-type M-MuLV and Mo+HTLV-II+M-MuLVproviruses inNIH 3T3 cells. LanesA,B, and Crepresentwild-typeM-MuLVprovirus,and lanes D,E,andF represent Mo+HTLV-II+ M-MuLV provirus. Cells were infected with undiluted virus supernatants of producer cell lines, and nuclei wereisolated anddigested with different concentrations of DNase 1. DNA wasextracted,digested withXlhoI,and analyzed by electro-phoresis on a 1.0% agarose gel and blot hybridization with the
32P-labeled M-MuLV-specific PstI-Xhol probe (Fig. 1c}. DNase I concentrationswere 0.5(lanes A and D), 1.0 (lanes B and E), and 5.0 (lanes C and F) U/mI. Lane M is a marker lane containing four M-MuLV-specific restriction fragments (<')mixed with uninfected NIH3T3cellularDNAthat had beendigestedwithXlioI. Thesizes of these markers were 2,007, 1,792, 1.711,and 1,532bp. The three HS sites within the HTLV-II sequences are indicated (*). DNA samples that were not treated with DNase I consistently did not showrestrictionfragments in regionscorresponding tothese LTR sub-bands(datanotshown).Hybridizationtohigh-molecular-weight DNAin alllanes includingtheNIH3T3lanerepresented hybridiza-tionof the MuLVprobetoendogenous mouseretrovirussequences. (b) DNase I HS site mapping of wild-type M-MuLV and Mo+HTLV-11+ M-MuLV provirusesin15S-Sacells. LanesA',B', and C' representwild-typeM-MuLVprovirus,and lanesD', E', and F'represent Mo+HTLV-II+M-MuLVprovirus. The same experi-mental procedures as those usedfor panel a were used. DNase I concentrationswere0.7(lanesA'andD'), 1.0(lanesB'and E'), and 5.0 (lanes C' and F') U/ml. Lane M' was a marker lane similar to lane M ofpanel a. The apparently higher density of the HS site corresponding to the TATA sequence in lane C' has not been consistently observed.
enzymeis shown in Fig. 3a and b, lanes D to F. In addition to the HS sites associated with wild-type M-MuLV, Mo+HTLV-II+ M-MuLV LTRs showed four HS sites within theHTLV-IIsequences. Three of them (marked with asterisks in Fig. 3) were prominent. It was interesting that thepatternsof HS sites in infected NIH 3T3 and15S-5a cells werethe same, indicating that taxIIprotein did notaffect the chromatin structure ofMo+HTLV-I1 M-MuLV.
To mapthe HS sites in theMo+HTLV-II+M-MuLV LTR moreaccurately, we also used a second reference restriction enzyme,BstEII. BstEII cleaves M-MuLV proviral DNA at nucleotide 725 and will generate DNase I HS subfragments from the LTRthat are approximately half as large as those
generatedbyXlIol.This afforded better resolution during the
gelelectrophoresis. The location of theBstEII site and also
the M-MuLV-specific probe used are indicated in Fig. Ic
FIG. 4. DNase I HS site mappingofMo+HTLV-II+ M-MuLV in15S-Sacells. DNAsamples usedinthis experimentwerefromthe sameDNase Idigestionarrayas shownin Fig. 3b, lanes D' toF'. Procedureswere asdescribed forFig. 3, exceptthat the DNA was digested with BstEII and analyzed by electrophoresis on a 1.5% agarose gel and blot hybridization with the 32P-labeled M-MuLV-specific SnalI-BstEII probe (Fig. lc). DNase Iconcentrations were 1.0(laneA), 5.0(laneB), and 10(laneC) U/ml. Lane Misamarker lane containing four M-MuLV-specific restriction fragments (<) mixed with uninfected 15S-Sacellular DNA that had beendigested withBstEll. The sizes of these markers were 1,172, 957, 876, and 697bp. Lanes C' and M' are lighterexposuresoflanesC and M, respectively.Thethree HS sites within theHTLV-IIsequencesare indicated (*). ThetwoHS sites in the upstream M-MuLV sequences present at low DNaseconcentrations areindicated in lane A (0).
The reduction in these bands at higher DNase concentrations is
most evident in laneC'. in which thecorresponding positionsare also indicated(0).
(Sma-Bst probe). Figure 4 shows HS site mapping with BstEIIof thesameMo+HTLV-II+ M-MuLV DNAsamples
inFig. 3b. The higherresolution allowed accurate mapping of the three prominent HS sites inthe HTLV-II sequences. (Although in lane C the top two of the three bands with asterisksareunresolved, they arein fact separate bands, as showninalighterexposureof lane C in lane C'.) Thesesites had the same relative spacing as the 21-bp repeats in the HTLV-II sequences, with an HS site located about 50 bp upstream from each 21-bp repeat. This might suggest the presenceofafactor(s) boundtoeach21-bp repeat. Another interesting phenomenon wasobserved with the HS sites in the M-MuLV enhancer sequences. At the lowest DNase I concentration(lane A), there were twoadditional HS bands immediately upstream of the strong HS bands located in the tandem repeats (indicated by dots in Fig. 4; see also the dashedarrowsinFig. 6). Athigher DNase I concentrations, theupstreambands were reduced or absent (most evident in the lighter exposure in Fig. 4, lane C'). This could result from the presence oftwoupstream sites of extremeDNase I hypersensitivity. Alternatively, this might reflect an initial DNase I cut at the upstream site, followed by progressive
digestion tothe downstream site.
It was alsoimportant toexamine the chromatin structure
-b
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.386.499.87.321.2] [image:5.612.65.302.87.284.2]A B' C' D' E' M A B C D E
M-MuLV LTR
Xba I -150
-450 -340 -180 -8Q-30 +70 +145
i CAT TATA
U3
R
U5
I
I
Mo+HTLV 11 M-MuLV LTR
|
*3
f U5
---*
T*A
T
FIG. 5. DNase I HS site mapping of delta Mo+HTLV-II+ M-MuLV in 15S-Sacells versus NIH 3T3 cells. Lanes A' through E' represent mapping in 15S-Sacells, and lanes A through E represent mapping in NIH 3T3 cells. Procedures were the same as those described forFig. 4, withBstEII as thereference enzyme. DNase I concentrations were 0 (lanes A' and A), 0.5 (lanes B' and B), 2.5 (lanes C'and C), 10 (lanes D' andD),and50(lanes E' and E)U/ml. Lane Mis a marker lane containing four M-MuLV-specific restric-tion fragments (<) mixed with uninfected 15S-Sa cellular DNA digested with BstEII. The three HS sites within the HTLV-II sequences areindicated (*). Lane Mwasoverexposed in theoptimal photographic exposure of the blot for visualization of the relevant sub-bands. In a lighter exposure, the four markers were evident at theindicated positions(data not shown).
ofthedelta Mo+HTLV-1lI M-MuLV LTR in the presence and absence of tax.. The Mo+HTLV-II+ M-MuLV LTR examined inFig. 3 and 4also contained M-MuLV enhancer elements, and it was possible that they affected the chroma-tin structure of the HTLV-II sequences. The delta Mo+HTLV-II+ M-MuLV LTRlacks the M-MuLV
enhanc-ers. These experiments were more difficult, because the
infectivity ofdelta Mo+HTLV-II t M-MuLV is very low in NIH 3T3cells,resulting in weaker hybridization signals.The sameHSsites associated with theHTLV-II sequencesinthe Mo+HTLV-II+ M-MuLV LTR were also present in delta Mo+HTLV-II+ M-MuLV proviruses, either inNIH 3T3 or in15S-5a cells(Fig.5and 6). Thus, theM-MuLVenhancers did not influence the distribution of HS sites within the HTLV-II sequences.
DISCUSSION
Inthe results reported here, the in vivo chromatin struc-ture of tax 1I-responsive M-MuLV proviruses was investi-gated.DNase IHS sites associated withtheinserted HTLV-II sequencesweredetected, and the spacing of the HS sites was the same as the spacing ofthe 21-bp repeats in these sequences; however, the HS sites were displaced
approxi-mately 50 bp upstream ofthecorresponding 21-bp repeats. The most likely interpretation of this is that protein factors were bound to each 21-bp repeat and that they induced a
A
Mo+HTLV 11
M-MuLV LTR
(Mo enhancersdeleted) | , <
3
U51
** *
FIG. 6. Locations ofmajor HS sites. The major HS sites are shown in relation tothe important sequences within the wild-type, Mo+HTLV-I11 M-MuLV. and delta Mo+HTLV-11+ M-MuLV LTRs. Symbols: *, locationsofHTLV-1l 21-bp repeats; V, posi-tions of themobility markers in theSouthernblots;
T
, f,majorHS sites(thicker arrows correspond to stronger HS sites); *,the three mostprominent HS sites within HTLV-1l sequences: 1, positionsof two additional HS sites observed at low DNase I concentrations.
DNase HS site upstream of the region of factor binding. It was theoretically possible that sequences upstream of the 21-bp repeats were responsible for generating the DNase HS sites, althoughthereis nosequencesimilarity between these upstream sequences.
Themostimportant part oftheresults was thefact thatthe chromatin structure of the chimeric LTRs was notaffected by the presence or absence oftiax II protein, even though run-onassaysconfirmed that therewastranscriptional trans-activation. Thisindicated that theprotein factors responsible forinducingthe DNase IHSsites intheHTLV-II sequences were already present in standard NIH 3T3 cells and did not require tax II to interact with the HTLV-II sequences. Therefore, these results would not support a model for transactivation in which ta II protein directly binds to the 21-bp repeats. Otherwise, different HS site patterns in the NIH 3T3 and 15S-Sa cells would probably result.
It is interesting to compare these results with those obtained from mouse mammary tumor virus (MMTV), an-other retrovirus. MMTV is transcriptionally transactivated by glucocorticoid hormones, and this results from direct interaction of hormone-bound glucocorticoid receptor pro-teinwithtarget DNAsequences in the MMTV LTR(25). In this system,activation ofMMTVtranscription by glucocor-ticoidsresultsinthe appearance ofa newDNaseI HSsitein the glucocorticoid response element (44). Changes in chro-matinstructureupon hormone inductionwerealso observed inchimeric M-MuLVswith LTRscontaining MMTV gluco-corticoid response sequences (39a). These results contrast
with those shown here for tax II transactivation and also suggestthat the mechanism fortaxII action is different from thatforproteins that directly bind totarget sequences such asthe glucocorticoid receptor.
These in vivo experiments support and complement in vitroDNA-binding experiments reported by others. Factors
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.83.271.80.322.2] [image:6.612.315.550.80.294.2]from cell extracts, which bind to the 21-bp repeats in the HTLV-I or HTLV-II
LTRs,
have been demonstrated byDNase I
footprinting (1, 17).
No difference inthe natureofthe
footprints
was observed when extracts from uninfectedand infected cells were
used, establishing
that taxproteindoes not
directly
bind the21-bp
repeat sequence DNAs invitro. The DNase
footprinting
also did not reveal proteins that bound to sequences upstream of the 21-bp repeats. These cellularproteins
from the in vitro studies are likelycandidates for those that induced the DNase HS sites inthe chromatin studies
reported
here.Recently,
it has been shown that a cellularprotein (CREB)
that binds to a core consensus sequence ofcyclic AMP-responsive
genes alsobinds to the HTLV-I
21-bp
repeats (T. H. Tan and R. G.Roeder,
personal communication).
If the DNase I HS sitesin the
HTLV-IT
sequencesresultedfrom cellular
proteins
bound at the21-bp
repeats aspro-posed here,
there was one somewhatunexpected result,
namely,
that themiddle HS siteactually mapped
coincident withthefarthestupstream21-bp
repeat.Thein vitro DNasefootprinting experiments
suggest that thebinding
of factorsat the
21-bp
repeats would prevent DNasedigestion,
al-though
in vitro DNase HS sites inimmediately adjacent
sequences can sometimes result frombinding
ofproteins.
One
explanation
could bethat,
invivo,
notnecessarily
all21-bp
repeats on aprovirus
are bound withprotein
at thesametime.
Thus,
aproviral
LTR withprotein
bound atthe middle21-bp
repeatbutnotthe upstream21-bp
repeatcouldgive
rise tothe DNAse I sub-bandmapping
coincident with theupstream21-bp
repeat. Analternativeexplanation
is thatprotein
bound at one21-bp
repeat could induce aDNase I HS site in upstream DNA sequences, even if thosese-quences arealso
complexed
withprotein.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants CA-32454andCA-32455 from the National CancerInstitute.
LITERATURE CITED
1. Altman,R., D.Harrich, J.A.Garcia,and R. B. Gaynor. 1988. Human T-cell leukemia virus types I and II exhibit different DNase I protection patterns. J. Virol. 62:1339-1346.
2. Bacheler,L.T.,and H. Fan.1979. Multiple integrationsites for Moloneymurine leukemia virusinproductivelyinfectedmouse
fibroblasts. J. Virol. 30:657-667.
3. Berns, A. J. M., M. H. T. Lai, R. A. Bosselman, M. A.
McKennett, L. T. Bacheler, H.Fan, E. C.Robanus-Maandaag,
H.vanderPutten, andI.M.Verma.1980. Molecularcloningof unintegratedandaportionofintegratedMoloneymurine leuke-mia viralDNA inbacteriophagelambda.J. Virol.36:254-263. 4. Brady, J., K. T. Jeang, J. Duvall, and G. Khoury. 1987.
Identification of p4Ox-responsive regulatory sequences within thehumanT-cell leukemia virustype I longterminalrepeat. J. Virol. 61:2175-2181.
5. Cann,A.J., J.D. Rosenblatt, W.Wachsman,N. P.Shah, and I. S. Y. Chen. 1985. Identification of the gene responsiblefor human T-cell leukemia virustranscriptionalregulation. Nature (London)318:571-574.
6. Chen,I. S. Y.,A.J.Cann,N.P.Shah,and R. B.Gaynor. 1985. Functional relation between HTLV-II x and adenovirus ElA proteinsintranscriptional activation. Science230:570-573. 7. Chen, I. S. Y., S. G. Quan, and D. W. Golde. 1983. Human
T-cellleukemia virus type II transforms normal human lympho-cytes. Proc. Natl. Acad. Sci. USA80:7006-7009.
8. Feinberg, A. P., and B. Vogelstein. 1984. A technique for radiolabelingDNA restriction endonuclease fragments to high
specific
activity.Anal. Biochem. 137:266-267.9. Felber,B. K.,H. Paskalis,C. Kleinman-Ewing,F. Wong-Staal,
and G. N. Pavlakis. 1985. The pX protein of HTLV-1 is a
transcriptional activator of its long terminal repeats. Science 229:675-679.
10. Firzlaff, J. M., and H. Diggelmann. 1984. Dexamethasone increases the number of RNA polymerase II molecules
tran-scribing integrated mouse mammary tumor virus DNA and flankingmousesequences.Mol. Cell. Biol. 4:1057-1062. 11. Fujisawa, J., M. Seiki, M. Sato, and M. Yoshida. 1986. A
transcriptional enhancer sequence ofHTLV-I is responsible for trcans-activation mediated by p4Gt of HTLV-I. EMBO J. 5: 713-718.
12. Goh, H. G., J. Sodroski, C. Rosen, M. Essex, and W. A. Haseltine. 1985. Subcellular localization of theproduct of the long openreadingframe of human T-cell leukemia virus type I. Science 227:1227-1229.
13. Greene, W. C., W. J. Leonard, Y. Wano, P. B. Svetlik, N. J. Peffer, J. G. Sodroski, C. A. Rosen, W. C. Goh, and W. A. Haseltine. 1986. trans-Activator geneofHTLV-II induces IL-2 receptor and IL-2 cellular gene expression. Science 232:877-880.
14. Gunning, P., P. Ponte, H. Okayama, J. Engel, H. Blu, and L. Kedes. 1983. Isolationand characterizationof full-length cDNA clones for human alpha-, beta-, and gamma-actin mRNAs: skeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol. Cell. Biol. 3: 787-795.
15. Hanecak, R., P. K. Pattengale, and H. Fan. 1988. Additionor substitution of simian virus 40 enhancer sequences into the Moloney murine leukemia virus (M-MuLV)long terminal repeat yields infectious M-MuLV with altered biological properties.J. Virol. 62:2427-2436.
16. Hinuma, H., K. Nagata, M. Hanaoka, M. Nakai, T.Matsumoto, K. Kinoshita, S. Shirakawa, and I. Miyoshi. 1981. Adult T-cell leukemia:antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc. Natl. Acad. Sci. USA 78:6476-6480.
17. Hyborg, J. K., W. S. Dynan, I. S. Y. Chen, and W. Wachsman. 1988.Binding of host-cell factors to DNA sequences in thelong terminal repeat of human T-cell leukemia virus type I: implica-tions for viral gene expression. Proc. Natl. Acad. Sci. USA 85:1457-1461.
18. Inoue, J., M. Seiki, T. Taniguchi, S. Tsuru, and M. Yoshida. 1986. Induction of interleukin 2 receptor gene expression by p40Yencodedbyhuman T-cell leukemia virustype 1. EMBO J. 5:2883-2888.
19. Inoue, J.,M.Yoshida,and M.Seiki.1987.Transcriptional (p40X) andpost-transcriptional (p27x-11) regulatorsarerequiredfor the expressionandreplicationof humanT-cell leukemia virus type Igenes. Proc. Natl. Acad. Sci. USA 84:3653-3657.
20. Kalyanaraman, V.S., M.G.Sarngadharan, M. Robert-Guroff, I.Miyoshi,D.Blayney, D. Golde, and R. C. Gallo. 1982. A new subtypeof humanT-cell leukemia virus(HTLV II) associated with a T-cell variant of hairy cell leukemia. Science 218: 571-573.
21. Kitado, H., I. S. Y. Chen, N. P. Shah, A. J. Cann, K. Shimo-tohno, and H. Fan. 1987. U3 sequencesfrom HTLV-I and -II LTRs confer pxprotein response to a murine leukemia virus LTR.Science 235:901-904.
22. Ohtani, K., M. Nakamura, S. Saito, T. Noda, Y. Ito, K. Sugimura, and Y. Hinuma. 1987. Identification of two distinct elements in thelongterminal repeat ofHTLV-I responsible for maximumgeneexpression.EMBO J. 6:389-395.
23. Overhauser,J., and H. Fan. 1985.Generationof glucocorticoid-responsive Moloney murine leukemia virus by insertion of regulatory sequences from murine mammary tumor virus into thelongterminal repeat. J.Virol. 54:133-144.
24. Paskalis,H.,B. K.Felber, and G. N. Pavlakis. 1986. Cis-acting sequences responsiblefor the transcriptionalactivation of hu-man T-cell leukemia virus type I constitute a conditional en-hancer. Proc. Natl. Acad. Sci. USA 83:6558-6562.
25. Payvar, F.,D.DeFranco, G. L. Firestone, B. Edgar,0. Wrange, S. Okret, J. A. Gustafsson, and K. R. Yamamoto. 1983. Se-quence-specific binding of glucocorticoid receptor to MTV
on November 10, 2019 by guest
http://jvi.asm.org/
DNA atsites within and upstream of the transcribed region. Cell 35:381-392.
26. Poiesz, B. J., F. W. Ruscetti, A. D. Gazdar, P. A. Bunn, J. D. Minna, and R. C. Gallo. 1980. Detection andisolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 77:7415-7419.
27. Rosen, C. A., J. G. Sodroski, and W. A. Haseltine. 1985. Location of cis-acting regulatory sequences in the human T-cell leukemia virus type I long terminal repeat. Proc. Natl. Acad. Sci. USA 82:6502-6506.
28. Rosenblatt, J. D., A. J. Cann, D. J. Slamon, I. S. Smalberg, N. P. Shah, J. Fujii, W. Wachsman, andI. S. Y. Chen. 1988. HTLV-Il transactivation is regulated by the overlapping taxlrex nonstructural genes. Science 240:916-919.
29. Rowe, W. P., W. E. Pugh, and J. W. Hartley. 1970. Plaque assay techniques formurineleukemia viruses. Virology 42:1136-1139. 30. Sagata, N., T. Yasunaga, and Y. Ikawa. 1985. Two distinct polypeptides maybe translatedfromasingle splicedmRNAof the X genes of human T-cell leukemia and bovine leukemia viruses. FEBS Lett. 192:37-42.
31. Seiki, M., S. Hattori, Y. Hirayama, and M. Yoshida. 1983. Human adult T-cell leukemia virus: complete nucleotide se-quenceof the provirus genomeintegrated in leukemiacell DNA. Proc. Natl. Acad. Sci. USA80:3618-3622.
32. Seiki, M., J. Inoue, T. Takeda, and M. Yoshida. 1986. Direct evidence thatp4GW of human T-cell leukemia virus type I is a trans-acting transcriptional activator. EMBOJ. 5:561-565. 33. Shimotohno, K., D. W. Golde, M. Miwa, T. Sugimura, and
I. S. Y. Chen. 1984. Nucleotide sequence analysis of the long terminal repeat ofhumanT-cell leukemia virus type lI. Proc. Natl.Acad. Sci. USA81:1079-1083.
34. Shimotohno,K., M.Takano, T. Teruuchi, and M. Miwa. 1986. Requirement ofmultiple copies ofa 21-nucleotide sequence in theU3regions ofhuman T-cellleukemiavirus type I and type II longterminalrepeatsfortrans-acting activationof transcription. Proc. Natl. Acad. Sci. USA 83:8112-8116.
35. Siekevitz, M., M. B. Feinberg,N.Holbrook,F.Wong-Staal, and
W.C. Greene.1987. Activationof interleukin 2 and interleukin 2 receptor (Tac) promoterexpressionby thetrans-activator (tat) geneproduct of human T-cell leukemia virus, type I. Proc. Natl. Acad. Sci. USA 84:5389-5393.
36. Slamon, D. J., M. F. Press, L. M. Souza, D. C. Murdock, M. J. Cline, D. W. Golde, J. C. Gasson, and I. S. Y. Chen. 1985. Studies of theputative transformingprotein of the type I human T-cell leukemia virus. Science 228:1427-1430.
37. Sodroski, J., C. Rosen, W. C.Goh, and W. A. Haseltine. 1985. A transcriptional activator protein encoded by the x-lor region of thehuman T-cell leukemia virus. Science 228:1430-1434. 38. Southern, E. M. 1975. Detection ofspecific sequences among
DNAfragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517.
39. Thompson, T., and H. Fan. 1985. Mappingof DNase I-hyper-sensitive sitesin the 5' and 3'longterminal repeats ofintegrated Moloneymurine leukemia virusproviralDNA. Mol. Cell. Biol. 5:601-609.
39a.Thompson, T., and H. Fan. 1988. Chromatin structure of hor-mone-responsive Moloney murine leukemia virus proviruses that contain sequences from mouse mammary tumor virus. VirusGenes 2:83-98.
40. Todaro, G. J., and H. Green. 1963. Quantitativestudies of the growth ofmouseembryo cellsin culture and theirdevelopment intoestablishedcell lines.J. Cell. Biol. 7:299-313.
41. Weintraub, H., andM.Groudine. 1976.Chromosomal subunits in active genes have an altered conformation. Science 193: 848-856.
42. Wu, C. 1984. Twoproteinbinding sitesinchromatinimplicated in the activation of heat-shock genes. Nature (London) 309: 229-234.
43. Yamamoto, N., M. Okada, Y. Koyanagi, M. Kannagi, and Y. Hinuma. 1982.Transformation ofhuman leukocytes by cocul-tivation withanadult T cell leukemia virus producercell line. Science 217:737-739.
44. Zaret, K. S., and K. R. Yamamoto. 1984. Reversible and persistent changes in chromatinstructureaccompanyactivation ofaglucocorticoid-dependentenhancerelement.Cell 38:29-38.