JOURNAL OF VIROLOGY, Jan. 1990, p. 445-449 0022-538X/90/010445-05$02.00/0
Copyright © 1990, American Society forMicrobiology
Expression
of the
Herpes
Simplex
Virus
Type
1Glycoprotein
C
Gene
Requires Sequences in the
5'
Noncoding Region of the
Gene
JERRY P. WEIR'* ANDP. R. NARAYANAN2
Department ofMicrobiology, Universityof Tennessee, Knoxville, Tennessee37996-0845,1 andDepartment of Immunology, Tuberculosis Research Center, Madras 600 031, India2
Received 5 June1989/Accepted 20 September 1989
The role of the 5'noncodingregionof theherpessimplexvirustype1glycoproteinC (gC)genein viralgene
expressionwasinvestigated with recombinant herpesviruses that contained the bacterial 13-galactosidasegene
underthecontrol of the gC promoter-regulatory region. Each of these viruses had thesameDNAsequences
from the start of gC transcription upstream to -114 but had variable segments of the downstream 140-base-pairsequencethat is between thestartofgC transcription and translation. Analysis of 13-galactosidase expression and mRNA synthesis from these viruses demonstrated the importance of DNAsequencesfrom the
startof gCtranscriptiondownstreamto +38 for optimal expression from the gCpromoter.
Todefine the DNA sequences that are involvedin
regu-lation and expression of viral late genes, we have
con-structed recombinantherpesviruses thatexpress the bacte-rialenzyme 1-galactosidase (13-Gal) fromthepromoterofthe glycoprotein C (gC)gene, a well-characterized herpesvirus
late gene. Recombinant viruses are advantageous in the
study of latepromoter-regulatory regions because herpesvi-rus late genes that are removed from the viral genome no longerrequire viral DNA replication for their expression (3, 9, 11, 12). In previousstudies using recombinant viruses,we demonstratedthat sequencesbetween -1350and +30
rela-tivetothestartof gCtranscriptionregulate 13-Galasaviral
lategene and thatupstream sequences canbe deleted toat least -109 without affecting expression (14). Other studies (5, 6)haveshown thatupstreamgCsequences canbedeleted to -34 without affecting expression and thata 15-base-pair (bp) TATA boxpromoterelement isabsolutely required for gC expression.
Although gC sequences between -109 and +30 regulate 13-Galexpressionasalate viralgeneinrecombinantviruses, itisnotclear whethergC sequencesdownstream from +30 have a role in gC expression. Homaet al. (5) noticed that deletionsof DNAsegmentscontaining thestartof transcrip-tion and downstream sequences lower the relative level of gC mRNA, and they suggested that this was due to a shortening in the length of the nontranslated leaderthat is
non-sequence specific. In this report, wedescribe the
con-structionofa setof recombinantherpesviruses designed to determine whether sequences involved inthe regulation of herpes simplex virus type 1 (HSV-1) gC expression are located in the 5' untranslated region ofthe gene. Each of these recombinant viruses had the p-Gal gene under the control of thepromoterfor the late viralgCgene. EachgC promotercontainedsequencesfromthestartoftranscription
upstream to -114butdifferentsegmentsof the downstream
140-bp nontranslated sequence between the start of tran-scriptionand translation.
The DNA sequences ofthe gC gene containing the 5'
nontranslated leader of thegene wereisolatedbyinsertion of aunique SaIrestrictionenzymesite in theclonedgCgeneat position +141 relative to the start of transcription. This changedthe ATGGCC thatbeginsthe gC codingsequence
*Correspondingauthor.
(4)toGTCGAC but left the 140 bp between the startof gC transcription and translation unaltered. Promotersequences of thegCgenefrom -114to +140wereinsertedupstreamof the ,B-Gal gene in the HSV-1 insertion vector pGal8 (J. P. Weir, K. R. Steffy, and M. Sethna, submitted for publica-tion)to generate the plasmid pgCL1. In additiontothe gC
sequences upstream of the 1-Gal gene, gCL1 containedan
additional 35 bp from the Sall siteto the ATG thatbegins 1-Galtranslation(Fig. 1). The construction gCL3contained
the same gC promotersequences, -114to +140, upstream
of the ,B-Gal gene as gCL1, but the 35 bp between the gC promoter and the ATG that begins 1-Gal translation was eliminated. Thus, the coding sequences for ,B-Gal began at
+141, in thesameposition in which the codingsequencesfor gCarenormally locatedintheauthenticgCgene.Sequential 3'-to-5' deletionsweremade in thegC leader region thatleft
sequences to +71 (gCL5), +38 (gCL6), +16 (gCL6.1), and
-3(gCL7).Twootherconstructions with deletedsequences between +28 and +99 (gCL3.1) and +2 and +39 (gCL8) were made. Recombinant viruses were made from each plasmid constructionasdescribedpreviously (14).
When Verocellswere infectedwitheachofthe recombi-nant virusesin thepresence orabsenceofphosphonoacetic acid(PAA), aninhibitorof viral DNAreplication, all ofthe virusesthatexpressed ,B-Gal didsointhe mannerofalate viralgene.Expressionwasfirstdetectableatapproximately 6hafterinfection andcontinuedtoriseforatleast 24 h(data notshown). Figure1 showsthe expressionof 1-Galat24 h postinfectioninthepresenceand absence of PAA. Elimina-tion ofthe 35 bp between the gCleader sequences and the ATG that begins the 1-Gal open reading frame (Fig. 1) resultedinahigher level of,B-Gal expressioninvgCL3-than invgCL1-infectedcells.Since thelocation of the 13-Galopen
reading frame has perfectly replaced the gC open reading frameinvgCL3, the p-Galactivity ofvgCL3-infected cells
wasset at 100%.Althoughthe lower level of,B-Gal
expres-sioninvgCL1-infected cells could be duetothe additionof non-gCsequences,wethink thatamorelikely explanationis that the sequence around the ATG in vgCL1 contains a pyrimidine at -3 relative to the A of the ATG. Such a
sequenceisnotpredictedtobeasfavorablefor translationas
thesequencethat exists ifapurineispresentat thisposition
(7). In each of our other constructions, the sequences
445
Vol. 64, No. 1
on November 10, 2019 by guest
http://jvi.asm.org/
446 NOTES
mRNA
-114 +1 +140
mRNA
_ _ 4AT |
-114 +1 +140
mRNA
.114 +1 +71
mRNA
1. {ge
-114 +1 .36
mRNA
.114 +1 +16
mRNA
EG
-114 .3
mRNA &
-114 +1 +26 +99 +140
mRNA t
I..14*
...9 ..14.114 +2 +3 140
.&GALACTIVITY
noPA EM
gCLI
gCL3
gCL5
gCLS
gCLA.1
gCL7
gCL3.1
gCLS
69 1.5
100 2.9
77 1.2
66 1.1
26 2.1
2.2 0.5
25 0.6
31 0.4
FIG. 1. Constructionof HSV-1 recombinant viruses and expressionofp-Gal. A2,000-bp SaiI-EcoRI fragmentof HSV-1(KOS)DNA containingthefirst500bp of thecoding sequence forgC andapproximately 1,500bpupstreamfrom the startofgCtranslationwascloned into thephagemid vector BluescribeM13+ (Stratagene). ASalIrestriction sitewasgeneratedbyoligonucleotidemutagenesis (8)at +141 relativetothestartoftranscription.AHindIlI sitewasinsertedbymutagenesisat-118(leaving gCpromotersequencesunchangedto-114), and thegC sequences from-114to +141 werecloned into theHSV-1 insertionvectorpGal8.This insertionvectorhas thecodingsequences ofthe Escherichia colip-Galgeneflankedby sequences from the HSV-1thymidine kinase genetodirecthomologous recombination intothe thymidine kinase gene of the viral genome. Recombinant viruseswereisolatedasdescribedpreviously (14). The promoter sequences from gCL1 were recloned into the phage vector M13mpl8, and the indicated deletions were made by oligonucleotide mutagenesis with oligonucleotidesthatspannedthedesireddeletion. Aftermutagenesis, the promoter sequenceswererecloned intopGal8forinsertion into the viral genome. The additional 35-bp segment ofDNApresentingCL1 between thegC leader andthep-Galcoding sequence
(U)
isfrom the original fragment ofDNAthat contained theP-Galgene usedtoconstructpGal8. Verocellswereinfectedat amultiplicityof infection of 10 in the presence or absence of300 ,ug of PAA per ml with each of the recombinant viruses. The p-Gal activity was measured at24 h postinfectionasdescribedpreviously (14) andexpressedrelative to that ofvgCL3.brought adjacenttothe ATG are predicted to be favorable fortranslation.
Progressive deletions from the startof translation toward the start of transcription resulted in decreasing levels of p-Gal expression (Fig. 1). Deletion of sequences to +38 (vgCL6) reducedp-Gal expression to70% of that exhibited
by
vgCL3. Deletion of another 22 bp to +16 (vgCL6.1) resultedin amoredramatic reduction of expression to 26% of maximalexpression, andelimination oftheentire 140-bp leader and mRNA start site (vgCL7) abolished detectablep-Gal
expression. None ofthese viruses expresseddetect-able
p-Gal
when viral DNA replication was inhibited withPAA,indicatingthatelimination ofgCleader sequences did notaffecttemporal regulation.
A progressive decrease in observed
p-Gal
expression could be due to the elimination of the specific DNA se-quencesthatareimportant forexpressionor to aprogressive reduction in thelengthof thegCleader that is non-sequence specific.Twodeletions, gCL3.1andgCL8,wereconstructed toaddressthisquestion. InvgCL3.1,70bpof thegCleader sequence between +28 and +99 was eliminated, with a resulting reduction ofp-Gal expression to25%. Elimination of36 bp of the gC leader sequence in vgCL8 (sequences between +2and +39) reducedp-Gal
expression in infectedJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.157.469.81.481.2]NOTES 447
A
wJ
a%
-iG
AGT 4
i
Z
- _A C G T PE
B
C
.w
GGG a
A -M
-ft4
.9
0-f
A C G T PE
w...-5..6
GG
A,....,
.4
AC
G TiP
D
'I!
A:
A, a,
A C G T PE PE 12
FIG. 2. Analysis of the 5' ends ofp-GalmRNAs. Verocells were infected with recombinant viruses at amultiplicity of infection of 10,
andtotalRNA wasisolated24 h later. Afterhybridization with a 5'-end-labeled oligonucleotide complementary to the 5' end of the p-Gal
gene, primer-RNA hybridswere extended with reverse transcriptase (14). The extended products (lanes PE) were separated on a 6%
sequencing gel alongsideasequence ladder prepared with the same primer and thecorrespondingplasmid DNA. (A) Primer extension analysis ofRNAfrom vgCL3-infectedcells; (B) RNA fromvgCL6.1-infectedcells;(C) RNA fromvgCL8-infected cells; (D) RNA from cells infected with vgCL7in the presence (lane PE2) or absence (lanePE1)ofPAA. The short sequence shown is that of the coding strand around the start of transcription.
cells to 31% of maximal expression. The remaining leader sequence invgCL3.1 and vgCL8 is substantially longer than thatremaininginvgCL5 and vgCL6, yet the levels of
p-Gal
expressionaredramatically lower. The results obtained with vgCL3.1 and vgCL8 indicate that the elimination of gC leader sequences upstream of +39 had a much more dra-matic effect on expressionthan the actual length of the gC leader sequencedid.
Inordertodetermine whether deletionsin the 5' nontrans-lated region of gC that resulted in lower levels of
P-Gal
A
:.
B..'''.'=k
:.
21Wg
i
!S:
... ,fg
.:
_. tF
t ...
t
*' ';11s:' ...
_
.>,.._ ;j. ... i,...
...^.... ^..
b...
*..:
expression altered thenormal start of mRNAtranscription, the 5' ends ofRNA from infected cells were analyzed by primer extension. The major extension product observed when RNA from vgCL3-infected Vero cells was used mi-grated alongsideaG residue(coding strand) in the sequence ladder (Fig. 2A) that was previously designated +1 in an analysis ofthe 5' end ofthe gC mRNA(14). As previously observed, minor extension products migrated
alongside
res-idues -2, -3, and +2 relative to the major extension product. Extension products obtained when we used RNA from cells infected with each of the other recombinant viruses that expressedp-Gal
were observed at the samepositions
asthose fromvgCL3. Primer extension analysesinwhich RNA from vgCL6.1- and vgCL8-infected cells was usedare showninFig. 2B and C,respectively. Thus, each
9.4
-6.6 -.
4.4
-2.3
-2.0
M
3
8
ACGT83
FIG. 3. Accumulation of p-Gal mRNA in cells infected with vgCL3 (lanes 3) andvgCL8 (lanes 8). (A)RNAs(40 ,ug) from vgCL3-andvgCL8-infected cellswereanalyzed forthe presence ofp-Gal mRNA byNorthern blotting. Nitrocellulose membranes were hy-bridizedto a 32P-labeled p-Gal-specific DNA probe, washed, and subjectedtoautoradiography.Therelativeamountsofp-GalmRNA weredetermined by densitometry of theautoradiographs. Labeled
DNAmarkers(lane M)weredenatured in thesamewayasthe RNA
andwere runonthegelforcomparison.Sizes shownareinkilobase pairs.Therelativep-GalmRNAratio(vgCL8/vgCL3)was0.30.(B) Primer extension analysis of p-Gal mRNAs from vgCL3- and vgCL8-infectedcellswasas described in thelegendto Fig.2. The
sequence ladderwasgeneratedfrom the p-Gal primerandpgCL8
DNA. The relative amounts of p-Gal primer extension products
were determined by densitometry of the autoradiograph. The
amountofgCmRNAwasquantitatedineachsampleto serve asan internal control for the amountofRNA used in each assay. The relativep-GalmRNAratio(vgCL8/vgCL3)was0.38.
VOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.136.488.79.306.2] [image:3.612.80.289.514.730.2]448 NOTES
A
...w...*
. *
*_.
...
..::. .:
3
6 12
100
c 75
._
4
50-z
E
25
100
'5
IN.3
P..,
_ 75
a:
tS 50
M
.- . 8,
G 4 6 12
B
.6
3
6
12
I1
4 8 12
Time AfterActinomycinD Time AfterActinomycin D
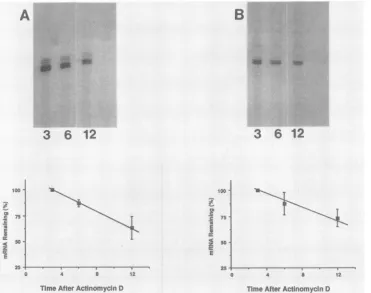
FIG. 4. Stability ofP-GalmRNAin cells infectedwithvgCL3 (A)andvgCL8 (B).Verocells(1 x 10')wereinfected withrecombinant virusat amultiplicityof infection of10. At12hpostinfection,dactinomycin(actinomycinD)wasaddedto afinal concentrationof 10
jig/ml.
RNA wasisolated 3, 6, and12h laterand used forprimer extensionanalysis (lanes3, 6, and12,respectively).The relativeamountsofp-Gal
primerextension productswere determinedbydensitometry ofautoradiographs. recombinant virus that expressed p-Galretained theoriginal
mRNAstartsite, regardless of the level ofp-Galexpression.
Because the start of p-Gal translation in vgCL7 was positioned upstream from the normalstart ofgC transcrip-tion, the lack ofp-Galexpression didnotnecessarily reflect the lack of mRNA expression from the -114 to -3 gC
promotersequences. The lack of
p-Gal
expression might bedue to the inability ofan expressed mRNA to translate a functional p-Gal protein. To address this possibility, RNA wasalso isolated from vgCL7-infected cells in thepresence orabsence of PAA and was analyzed by primer extension (Fig. 2D). A faint primer extension productwasobservedto migrate alongside the G residue (coding strand)at -3 (lane PE1).However,neither the major extension productnorthe other minor extension products were observed.
Further-more,in thepresenceof PAA,noprimer extension products
were seen (lane PE2). Therefore, deletionof the entire gC leaderregion and normal mRNAstartsite eliminated mRNA synthesis from the major gC startsite.
Thereducedp-Galexpression that resulted from deletions inthegC noncoding leader could be the result ofadecrease in thelevel ofp-Gal mRNA or adecrease in translation of
p-GalmRNA. Toevaluate thesetwopossibilities,RNAwas isolatedfrom cells infected with either vgCL3orvgCL8 and analyzed by Northern (RNA) blot hybridization. RNA from infected cells was probed initially for gC mRNA with a
32P-labeled DNAfragment specific for gC. The size of the major RNA species detected by the gC probewas approxi-mately 2.5 kilobases (datanotshown), which corresponded to the size previously reported for the gC mRNA (4). The amountofgC mRNAwasquantitated by densitometry of the
autoradiographs to serve as an internal control for the amount of RNA loaded onto each well of the gel. The gC probe was then removed, and the membrane was probed with a second 32P-labeled fragment that was specific for
P-Gal.
The size ofthe majorRNA species detected by thep-Gal
probe was approximately 4.3 kilobases (Fig. 3A),which corresponded to the size expected for the
p-Gal
transcript. The relative amounts of
p-Gal
mRNA in vgCL8-andvgCL3-infected cells werequantitated
by densitometry oftheautoradiographs.Theaccumulation of
p-Gal
mRNAinvgCL8-and vgCL3-infectedcellswasalsodeterminedby primer extension (Fig. 3B). After autoradiography of the sequencing gel with thep-Gal
primer extension products of vgCL8andvgCL3 RNA,therelativeamountsof
p-Gal
mRNA wereagain quantitated by densitometry. The relative steady-state levels ofp-Gal
mRNA produced in vgCL3- and vgCL8-infected cells re-flected the relative
p-Gal
activity observed(Fig. 1 and3).The decreasein
p-Gal
mRNAaccumulation thatresulted from the deletion of sequences between +2 and +39 could be due to either a decrease in the rate of mRNAsynthesisor a decrease in mRNA stability. To determine the relative stability ofthep-Gal
mRNAinvgCL3-andvgCL8-infected cells, RNA wasisolated 3,6, and 12 h aftertheaddition of dactinomycinandquantitated by primer extension.Thelevelof
p-Gal
mRNAinvgCL3-and vgCL8-infectedcellsgradu-ally diminishedin the 12 h afterdactinomycin addition,with anestimated half-lifeof
p-Gal
mRNAof >10 h(Fig. 4).For comparison, the relative stability of gC mRNA was deter-minedby primer extensionanalysiswith the same RNAs. In both vgCL3-andvgCL8-infected cells, gC mRNAdecayedJ. VIROL.
1-1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.120.491.86.379.2]NOTES 449
atsimilar rates with anestimated half-life similar to that of
p-Gal
mRNA (data not shown). Thus, the decrease inrelative ,-Gal mRNA levels in vgCL8- and vgCL3-infected cells cannot be accounted for by a decrease in mRNA stability. Although these results suggest that sequences between +2 and +39 are important for mRNA synthesis, this has not been shown directly. Unfortunately, results from direct measurement of HSV-1 initiation at late times during infection are difficult to interpret (13).
Althoughthe resultsfrom recombinant virus construction show the importance of gC leader sequences upstream from +39, the actual role ofthese sequences ingeneexpression remains to be resolved. A recent report showed that the immediate-early 175-kilodalton protein (a4) of HSV-1 binds to DNA fragments derived from the 5' noncoding leader region of two viral late genes (10). It remains to be deter-minedwhether thegCleader sequences determined here to beimportantfor gC expression will also bind a4. Inspection of theleadersequencesof the three genes did not reveal any obvious similarities.
Other reports have indicated a role for the 5' leader regions in the expression of HSV-1 genes other than gC. Using transientassays,Blair etal.(1) showed that sequences from -3 to +77 in the leader region of the viral gene VP16 arenecessaryforvirus-induced transcription. Coen et al. (2) observed that a mutation in the +5-to-+15 region of the thymidine kinase gene, when introduced into the viral ge-nome, significantlylowered thymidine kinase mRNA accu-mulation and transcription rate. Further studies, including mutational analyses, of the 5' nontranslatedregionof imme-diate-early, early, and late HSV-1 genes should lead to a better understanding ofthe role of this partofthe gene in viral gene expression and regulation.
This workwas supported in partby PublicHealthServicegrant A124471 from the National Institutes of Health, aJunior Faculty Research Awardfrom the American CancerSociety (J.P.W.), anda fellowship from the National Biotechnology Board,Governmentof India(P.R.N.).
We thankMarkChallberg forhelpful discussions. LITERATURECITED
1. Blair, E. D., C. C. Blair, and E. K. Wagner. 1987. Herpes simplex virus virionstimulatory proteinmRNAleadercontains sequence elements which increase bothvirus-induced
transcrip-tion and mRNAstability. J. Virol. 61:2499-2508.
2. Coen, D. M., S. P.Weinheimer, and S. K. McKnight. 1986. A genetic approach to promoter recognition during trans induction ofviralgene expression. Science 234:53-59.
3. Dennis, D., and J. R. Smiley. 1984. Transactivation of a late herpessimplex virus promoter. Mol. Cell. Biol. 4:544-551. 4. Frink,R.J.,R.Eisenberg,G. Cohen,and E. K.Wagner. 1983.
Detailedanalysis of the portion of the herpes simplex virus type 1genomeencoding glycoproteinC. J. Virol.45:634-647. 5. Homa, F. L., J. C. Glorioso, and M. Levine. 1988. Aspecific
15-bpTATAbox promoter element is required forexpression of aherpes simplex virus type1late gene. GenesDev. 2:40-53. 6. Homa,F. L.,T. M.Otal, J.C.Glorioso,and M. Levine. 1986.
Transcriptional controlsignals ofaherpessimplex virus type 1 late(Y2) gene lie within bases -34 to +124relative to the 5' terminus of the mRNA. Mol. Cell. Biol. 6:3652-3666. 7. Kozak, M. 1986. Point mutations define a sequenceflanking the
AUG initiator codon thatmodulates translation byeukaryotic ribosomes. Cell 44:283-292.
8. Kunkel, T. A.,J. D. Roberts, and R. A. Zakour.1987.Rapidand
efficientsite-specific mutagenesis without phenotypicselection. MethodsEnzymol. 154:367-382.
9. Mavromara-Nazos, P., S. Silver, J. Hubenthal-Voss, J. L. C.
McKnight, and B. Roizman. 1966.Regulation of herpes simplex
virus1genes: gene sequencerequirements for transient induc-tion of indicatorgenes regulatedby
P
or late(y2) promoters. Virology149:152-164.10. Michael, N., D. Spector, P. Mavromara-Nazos, T. M. Kristie,
and B.Roizman.1988. TheDNA-binding properties of the major
regulatory protein ca4 of herpes simplex virus. Science 239: 1531-1534.
11. Shapira,M.,F. L. Homa, J. C. Glorioso, and M. Levine. 1987.
Regulation of the herpes simplex virustype 1late(y2)
glycopro-teinCgene:sequences betweenbasepairs -34and +29 control
transientexpression andresponsiveness to transactivation by theproducts of the immediate early (a) 4 and0 genes. Nucleic AcidsRes. 15:3097-3111.
12. Silver, S.,and B. Roizman. 1985.-y2-Thymidine kinase chimeras areidentically transcribed but regulated as Y2genes inherpes simplex virus genomes andas
P
genesin cell genomes. Mol. Cell. Biol. 5:518-528.13. Weinheimer, S. P., andS. L. McKnight. 1987. Transcriptional
andposttranscriptional controls establish the cascade of herpes simplex virusproteinsynthesis. J. Mol. Biol. 195:819-833. 14. Weir, J. P.,and P. R.Narayanan. 1988. Theuseof
P-galactosi-daseas amarkergene todefine theregulatorysequencesof the herpes simplex virustype 1glycoprotein Cgenein recombinant herpesviruses. Nucleic AcidsRes. 16:10267-10282.
VOL.64, 1990