Viral Replicative Capacity, Antigen Availability via
Hematogenous Spread, and High T
FH
:T
FR
Ratios Drive
Induction of Potent Neutralizing Antibody Responses
Preethi Eldi,a*Geeta Chaudhri,a*Stephen L. Nutt,b,cTimothy P. Newsome,d Gunasegaran Karupiaha,e
aJohn Curtin School of Medical Research, The Australian National University, Canberra, Australian Capital Territory, Australia
bThe Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia
cDepartment of Medical Biology, University of Melbourne, Parkville, Victoria, Australia
dSchool of Life and Environmental Sciences, The University of Sydney, Sydney, New South Wales, Australia
eSchool of Medicine, College of Health and Medicine, University of Tasmania, Hobart, Tasmania, Australia
ABSTRACT Live viral vaccines elicit protective, long-lived humoral immunity, but the underlying mechanisms through which this occurs are not fully elucidated. Gen-eration of affinity matured, long-lived protective antibody responses involve close in-teractions between T follicular helper (TFH) cells, germinal center (GC) B cells, and T
follicular regulatory (TFR) cells. We postulated that escalating concentrations of
anti-gens from replicating viruses or live vaccines, spread through the hematogenous route, are essential for the induction and maintenance of long-lived protective anti-body responses. Using replicating and poorly replicating or nonreplicating orthopox and influenza A viruses, we show that the magnitude of TFH cell, GC B cell, and neutralizing antibody responses is directly related to virus replicative capacity. Fur-ther, we have identified that both lymphoid and circulating TFH:TFRcell ratios during
the peak GC response can be used as an early predictor of protective, long-lived an-tibody response induction. Finally, administration of poorly or nonreplicating viruses to allow hematogenous spread generates significantly stronger TFH:TFRratios and
ro-bust TFH, GC B cell and neutralizing antibody responses.
IMPORTANCE Neutralizing antibody response is the best-known correlate of long-term protective immunity for most of the currently licensed clinically effective viral vaccines. However, the host immune and viral factors that are critical for the induc-tion of robust and durable antiviral humoral immune responses are not well under-stood. Our study provides insight into the dynamics of key cellular mediators of ger-minal center reaction during live virus infections and the influence of viral replicative capacity on the magnitude of antiviral antibody response and effector function. The significance of our study lies in two key findings. First, the systemic spread of even poorly replicating or nonreplicating viruses to mimic the spread of antigens from replicating viruses due to escalating antigen concentration is fundamental to the in-duction of durable antibody responses. Second, the TFH:TFRratio may be used as an
early predictor of protective antiviral humoral immune responses long before mem-ory responses are generated.
KEYWORDS germinal center B cells, inactivated vaccines, influenza A viruses, live vaccines, long-lived antibody response, neutralizing antibodies, orthopoxviruses, T follicular helper cells, T follicular regulatory cells, hematogenous viral spread
S
mallpox eradication through the use of a live-virus vaccine is one of the most successful public health endeavors of modern medicine. Humoral immunity against smallpox in vaccinated individuals, characterized by neutralizing antibody, is stable,CitationEldi P, Chaudhri G, Nutt SL, Newsome TP, Karupiah G. 2019. Viral replicative capacity, antigen availability via hematogenous spread, and high TFH:TFRratios drive induction of
potent neutralizing antibody responses. J Virol 93:e01795-18.https://doi.org/10.1128/JVI .01795-18.
EditorJoanna L. Shisler, University of Illinois at Urbana Champaign
Copyright© 2019 American Society for Microbiology.All Rights Reserved. Address correspondence to Gunasegaran Karupiah, [email protected].
*Present address: Preethi Eldi, University of South Australia, Adelaide, South Australia, Australia; Geeta Chaudhri, Research School of Population Health, The Australian National University, Canberra, Australian Capital Territory, Australia.
Received9 October 2018
Accepted19 December 2018
Accepted manuscript posted online9 January 2019
Published
crossm
5 March 2019
on November 6, 2019 by guest
http://jvi.asm.org/
lasts for decades (1, 2), and is considered a valuable benchmark of the functional attributes of a good vaccine. Indeed, neutralizing antibody is the best correlate of long-term protective immunity for all the currently licensed clinically effective viral vaccines (3). Despite the success of attenuated live vaccines in preventing disease, very little is known about viral and host factors that drive induction of protective antibody responses that are long-lived. A potential clue resides in findings that humoral immu-nity following natural infection with variola (4), measles (5), polio (6), or yellow fever (7) viruses persist for decades even in the absence of reexposure to virus. These viruses cause acute generalized infections, with hematogenous spread of the virus and viral antigens to numerous secondary lymphoid organs, including the spleen.
Central to antiviral antibody and immune memory generation is the germinal center (GC) response in secondary lymphoid organs. Here, specialized CD4 T cell subsets, T follicular helper (TFH), and T follicular regulatory (TFR) cells provide survival, proliferative,
and differentiation cues to B cells, culminating in the production of somatically mu-tated, high-affinity antigen-specific neutralizing antibody (8, 9). Dysregulation of TFH
cells (10–13), TFH-B cell interaction (14, 15), or TFRcells (16–18) has detrimental effects
on the GC and subsequent antibody response. The cross talk between the TFH, TFR, and
B cells dictates the outcome of the GC reaction.
Much of our current understanding of high-affinity neutralizing antibody production is largely based on studies using nonreplicating model antigens (19, 20), inactivated viral vaccines that do not replicate in mice (21), persistent viral infection models (22), and models that do not have a natural host-pathogen relationship (23). For these reasons, we have used the mouse pathogen ectromelia virus (ECTV), which causes a smallpox-like disease termed mousepox, an excellent surrogate for smallpox, and induces long-lived neutralizing antibody responses. The importance of antibodies as a primary correlate of protection in ECTV infection has been established by studies using mice lacking B cells, CD40, or major histocompatibility complex class II (24–27). How-ever, very little is known about the induction or dynamics of the TFH, TFR, and GC B cell
responses associated with protective neutralizing antiviral antibody generation. We hypothesized that the escalating concentration of antigens during virus repli-cation or live viral vaccine immunization must reach sufficient concentrations for hematogenous spread to drive TFH differentiation in secondary lymphoid organs,
including the spleen, for induction of potent and durable humoral immunity. Using replication-efficient and -inefficient versions of ECTV and the closely related vaccinia viruses, Western Reserve strain (VACV-WR) and Chorioallantois vaccinia Ankara (CVA), we report here that virus replicative capacity has a significant impact on the magnitude of the TFH, GC B cell, and protective neutralizing antibody responses. Similar findings
were made with live and inactivated influenza A virus (IAV) strains. Increasing the dose of replication-inefficient or replication-deficient poxviruses or IAV strains significantly augmented TFH, GC, and protective neutralizing antibody titers only when combined
with an intravenous (i.v.) route of immunization. Overcoming the dissemination barrier to allow hematogenous spread and optimal antigen availability in secondary lymphoid organs may thus be a key to improving the immunogenicity and protective humoral immunity induced by replication-deficient virus vaccines. Our data also show that TFH:TFRratios at the peak of the GC response in both the spleen and blood may be used
as universal biomarkers that are predictive of protective long-lived antibody genera-tion. Manipulation of TFH:TFRratios may thus be a vital step in optimizing the efficiency
of viral vaccines and vaccination regimes in generating a potent antiviral antibody response.
RESULTS
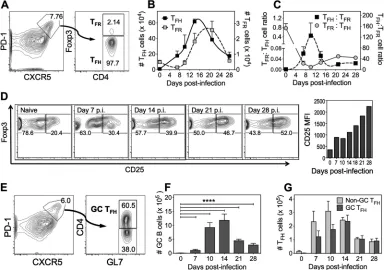
A strong TFH response drives robust GC B cell and neutralizing antibody re-sponses.To understand the dynamics of the key cellular players of antibody response generation against ECTV infection, we measured total TFH(B220–CD4⫹CD44hiCXCR5hi
PD-1hiFoxp3–), T
FR(B220–CD4⫹CD44hiCXCR5hiPD-1hiFoxp3⫹), and GC B (B220⫹Fas⫹
GL7⫹) cell responses over 4 weeks following subcutaneous (s.c.) infection of wild-type
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 2
on November 6, 2019 by guest
http://jvi.asm.org/
C57BL/6 mice. TFHcells (Fig. 1A) expanded rapidly as evident on day 7 postinfection
(p.i.), peaked in numbers between days 14 and 16 p.i. (Fig. 1B, leftyaxis) and accounted for about 4 to 6% of all splenic CD4⫹T cells. Although numbers contracted after this
period, the response was still ongoing at day 28 p.i. In contrast to TFHcells, there was
an initial significant 3-fold drop in total numbers (Fig. 1B, rightyaxis) of TFRcells at day
7 p.i. This was followed by a 12- to 16-fold increase in TFRcell numbers, coincident with
the TFHcontraction phase between days 14 and 21 p.i. These changes in numbers of
cells, also depicted by TFH:TFR and TFR:TFH cell ratios (Fig. 1C), revealed an inverse
relationship between the two cell subsets from about days 7 to 10 p.i. The TFH:TFRratio
was about 1:1 in naive animals but increased to 120:1 at the peak of the TFHresponse.
The proportion of TFR cells that expressed CD25, the IL-2 receptor␣ (IL-2R␣) chain,
progressively increased during the course of infection, suggesting a possible IL-2-IL-2-R␣-mediated layer of regulation on TFHand/or GC B cells (Fig. 1D). GL7⫹GC TFHcells
(B220–CD4⫹CD44hiCXCR5hiPD-1hiGL7⫹; Fig. 1E), reported to have enhanced B-cell
help capabilities (28), followed similar kinetics of expansion and contraction as the total TFHcell response (Fig. 1F), accounting for 50% of all TFHcells at the peak of the response
at day 14 p.i. and beyond (Fig. 1G).
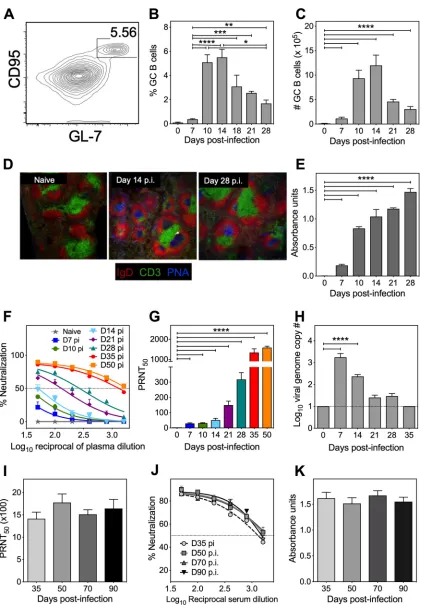
The GC B cell response (Fig. 2A) was also similar in kinetics to that of TFHcells, with
a peak proliferative response observed at day 14 p.i. (Fig. 2B and C). Histological analysis revealed larger and more GC per spleen section at day 14 p.i. and that GC persisted even at day 28 p.i. (Fig. 2D). Anti-ECTV IgG antibodies were detectable as early as day 7 p.i., with IgG absorbance units increasing progressively over time (Fig. 2E), contem-poraneous with increases in the virus-neutralizing activity (Fig. 2F) and the 50% plaque reduction neutralization test (PRNT50) titers (Fig. 2G). The ongoing TFHand GC B cell
FIG 1Kinetics of TFHand TFRcells during ECTV-WT infection. C57BL/6 mice (n⫽3 to 7 mice per group) infected s.c. with
103PFU of ECTV-WT were sacrificed at the time points indicated. (A) Flow cytometry contour plots of splenic T FH(CD4⫹
CD44hiCXCR5hiPD-1hiFoxp3–) and T
FR(CD4⫹CD44hiCXCR5hiPD-1hiFoxp3⫹) cells. (B) Total numbers of TFH(leftyaxis) and
TFR(rightyaxis) cells per spleen. (C) Splenic TFH:TFRratio during the course of infection. The data represent means⫾the
standard errors of the mean (SEM). (D) Concatenated flow cytometric contour plots of CD25-expressing TFRcells during the
course of infection with a graphical representation of CD25 median fluorescent intensity at the indicated time points. (E) Flow cytometry contour plot of GL7-expressing GC TFH(CD4⫹CD44hiCXCR5hiPD-1hi) cells. (F) Total GC TFHcell numbers
per spleen. (G) Comparative analysis of GL7⫹and GL7–CXCR5hiPD-1hiT
FHcells. The data represent means⫾the SEM; data
were log transformed, and the statistical significance was determined by one-way ANOVA (****,P⬍0.0001).
on November 6, 2019 by guest
http://jvi.asm.org/
[image:3.585.42.427.73.343.2]FIG 2Kinetics of GC B cell and antibody responses during ECTV-WT infection. C57BL/6 mice (n⫽3 to 7 mice per group) were infected as described in Fig. 1 and sacrificed at the time points indicated. (A) Flow cytometry contour plots of GC B cells (B220⫹GL7⫹CD95⫹)
cells at day 14 p.i. (B) Percentage of GC B cells. (C) GC B cell numbers per spleen. (D) Immunofluorescent staining (IgD, red; CD3, green; peanut agglutinin [PNA], blue) of 10-m frozen spleen sections from uninfected (naive) and ECTV-WT-infected (days 14 and 28 p.i.) mice (magnification, ⫻10). (E) ECTV-specific IgG absorbance units determined by ELISA (1:200 serum dilution). (F) Kinetics of virus-neutralizing antibody response, curve fitted using four-parameter, nonlinear regression analysis. The dotted horizontal line
(Continued on next page)
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 4
on November 6, 2019 by guest
http://jvi.asm.org/
[image:4.585.42.464.66.676.2]responses detected at day 28 p.i. were likely responsible for the continued increase in neutralizing antibodies at days 35 and 50 p.i., which inversely correlated with the viral load in blood (Fig. 2H). Virus-specific antibody levels, neutralization, and PRNT50titers
stabilized between 5 and 6 weeks p.i. and were maintained at 3 months p.i. (Fig, 2I, J, and K, respectively).
Viral replicative capacity influences TFH, TFR, GC B cell, and long-lived protec-tive antibody responses. We posited that viral replicative capacity was key to the induction of long-lived humoral immunity and that the escalating concentration of replicating viral antigen provides essential cues for priming TFHcell differentiation that
drives robust GC and long-lived neutralizing antibody response generation. We tested our hypothesis by comparing TFH, TFR, GC B cell, and antibody responses in mice
infected with ECTV-WT or ECTV-TKΔ. The latter is a highly attenuated,
replication-inefficient virus due to deletion of the thymidine kinase (TK) gene in the parent ECTV-WT (29) and its replication is restricted to the site of inoculation (30). Even when very high doses are used to establish systemic infection, ECTV-TKΔdoes not cause any
morbidity in gene knockout mice deficient in type I or type II interferon (IFN), IFN regulatory factor 1, perforin, or granzymes A and B (31, 32). Most of these strains are highly susceptible to ECTV-WT and die from infection with just 1 PFU of virus. ECTV-TKΔ
is attenuated to such an extent that it does not persist in the bone marrow of the ECTV-susceptible BALB/c mice, whereas the other attenuated deletion mutant viruses lacking virus-encoded IFN-␥-binding or IFN-␣/-binding proteins do (33).
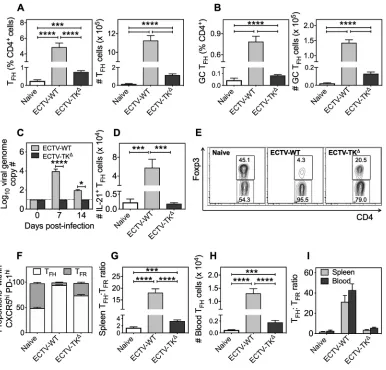
At comparable doses (103 PFU), ECTV-WT-infected mice generated significantly
higher proportions and numbers of TFH(Fig. 3A) and GC TFH(Fig. 3B) cells compared to
ECTV-TKΔ-infected mice. Viral replicative capacity and viral load were driving the
significantly higher TFHand GC TFHresponses as viral genomes were detected in the
blood of mice infected with ECTV-WT at days 7 and 14 p.i. but not in ECTV-TKΔ-infected
mice (Fig. 3C). We also used IL-21 GFP transgenic mice (34) to further establish that replication competent ECTV-WT induced significantly higher numbers of TFHcells that
produced IL-21, the hallmark cytokine associated with TFHcells (Fig. 3D). Notably, the
proportion of TFH and TFRcells within the parent CXCR5hi PD-1hi population varied
distinctly between the two viral infections. Although naive mice had similar proportions of TFHand TFRcells (Fig. 3E and F, left panel), there was a clear bias toward a TFHcell
response in ECTV-WT infected mice (Fig. 3E and F, middle panel). In contrast, TFRcell
proportions remained high in ECTV-TKΔ-infected animals with a small increase in T FH
cell proportions (Fig. 3E and F, right panel). The stronger TFHcell response to ECTV-WT
infection resulted in a⬎10-fold increase in splenic TFH:TFRratio (Fig. 3G),
contempo-raneous with a statistically significant 7-fold increase in blood TFHcell numbers
com-pared to the replication-inefficient virus (Fig. 3H). The circulating TFH:TFRratios in blood
paralleled those detected in the spleen (Fig. 3I).
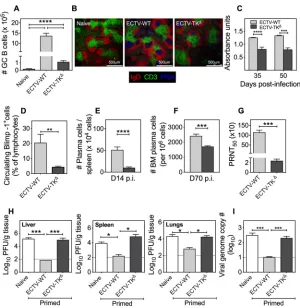
Replication-competent virus infection was associated with significant 10- to 12-fold-higher GC B cell responses (Fig. 4A), substantially larger and 12-fold-higher numbers of GC per spleen section (Fig. 4B) and significantly higher virus-specific IgG absorbance units (Fig. 4C) compared to ECTV-TKΔ-infected mice. We used B lymphocyte-induced maturation
protein 1 (Blimp-1) green fluorescent protein (GFP) transgenic mice (35) to establish that the replication-competent ECTV-WT-infected mice generated a significantly higher frequency of circulating Blimp-1⫹antibody-secreting cells compared to
replication-inefficient ECTV-TKΔ-infected mice (Fig. 4D). In addition, ECTV-WT induced significantly
higher splenic plasma cell (B220–, CD138⫹) numbers compared to ECTV-TKΔat the peak
FIG 2Legend (Continued)
indicates 50% neutralization. (G) PRNT50 titer calculated as the reciprocal of the plasma dilution at which 50% of the virus is
neutralized. (H) Blood viral genome copy numbers determined by qRT-PCR. The dotted horizontal line indicates the minimum level of detection (10 viral genome copies). All graphical data represent means⫾the SEM. Data were log transformed, and the statistical significance determined by one-way ANOVA (**,P⬍0.005;***,P⬍0.0005;****,P⬍0.0001). For long-term antibody response analyses, C57BL/6 mice (n⫽3 mice per time point) were infected s.c. with 103PFU of ECTV-WT and bled at time points indicated, and
the ECTV-specific IgG levels (I), virus-neutralizing activity (J), and PRNT50 titers (K) were determined. The data represent two
independent experiments expressed as means⫾the SEM. No statistical difference was detected by one-way ANOVA.
on November 6, 2019 by guest
http://jvi.asm.org/
of the GC response (Fig. 4E). These differences were maintained at day 70 p.i. in the bone marrow, a survival niche for long-lived plasma cells (Fig. 4F) and, in serum, the PRNT50titers were 45-fold higher in ECTV-WT-infected mice compared to ECTV-TKΔ
-infected mice (Fig. 4G). Naive mice passively transferred with immune serum from ECTV-WT-infected mice exhibited far more significant reductions in viral load in liver (Fig. 4H, left panel), spleen (Fig. 4H, middle panel), lungs (Fig. 4H, right panel), and blood (Fig. 4I) compared to mice that received sera from ECTV-TKΔ-primed mice. Virus
replicative capacity is thus very strongly associated with induction of long-lived pro-tective antibody responses. In addition to its effects on humoral immunity, virus replicative capacity is also strongly associated with a greater magnitude of increased numbers of effector memory (CD44hi, CD62Llo, and CD127hi) CD4 and CD8 T cells in the
spleen and bone marrow (data not shown).
FIG 3Influence of viral replicative capacity on splenic and blood TFHcell populations. C57BL/6 mice (n⫽5 to 6 mice per
group) infected s.c. with 103PFU of ECTV-WT or 103PFU of ECTV-TKΔor left uninfected (naive) were sacrificed on day 14
p.i., and TFHcell populations were analyzed by flow cytometry. (A and B) Percentages and total numbers of TFH(A) and GC
TFH(B) cells per spleen. (C) IL-21 GFP transgenic mice (n⫽4 mice per group) were infected as described for panel A, and
IL-21-producing (IL-21⫹) T
FH(CD4⫹, CD44hi, CXCR5hi, PD-1hi, Foxp3–) cells were enumerated by flow cytometry. (D) C57BL6
mice infected as described for panel A were bled on days 0, 7, and 14 p.i. DNA was isolated, and viral genome copy numbers were determined by qRT-PCR. The dotted horizontal line indicates the minimum level of detection (10 viral genome copies). (E) C57BL6 mice infected as described for panel A were sacrificed at day 14 p.i., and proportions of Foxp3⫹T
FRand Foxp3–
TFHcells, gated on CD4⫹, CD44hi, CXCR5hi, and PD-1hicells, are represented by flow cytometric contour plots and as a
graphical representation (F). (G) Splenic TFH:TFRratio. (H) Circulating CD4⫹, CD44hi, CXCR5⫹, PD-1⫹, Foxp3–TFHcells in the
blood. The data shown are from one of two independent experiments with comparable outcomes. Data are expressed as means⫾the SEM; data were log transformed, and the statistical significance was determined by one-way ANOVA (***,P⬍
0.0005;****,P⬍0.0001). (I) In a separate experiment, C57BL/6 mice (n⫽3 to 5 mice per group) infected s.c. with 103PFU
of ECTV-WT or 103PFU of ECTV-TKΔor left uninfected were sacrificed, and a comparative analysis of the splenic and blood
TFH:TFRratio was performed. Data are expressed as means⫾the SEM.
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 6
on November 6, 2019 by guest
http://jvi.asm.org/
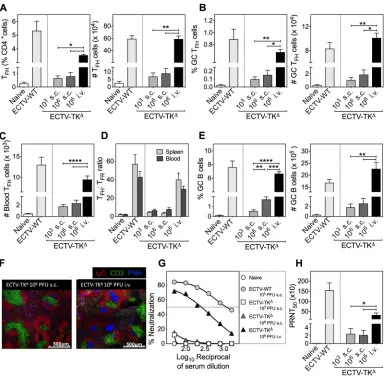
[image:6.585.46.432.70.440.2]Increased dose combined with the hematogenous spread of replication-inefficient virus augments the magnitude of TFH, GC B cell, and antibody responses.We pre-dicted that increasing the dose of replication-inefficient virus to escalate antigen availability and overcoming the dissemination barrier by using the i.v. route of admin-istration for hematogenous spread would augment TFH, GC B cell, and antibody
responses generated against attenuated viruses. C57BL/6 mice were therefore infected with replication-inefficient ECTV-TKΔvia the s.c. route at low (103PFU) or high (106PFU)
doses or via the i.v. route at a high dose (106 PFU) to investigate the influence of
increased inoculum dose and hematogenous viral antigen spread on TFHand GC B cell
responses at day 14 p.i.
FIG 4Viral replicative capacity determines the magnitude of GC B cell and protective antibody re-sponses. (A) C57BL/6 mice infected as described in Fig. 3 were sacrificed at day 14 p.i., and the total number of GC B (B220⫹GL7⫹CD95⫹) cells was enumerated by flow cytometry. (B) Immunofluorescent
PNA⫹GC staining of 10-m frozen spleen sections from naive mice or ECTV-WT- or ECTV-TKΔ-infected
mice at day 14 p.i. (magnification,⫻10; scale bar, 500m). (C) C57BL/6 mice (n⫽4 mice per group) infected with 103PFU of ECTV-WT or ECTV-TKΔwere bled at the times indicated, and the ECTV-specific
IgG absorbance units were determined by ELISA (1:200 serum dilution). Statistical significance was determined by two-way ANOVA. (D) Blimp-1 GFP transgenic mice (n⫽4 mice per group) were infected as described for panel A and sacrificed at day 14 p.i., and the frequency of circulating Blimp-1 GFP⫹
(Blimp-1⫹) antibody-secreting cells was determined by flow cytometry. (E) C57BL/6 mice infected as in
panel A (n⫽9 mice per group) were sacrificed at day 14 p.i., and the splenic plasma cells (B220–ve,
CD138⫹ve) were enumerated by flow cytometry. (F) C57BL/6 mice infected as in panel A were sacrificed
at day 70 p.i. (n⫽5 to 6 mice per group), and the bone marrow plasma cells were enumerated by flow cytometry. Data are presented as means⫾the SEM; the data were log transformed, and the statistical significance was determined by a Studentttest. (G) Virus-neutralizing activity and PRNT50titers in the
sera of ECTV-WT- or ECTV-TKΔ-infected C57BL/6 mice at day 50 p.i. Statistical significance was determined
by a two-tailedttest. (H) C57BL/6 (n⫽4 mice per group) naive, ECTV-WT, or ECTV-TKΔ-primed mice were
sacrificed at day 50 p.i., and 200l of extracted sera from each mouse was passively transferred into a new naive recipient mouse and challenged 24 h later with 103PFU of ECTV-WT. Mice were sacrificed at
day 5 postchallenge, and the viral load in the liver, spleen, and lungs was determined by a viral plaque assay. (I) DNA isolated from blood was used to quantitate viral genome copy numbers by qRT-PCR. The data shown are from one of two independent experiments with comparable outcomes. Data are expressed as means⫾the SEM. Dashed lines indicate the limit of detection. Data were log transformed and, unless indicated otherwise, the statistical significance was determined by one-way ANOVA (*,P⬍
0.05;***,P⬍0.0005;****,P⬍0.0001).
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.55.355.68.376.2]Flow cytometry analyses revealed that a 1,000-fold increase in ECTV-TKΔgiven s.c. to
mice did not result in any significant changes in the frequencies or numbers of splenic TFH(Fig. 5A), GC TFH(Fig. 5B), blood TFH(Fig. 5C), splenic and blood TFH:TFRratios (Fig.
5D), or GC B (Fig. 5E) cells compared to 103 PFU of virus. However, when 106 PFU
ECTV-TKΔwas injected i.v., the T
FH, GC TFH, blood TFH, GC B cells, and TFH:TFRratios were
all significantly increased (Fig. 5A to E). Immunohistochemistry revealed that i.v. inoc-ulation with 106 PFU ECTV-TKΔ induced more and larger GC responses per spleen
section compared to infection with a similar dose through the s.c. route (Fig. 5F). Importantly, there was a positive correlation (r⫽0.9916; Pearson correlation coefficient andP⬍0.0001) between high dose ECTV-TKΔadministered i.v. and the magnitude of
TFHand GC B cell responses. Concordantly, at day 50 p.i., significantly higher
virus-neutralizing activity (Fig. 5G) and PRNT50titers (Fig. 5H) were detected in mice infected
i.v. with a high dose of ECTV-TKΔ. These results suggest that i.v. inoculation of the highly
attenuated virus, resulting in hematogenous spread to overcome the dissemination barrier, can augment humoral immune responses. In addition, the strong correlation
FIG 5Effect of virus dose and route of infection on TFH, TFR, GC B cell, and neutralizing antibody responses. C57BL/6 mice
(n⫽3 per group) left uninfected or infected with either ECTV-WT (103PFU s.c.) or ECTV-TKΔ(103PFU s.c., 106PFU s.c., or
106PFU i.v.) were sacrificed at day 14 p.i. (A) Percentage and total number of T
FHcells. (B) Percentage and total number
of GC TFH cells. (C) Circulating TFHcell numbers. (D) Comparative analysis of the splenic and blood TFH:TFR ratio. (E)
Percentage and total GC B cell numbers. (F) Immunofluorescent GC staining (IgD; red, CD3; green, PNA; blue) of 10-m frozen spleen sections from mice infected with ECTV-TKΔat 103PFU s.c., 106PFU s.c., or 106PFU i.v. (magnification,⫻10;
scale bar, 500m). (G and H) Virus-neutralizing activity (G) and PRNT50titers (H) in sera at day 50 p.i. Data are shown from
one of two independent experiments with comparable outcomes. Data are expressed as means⫾the SEM; data were log transformed, and the statistical significance was determined by one-way ANOVA (***,P⬍0.0005;****,P⬍0.0001).
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 8
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.42.429.70.447.2]between GC responses and TFH:TFRratios support our idea that splenic and circulating
TFH:TFRratios are robust indicators of durable, protective antiviral antibody responses.
TFH:TFRratios can predict the induction of durable humoral immunity against viruses.We determined whether the TFH: TFRratio might be used to predict induction
of durable humoral immunity more broadly by extending our observations with the ECTV model to other viruses.
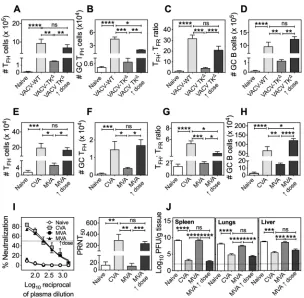
We first analyzed humoral immunity to the closely related orthopoxvirus, vaccinia virus (VACV). Unlike ECTV, VACV is not a mouse pathogen, and replication of even the mouse-adapted, neurovirulent VACV-WR strain (referred to as VACV-WT) is restricted to the site of infection after cutaneous inoculation of immunocompetent mice (36). The i.v. route of infection was therefore used to overcome the dissemination barrier and to simulate blood-borne systemic viral spread. Replication-competent VACV-WT induced significantly higher splenic TFH(Fig. 6A), GC TFH(Fig. 6B), TFH:TFRratio (Fig. 6C), and GC
B (Fig. 6D) cell numbers compared to the replication-inefficient VACV-TKΔat a
compa-rable dose. Altering the dose and antigen availability by a 1,000-fold increase in VACV-TKΔdose significantly increased the magnitude of T
FH, GC TFH, TFH:TFRratios, and
GC B cells (Fig. 6A to D). We also determined that there was a positive correlation FIG 6TFH:TFRratios are predictive of protective antibody responses: VACV and MVA infection models.
C57BL/6 mice (n⫽3 mice per group) left uninfected or infected i.v. with VACV-WT (103PFU) or VACV-TKΔ
(103or 106PFU) were sacrificed at day 10 p.i. Blood and spleens were harvested for flow cytometric
analysis of TFH(A), GC TFH(B), the TFH:TFRratio (C), and GC B cells (D). The data are representative of two
independent experiments expressed as means⫾the SEM; the data were log transformed, and statistical analysis was performed by one-way ANOVA (*,P⬍0.05;**,P⬍0.005;***,P⬍0.0005;****,P⬍0.0001). C57BL/6 mice (n⫽4 mice per group) left uninfected or infected i.v. with replication-competent CVA (103
PFU) or replication-inefficient MVA (103PFU or 108PFU) were sacrificed at day 10 p.i. The total T FH(E),
GC TFH(F), TFH:TFRratio (spleen) (G), and GC B cell (H) numbers per spleen were determined. (I) C57BL/6
mice (n⫽4 to 5 mice per group) infected as in panels E to H were bled at day 50 p.i., and the virus-neutralizing activity and PRNT50 titers were determined. (J) C57BL/6 mice (n⫽4 to 5 mice per
group) left unprimed or primed as described previously (see panels E to H) were challenged with a lethal dose of ECTV-WT (106PFU i.v.) 50 days later. Mice were sacrificed 5 days p.c., and the viral loads in the
spleen, lungs, and liver were determined by viral plaque assay. All data represent means⫾the SEM; data were log transformed, and statistical analysis was performed by one-way ANOVA (*,P⬍0.05;**,P⬍
0.005;***,P⬍0.0005;****,P⬍0.0001).
on November 6, 2019 by guest
http://jvi.asm.org/
[image:9.585.54.358.68.366.2](r⫽0.8123; Pearson correlation coefficient andP⬍0.05) between increased dose of VACV-TKΔand the magnitude of T
FHand GC B cell responses.
We next used modified vaccinia virus Ankara (MVA), a highly attenuated third-generation smallpox vaccine (37), which is currently being considered as a recombinant vaccine vector for infectious diseases and cancer (38–42). MVA does not cause a productive infection in the mammalian host and is therefore considered safe. However, MVA-based expression vectors have required more than one immunization or combi-nation with prime-boost regimes involving different vector systems or recombinant protein antigens to induce effective levels of protective immunity (43). We posited therefore that a single low dose of MVA would elicit poor GC and protective antibody responses compared to the replication-competent parental CVA but that the MVA response could be augmented by administration of a higher dose and given i.v.
At a dose of 103PFU, CVA elicited significantly more robust splenic T
FH(Fig. 6E) and
GC TFH(Fig. 6F) responses, higher TFH:TFRratios (Fig. 6G), stronger GC B cell responses
(Fig. 6H) at day 14 p.i., and 25-fold higher PRNT50titers at day 50 p.i. than a comparable
dose of MVA (Fig. 6I). Increasing the dose of replication-incompetent MVA by 100,000-fold (108PFU; MVA1dose) positively correlated (r⫽0.7948; Pearson correlation
coef-ficient andP⬍0.02) with the significantly increased TFHand GC responses (Fig. 6E to
H), and there was a clear concomitant increase in virus neutralizing PRNT50titers (Fig.
6I). At 50 days postpriming with CVA (103PFU) or MVA (103PFU or 108PFU), mice were
challenged with a high dose of ECTV-WT, sacrificed at day 5 postchallenge (p.c.) and viral load in organs determined. CVA-primed or high-dose-MVA-primed mice had a significantly lower viral burden in the spleen (Fig. 6J, left panel), lung (Fig. 6J, middle panel), and liver (Fig. 6J, right panel) compared to naive or low-dose-MVA-primed mice. These results established that a single high dose of replication-deficient MVA given i.v. to allow hematogenous spread induced protective antibody responses that were comparable to responses generated by the replication-competent CVA.
To exclude the possibility that our findings were unique to orthopoxviruses, we extended our studies to influenza A viruses (IAV). IAV is transmitted via the respiratory route causing infection of the upper and lower respiratory tract (44). We reasoned that hematogenous spread of inactivated IAV through the i.v. route of inoculation would also augment the antibody responses, as shown by the previous experiments. At comparable doses, live mouse-adapted influenza A/Puerto Rico/8/1934 (A/PR/8/34; H1N1) elicited significantly higher TFH(Fig. 7A), TFH:TFRratios (Fig. 7B), and GC B cell (Fig.
7C) numbers at day 10 and significantly higher hemagglutination inhibition (HAI) antibody titers at day 50 p.i. (Fig. 7D) compared to inactivated (replication incompetent) A/PR/8/34 immunization.
Finally, we measured TFHand GC B cell responses in mice infected 10 days previously
with live or inactivated seasonal vaccine strain A/Solomon Islands/3/2006 (A/SI/3/06; H1N1) i.v. Expectedly, live virus induced significantly higher numbers of TFH(Fig. 7E)
and GC B (Fig. 7F) cells and higher TFH:TFRratios in spleen and blood (Fig. 7G) compared
to inactivated virus at a comparable dose. Immunization with a 40-fold-higher dose of inactivated A/SI/3/06 (inactivated 1dose) augmented the TFH numbers (Fig. 7H),
TFH:TFRratios (Fig. 7I), and GC B cell numbers (Fig. 7J) to levels similar to those induced
by live virus. Since current inactivated IAV vaccines are administered via intramuscular (i.m.) or deep s.c. routes, we predicted that i.m. administration of inactivated virus, even at an increased dose (inactivated 1dose), would limit systemic antigen availability, thereby limiting the GC response. Indeed, compared to the i.v. route of immunization, the i.m. route of immunization with inactivated virus resulted in significant reductions in TFHnumbers (Fig. 7H), TFH:TFRratios (Fig. 7I), and GC B cell numbers (Fig. 7J). We
found a positive correlation (r⫽0.7692; Pearson correlation coefficient andP⬍0.03) between high-dose-inactivated IAV administered i.v. and the magnitude of TFHand GC
B cell responses.
Collectively, our data from experiments using orthopoxviruses and orthomyxovi-ruses confirm a direct correlation between systemic antigen availability and generation
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 10
on November 6, 2019 by guest
http://jvi.asm.org/
of durable antibody responses. They also strongly support the reliability of TFH:TFRratios
for predicting induction of robust GC and long-lived protective antibody responses.
DISCUSSION
The key goal of rational vaccine design is the effective induction and maintenance of immunological memory. This objective often eludes many viral vaccines that prog-ress to clinical trials. While there are no universal prognostic biomarkers which can predict whether a vaccine will induce long-lived immunity, the best correlate of long-term protection is neutralizing antibody (3). This attribute is integrally associated with live viral vaccines. Indeed, the most successful human vaccine, a live viral vaccine against smallpox, elicits stable and life-long humoral immunity (2, 45–47). Much of our understanding of antiviral antibody responses comes largely from studies using protein antigens, inactivated vaccines, viruses that are not natural pathogens, or viruses that cause chronic infection. As a consequence, it has not been entirely clear why replicating viruses or live virus vaccines are superior at eliciting robust and effective long-lived immunity. We have addressed this fundamental question using a model of generalized viral infection in its natural host. ECTV causes a smallpox-like disease termed mousepox FIG 7The magnitudes of TFHand GC responses to inactivated influenza viruses are governed by the dose
and route of administration. C57BL/6 mice (n⫽4 mice per group) either uninfected or infected i.v. with 105HAU of live or formalin-inactivated influenza A/PR/8/34 were sacrificed at day 10 p.i. (A) Total T
FHcells
per spleen. (B) Splenic TFH:TFRratio. (C) GC B cell numbers per spleen. (D) HAI titers at day 50 p.i. Data
represent two independent experiments expressed as means ⫾ the SEM; statistical analysis was performed using one-way ANOVA (**,P⬍0.005;****,P⬍0.0001). C57BL/6 mice (n⫽4 mice per group) left uninfected or infected i.v. with 105HAU of live or formalin-inactivated A/SI/3/06 were sacrificed at day
10 p.i., and the blood and spleens were harvested for flow cytometric analysis. (E) Total TFHcells per
spleen. (F) Comparison of the TFH:TFRratio in spleen and blood. (G) GC B cell numbers per spleen. C57BL/6
mice (n⫽4 mice per group) left uninfected or infected with live (105HAU i.v.) or formalin-inactivated
(4⫻106HAU1dose i.v. or i.m.) A/SI/3/06 were sacrificed at day 10 p.i. to evaluate the combined effects
of increased dose and route of administration on TFHand GC B cell populations. (H) Total TFHcells per
spleen. (I) TFH:TFRratio in the spleen. (J) GC B cell numbers per spleen. All data represent means⫾the
SEM; data were log transformed, and statistical comparison with the live virus-infected group was performed by one-way ANOVA (*,P⬍0.05;**,P⬍0.005;***,P⬍0.0005;****,P⬍0.0001).
on November 6, 2019 by guest
http://jvi.asm.org/
[image:11.585.56.356.70.372.2]and is an excellent surrogate for smallpox and for studying long-lived neutralizing antibody responses.
Using ECTV and other viral models, we have made three key findings in relation to induction of long-lived antiviral antibody responses. First, the replicative capacity of virus or viral vaccine vector shapes the evolution of the GC response and induction of durable humoral immunity. Second, virus replicative capacity has a significant impact on the TFH:TFRratio, a biomarker that can predict induction of protective, long-lived
antibody responses. Third, administration of poorly or nonreplicating viruses to allow hematogenous spread generates a significantly stronger TFH:TFRratio and robust TFH,
GC B cell, and neutralizing antibody responses.
In humans, an increase in circulating TFHcells in blood following IAV vaccination was
shown to correlate with increases in preexisting antibody titers but not with the induction of primary antibody responses (48). In another human study, the activation of circulating TFHcells induced by IAV vaccination was shown to correlate with the
development of memory B cells (49). The present study indicates that the magnitude of the splenic or blood TFH response and TFH:TFR ratio can predict the strength of
primary, memory, and long-lived antibody responses. In humans, the analysis of blood TFH subsets has permitted the assessment of ongoing TFH responses in secondary
lymphoid organs, thus making it feasible to monitor circulating TFHand TFRresponses
(48, 50–52). The use of TFH:TFR ratios as a predictive indicator of durable antibody
response generation would be of substantial value in a clinical vaccine trial, significantly reducing testing time and resources. TFH:TFR ratios can be determined within 10 to
14 days after vaccination, long before B cell memory forms and long-lived antibody responses are generated. While it may not be possible to use a single universal TFH:TFR
ratio value as predictive of durable antibody response generation to different vaccine types, our data from the ECTV-TKΔ, MVA, and inactivated IAV immunization models
indicates that a range between ratios of 5:1 and 10:1 would be sufficient. Although we have only measured total TFHand TFRcell responses, which may not necessarily all be
antigen specific, our data indicate that in specific pathogen-free mice, the TFH:TFRratios
based on total TFHcell responses to four strains of poxviruses and two strains of IAV are
predictive of induction of potent protective antibody responses following immuniza-tion/infection. One potential caveat to using total and not antigen-specific TFH:TFR
ratios in humans is when an individual in a clinical trial has contracted some other infection postvaccination; in that case the measurement of vaccine antigen-specific responses would be more relevant. Under such circumstances, measurement of antigen-specific TFHand TFRresponses would be more relevant.
A previous study in mice using nonreplicating antigens showed that the TFRcell
responses follow the kinetics of the TFHcells (16). However, using the natural mouse
pathogen ECTV, we have found that there is, in fact, an inverse relationship with respect to cell numbers and kinetics between the two subsets from about days 7 to 10 p.i., just prior to the peak GC response at day 14. TFHnumbers increase beginning around day
7, as shown in our study, as well as by others (48, 49, 53), whereas TFRnumbers are
reduced during this time. The fact that there is an inverse relationship between TFHand
TFR cell would suggest that the bigger the ratio, the more durable the antibody
response generated. The precise molecular mechanisms that control this inverse rela-tionship are not known but must be important in order for expansion of TFHcells just
before the peak GC response. In a recent study, high concentrations of IL-2 produced at the peak of IAV infection were reported to prevent TFR cell development by a
Blimp-1-dependent mechanism (54). When IL-2 levels were reduced following resolu-tion of IAV infecresolu-tion, TFRcells were found to differentiate and move to B cell follicles to
maintain B cell tolerance. In mice infected with ECTV-WT, IL-2 production rapidly increases from day 4 p.i., peaks at day 8, after which the levels are somewhat reduced but remain high even at day 12 (55). In addition to the early drop in TFRnumbers, we
observed an increase in the proportion of IL-2R␣-expressing TFRcells over the course of
infection, particularly during the contraction phase of the TFHand GC responses. It is
possible that the IL-2:IL-2R␣ axis contributes to the suppressive activity of TFRcells
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 12
on November 6, 2019 by guest
http://jvi.asm.org/
following the peak TFHand GC B cell responses. Although IL-2 can mediate inhibition
of early TFH differentiation (56, 57), it is conceivable that the cytokine could play a
context-dependent paradoxical role in TFHregulation by TFRcells.
The biosafety aspects of attenuated viruses, either replication inefficient or replica-tion defective, have prompted their use as viral vaccine vectors. However, our current understanding of the mechanisms of immune protection provided by attenuated viruses is confounded by the use of various doses, routes, animal models, and challenge strategies that have been reported. In this study, the antiviral humoral immune responses elicited by replication-efficient and -inefficient ECTV were compared, with a deliberate attempt to ensure that the comparisons were carried out under similar conditions. At the same dose and route of administration (s.c.), significant increases in the magnitude of TFHand GC responses and neutralizing antibody titers were observed
following infection with replication-competent virus compared to replication-inefficient virus. This finding can be primarily attributed to increased viral antigen availability, hematogenous spread, activation of potentially numerous secondary lymphoid organs, including the spleen, and most likely longer antigen retention by the GC follicular dendritic cells in its native form promoting sustained presentation and subsequent antibody affinity maturation (58). Our findings are consistent with previous studies examining the role of antigen levels in early TFHproliferation and terminal
differenti-ation to GC TFH, albeit using protein antigens (19), bacteria (23), or persistent viral
infection models (22). However, it should be noted that the chronic persistence of antigen leads to an abnormal TFH expansion, resulting in dysregulated antibody
responses (59).
In our experiments with poxvirus and IAV infection models, we found a direct correlation between attenuated/inactivated virus dose administered i.v. and the mag-nitude of TFHand GC B cell response. A previous study established that mice inoculated
i.v. with 108PFU of recombinant MVA expressing IAV HA and boosted 35 days later
generated significantly higher titers of HAI antibody compared to the s.c., i.m., or intranasal routes of inoculation with an equivalent dose (60). The findings from our study and that of Sutter et al. (60) are consistent with results from a clinical trial that used nonreplicating sporozoite vaccine against malaria which demonstrated superior protection when administered i.v. compared to s.c. or intradermal routes (61). Gener-alized viral infections such as variola, measles, and polio result in viremia and hema-togenous dissemination, which could explain the generation of robust and durable protective antibody responses.
In order to understand the molecular changes in antigen-specific B cells associated with virus replicative capacity, dose, and route of virus inoculation, we have utilized the SWHEL B cell receptor transgenic (Tg) mice (62) in combination with recombinant ECTV (WT and TKΔ) engineered to express the model protein antigen, hen egg lysozyme
(HEL). SWHEL Tg mice have HEL-specific Tg B cells that can class switch and can be tracked as they undergo affinity maturation and class switch recombination in response to virus-expressed HEL. We have found that systemic spread of a replication-inefficient virus through i.v. inoculation significantly increases the induction of somatic hypermu-tation events in antigen-specific B cells and affinity maturation of antibody (unpub-lished data).
Our data suggest that the TFH/TFR balance may shape the evolution of the GC
response. Neutralizing antibody responses were strongly correlated with lymphoid tissue resident TFH:TFR ratios across four virus infection models. Furthermore, the
circulating TFH:TFRratios paralleled those detected in the lymphoid tissue and may be
used reliably to predict induction of potent virus neutralizing antibody responses, provided that the terminal TFHdifferentiation and GC formation steps are not affected.
Modulating the TFH/TFRbalance could thus, represent one of the axes along which the
quality and duration of antibody response may be manipulated during or postvacci-nation. However, any strategy to regulate the TFRcells for optimal GC response must
consider the level and timing of the modulation so as to prevent generation of autoreactive antibodies. Altered TFR:TFHratios have been observed in several human
on November 6, 2019 by guest
http://jvi.asm.org/
and animal models of autoimmunity (63–65). Although there is evidence that TFRcells
can govern the GC response by suppressing TFH and B cells (16, 18), the exact
mechanism still remains unclear. A better understanding of the molecular mechanisms of IL-2R␣-mediated TFRsuppression of GC responses will be critical for augmenting
protective antibody responses following vaccinations.
Recently, transcriptomics, combined with systems biology using peripheral blood leukocytes from vaccinees have revealed molecular signatures of protective antibody responses associated with vaccine-induced immunity, but no universal signatures have been found (66–69). Another recent study has demonstrated a direct relationship between GC activity and levels of GC TFHexpressed CXCL13 in plasma (70). CXCL13 is
the ligand for CXCR5, and the authors have suggested it may be potentially used as a plasma biomarker of GC activity. We postulate that the judicious use of the circulating TFH:TFRratio, with or without plasma CXCL13 (as a marker for terminal TFH
differenti-ation), as a correlate that can predict antiviral antibody response generation within days or weeks of vaccination will facilitate efficient screening of the efficacy of new and novel viral vaccine formulations.
In summary, the results presented in this study demonstrate that the replicative capacity of virus or viral vaccine vector governs the induction of neutralizing antibody responses by influencing antigen availability. Our data clearly shows that allowing dissemination of highly attenuated (MVA) or inactivated (IAV) vaccines through the hematogenous route in mice can significantly increase the magnitude of TFHand GC B
cells and subsequent neutralizing antibody responses. Although the i.v. route may not be feasible for mass vaccination, it may be an important alternative for inducing better and long-lived antibody responses against pathogens, especially in instances where the other routes of vaccination are not effective.
MATERIALS AND METHODS
Ethics statement.All animal experiments were conducted in strict accordance with the good animal practice for care and use defined by the Australian code for National Health and Medical Research Council guidelines and protocols approved by the Animal Ethics and Experimentation Committee of the Australian National University.
Mice.Inbred, specific-pathogen-free C75BL/6J, Blimp-1 GFP, and IL-21 GFP transgenic mice on a C57BL/6 background were bred and maintained at the Australian Phenomics Facility, Australian National University, and used at 6 to 8 weeks of age.
Animal experiments.All animal infections were performed under intraperitoneal avertin anesthesia, which was freshly prepared before use. Mice were infected either subcutaneously (s.c.) in the flank of the left hind limb (hock), intramuscularly (i.m.) in the left thigh, or intravenously (i.v.) in the lateral tail veins. Mice were monitored daily during the entire experimental period and scored against a clinical matrix taking into account hair coat, posture, breathing, activity, foot swelling, and body weight.
Cell lines and culture.African green monkey kidney epithelial (B-SC-1),Canis familiariskidney cell line (MDCK), and Syrian hamster kidney fibroblast cell line (BHK21) were maintained in Eagle minimum essential medium (EMEM) and Roswell Park Memorial Institute (RPMI) 1640 medium, respectively, supplemented with 10% fetal calf serum, 2 mM L-glutamine, 120g/ml penicillin, and 200g/ml streptomycin and neomycin sulfate.
Virus stocks.The Moscow strain of ECTV (referred to as ECTV-WT; ATCC VR-1734), the TK deletion mutant of ECTV-WT (ECTV-TKΔ), the Western Reserve strain of VACV (referred to as VACV-WT; ATCC
VR-1354), and the TK deletion mutant of VACV-WT, VACV-TKΔ(71), were propagated in BS-C-1
mono-layers, and titers were determined by viral plaque assay as described previously (72, 73). CVA virus was kindly provided by Jürgen Hausmann at the Bavarian Nordic GmbH, Germany. Titration of MVA stocks was carried out on BHK-21 cell monolayers by immunostaining as described previously (74). IAV A/PR/8/34 and A/SI/3/06 stocks were propagated in specific-pathogen-free embryonated chicken eggs, and viral titers were determined by plaque assay using MDCK cells (75). Inactivation of influenza virus stocks was performed by incubation with formalin (0.02% final concentration) for 18 h at 37°C, followed by dialysis against four changes of PBS (76). Inactivation of the virus was confirmed by plaque assay.
Viral load in organs and blood.Organs harvested were snap-frozen and stored at – 80°C until processed. Briefly, serial dilutions of organ homogenates (Polytron homogenizer; Pro-Scientific, Inc.) were plated onto confluent BS-C-1 monolayers, and virus titers were determined using the conventional viral plaque assay as described previously (77). Viral titers in blood were measured by real-time qualitative PCR and expressed as viral genome copy numbers correlated with the copy numbers of ECTV156, a late gene encoding viral hemagglutinin (73).
Immunofluorescent staining protocol.Harvested spleen was frozen in O.C.T and stored at – 80°C until required. Air-dried cryosections (10m) were fixed in ice-cold acetone and blocked with PBS containing 3% bovine serum albumin (BSA) before staining with peanut agglutinin (PNA; Sigma-Aldrich), rabbit anti-mouse (clone 11.26 C; Southern Biotech), and rat anti-mouse CD4 (clone GK1.5; BioLegend) in
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 14
on November 6, 2019 by guest
http://jvi.asm.org/
PBS containing 0.5% BSA and 0.1% Tween 20. Sections were washed, mounted with Vectashield mounting medium (Vector Laboratories), and imaged using a Leica-SP5 confocal laser scanning micro-scope.
ELISA.Virus-specific IgG levels were measured by enzyme-linked immunosorbent assay (ELISA) as described elsewhere (31). Briefly, high-binding ELISA plates (Thermo Fisher Scientific) were coated with sucrose-cushion purified ECTV-WT virus, washed, and incubated with serial dilutions of the serum samples. Antibodies were detected using horseradish peroxidase conjugated to anti-mouse IgG or IgG subclass antibodies (Southern Biotech), and the colorimetric reaction (tetramethyl benzidine; Invitrogen Life Technologies) absorbance was measured at 650 nm using Tecan plate reader (Tecan).
Flow cytometry.Cells harvested from spleen or blood were incubated with Fc block (clone 2.4G2; BD Biosciences) and surface stained as required with anti-mouse CD4 (clone RM4-5; BD Biosciences), B220 (clone RA3-6B2; BD Biosciences), CD44 (clone IM7, BioLegend), CXCR5 (clone DG8; BD Biosci-ences), PD-1 (clone J43; eBioscience), GL7 (clone GL7; BD BiosciBiosci-ences), CD95 (clone Jo2; BD Biosciences), CD138 (clone 281.2; BD Biosciences), CD62L (clone MEL-14; BD Biosciences), or CD62L (clone H1.2F3; eBioscience). For Foxp3 staining, cells were fixed and permeabilized according to the manufacturer’s instructions (Foxp3 transcription factor buffer kit; eBioscience) and incubated with anti-mouse Foxp3 (clone FJK-16s; eBioscience). All samples were acquired on BD LSRII or BD LSR Fortessa and subsequently analyzed using FlowJo software (Tree Star, Inc.).
Statistical analysis.Statistical analyses of experimental data, as indicated in figure legends, were performed using Prism V6 (GraphPad Software, San Diego). Where indicated, single comparisons were analyzed using the unpaired student two-tailedttest with Welch correction, and multiple comparisons were analyzed using one-way or two-way analysis of variance (ANOVA), followed by the Holm-Sidak multiple-comparison test.
ACKNOWLEDGMENTS
This study was supported by grants from the National Health and Medical Research Council of Australia to G.K. and G.C. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Bernard Moss of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, for the gift of MVA and VACV-TKΔ; Jürgen Hausmann of
Bavarian Nordic GmbH for the gift of CVA; Ronald Jackson of the Australian National University for the gift of ECTV-TKΔ; and Patrick Reading, WHO Collaborating Centre for
Reference and Research on Influenza, Melbourne, Australia, for influenza A/SI/3/06 stocks. We also acknowledge the assistance of staff at the Australian National University Phenomics Facility and the John Curtin School of Medical Research Microscopy and Flow Cytometry Research Facility.
REFERENCES
1. Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. 2003. Cutting edge: long-term B cell memory in humans after smallpox vac-cination. J Immunol 171:4969 – 4973.
2. Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. 2003. Duration of antiviral immunity after smallpox vaccination. Nat Med 9:1131–1137.https://doi.org/10.1038/nm917. 3. Plotkin SA. 2010. Correlates of protection induced by vaccination. Clin
Vaccine Immunol 17:1055–1065.https://doi.org/10.1128/CVI.00131-10. 4. Fenner F. 1988. Smallpox and its eradication. World Health Organization,
Geneva, Switzerland.
5. Panum PL. 1847. Beobachtungen über das Maserncontagium. Archiv Pathol Anat 1:492–512.https://doi.org/10.1007/BF02114472.
6. Paul JR, Riordan JT, Melnick JL. 1951. Antibodies to three different antigenic types of poliomyelitis virus in sera from North Alaskan Eski-mos. Am J Hyg 54:275–285.
7. Sawyer WA. 1931. The persistence of yellow fever immunity. J Prevent Med 5:413– 428.
8. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. 2016. Follicular helper T cells. Annu Rev Immunol 34:335–368.https://doi.org/10.1146/annurev -immunol-041015-055605.
9. Tangye SG, Ma CS, Brink R, Deenick EK. 2013. The good, the bad and the ugly: TFH cells in human health and disease. Nat Rev Immunol 13: 412– 426.https://doi.org/10.1038/nri3447.
10. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, Ma J, Tezuka K, Yagita H, Okumura K. 2005. The role of ICOS in the CXCR5⫹follicular
B helper T cell maintenancein vivo. J Immunol 175:2340 –2348. 11. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD,
Wang YH, Dong C. 2009. Bcl6 mediates the development of T follicular
helper cells. Science 325:1001–1005. https://doi.org/10.1126/science .1176676.
12. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, Arkwright PD, Kreins AY, Averbuch D, Engelhard D, Magdorf K, Kilic SS, Minegishi Y, Nonoyama S, French MA, Choo S, Smart JM, Peake J, Wong M, Gray P, Cook MC, Fulcher DA, Casanova JL, Deenick EK, Tangye SG. 2012. Functional STAT3 deficiency compromises the generation of hu-man T follicular helper cells. Blood 119:3997– 4008.https://doi.org/10 .1182/blood-2011-11-392985.
13. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, Durandy A, Baumann U, Schlesier M, Welcher AA, Peter HH, Warnatz K. 2006. ICOS deficiency is associated with a severe reduction of CXCR5⫹
CD4 germinal center Th cells. J Immunol 177:4927– 4932.https://doi.org/ 10.4049/jimmunol.177.7.4927.
14. Cannons JL, Yu LJ, Jankovic D, Crotty S, Horai R, Kirby M, Anderson S, Cheever AW, Sher A, Schwartzberg PL. 2006. SAP regulates T cell-mediated help for humoral immunity by a mechanism distinct from cytokine regulation. J Exp Med 203:1551–1565.https://doi.org/10.1084/ jem.20052097.
15. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. 2008. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455:764 –769.https://doi.org/10.1038/nature07345.
16. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KG, Vinuesa CG. 2011. Foxp3⫹follicular regulatory T cells control
the germinal center response. Nat Med 17:975–982.https://doi.org/10 .1038/nm.2425.
17. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, Graca L. 2011. Regulation of the germinal center reaction by Foxp3⫹
on November 6, 2019 by guest
http://jvi.asm.org/
follicular regulatory T cells. J Immunol 187:4553– 4560.https://doi.org/ 10.4049/jimmunol.1101328.
18. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, Fan HM, Liu ZM, Neelapu SS, Dong C. 2011. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17:983–988.https://doi.org/10.1038/ nm.2426.
19. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. 2013. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 38:596 – 605.
https://doi.org/10.1016/j.immuni.2012.11.020.
20. Brink R, Phan TG, Paus D, Chan TD. 2008. Visualizing the effects of antigen affinity on T-dependent B-cell differentiation. Immunol Cell Biol 86:31–39.https://doi.org/10.1038/sj.icb.7100143.
21. Kamal RP, Blanchfield K, Belser JA, Music N, Tzeng WP, Holiday C, Burroughs A, Sun X, Maines TR, Levine MZ, York IA. 2017. Inactivated H7 influenza virus vaccines protect mice despite inducing only low levels of neutralizing antibodies. J Virol 91:e01202-17.https://doi.org/10.1128/JVI .01202-17.
22. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. 2011. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med 208:987–999.https://doi.org/10.1084/ jem.20101773.
23. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, Jenkins MK. 2013. Single naive CD4⫹T cells from a diverse
repertoire produce different effector cell types during infection. Cell 153:785–796.https://doi.org/10.1016/j.cell.2013.04.007.
24. Panchanathan V, Chaudhri G, Karupiah G. 2006. Protective immunity against secondary poxvirus infection is dependent on antibody but not on CD4 or CD8 T-cell function. J Virol 80:6333– 6338.https://doi.org/10 .1128/JVI.00115-06.
25. Panchanathan V, Chaudhri G, Karupiah G. 2010. Antiviral protection following immunization correlates with humoral but not cell-mediated immunity. Immunol Cell Biol 88:461– 467.https://doi.org/10.1038/icb .2009.110.
26. Chaudhri G, Panchanathan V, Bluethmann H, Karupiah G. 2006. Obliga-tory requirement for antibody in recovery from a primary poxvirus infection. J Virol 80:6339 – 6344.https://doi.org/10.1128/JVI.00116-06. 27. Fang M, Sigal LJ. 2005. Antibodies and CD8⫹T cells are complementary
and essential for natural resistance to a highly lethal cytopathic virus. J Immunol 175:6829 – 6836.
28. Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, Barnett B, Crotty S. 2010. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol 185:190 –202. https://doi.org/10.4049/ jimmunol.0903505.
29. Jackson RJ, Maguire DJ, Hinds LA, Ramshaw IA. 1998. Infertility in mice induced by a recombinant ectromelia virus expressing mouse zona pellucida glycoprotein 3. Biol Reprod 58:152–159.
30. Kochneva GV, Urmanov IH, Ryabchikova EI, Streltsov VV, Serpinsky OI. 1994. Fine mechanisms of ectromelia virus thymidine kinase-negative mutants avirulence. Virus Res 34:49 – 61.
31. Chaudhri G, Tahiliani V, Eldi P, Karupiah G. 2015. Vaccine-induced pro-tection against orthopoxvirus infection is mediated through the com-bined functions of CD4 T cell-dependent antibody and CD8 T cell responses. J Virol 89:1889 –1899.https://doi.org/10.1128/JVI.02572-14. 32. Panchanathan V, Chaudhri G, Karupiah G. 2005. Interferon function is
not required for recovery from a secondary poxvirus infection. Proc Natl Acad Sci U S A 102:12921–12926.https://doi.org/10.1073/pnas .0505180102.
33. Sakala IG, Chaudhri G, Scalzo AA, Eldi P, Newsome TP, Buller RM, Karupiah G. 2015. Evidence for persistence of ectromelia virus in inbred mice, recrudescence following immunosuppression and transmission to naive mice. PLoS Pathog 11:e1005342.https://doi.org/10.1371/journal .ppat.1005342.
34. Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, Nutt SL. 2012. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol 13:491– 498.https:// doi.org/10.1038/ni.2261.
35. Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL. 2004. Plasma cell ontogeny defined by quantitative changes in Blimp-1 expression. J Exp Med 200:967–977.https://doi.org/10.1084/jem .20040973.
36. Tscharke DC, Smith GL. 1999. A model for vaccinia virus pathogenesis
and immunity based on intradermal injection of mouse ear pinnae. J Gen Virol 80:2751–2755.https://doi.org/10.1099/0022-1317-80-10-2751. 37. Mayr A, Stickl H, Muller HK, Danner K, Singer H. 1978. [The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a de-bilitated defence mechanism (author’s transl)]. Zentralbl Bakteriol B 167:375–390.
38. Altenburg AF, Kreijtz JH, de Vries RD, Song F, Fux R, Rimmelzwaan GF, Sutter G, Volz A. 2014. Modified vaccinia virus Ankara (MVA) as produc-tion platform for vaccines against influenza and other viral respiratory diseases. Viruses 6:2735–2761.https://doi.org/10.3390/v6072735. 39. Cottingham MG, Carroll MW. 2013. Recombinant MVA vaccines:
dispel-ling the myths. Vaccine 31:4247– 4251.https://doi.org/10.1016/j.vaccine .2013.03.021.
40. Goepfert PA, Elizaga ML, Sato A, Qin L, Cardinali M, Hay CM, Hural J, DeRosa SC, DeFawe OD, Tomaras GD, Montefiori DC, Xu Y, Lai L, Kalams SA, Baden LR, Frey SE, Blattner WA, Wyatt LS, Moss B, Robinson HL. 2011. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J Infect Dis 203:610 – 619.https://doi.org/10.1093/infdis/jiq105.
41. Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J, Dahut WL, Arlen PM, Gulley JL, Godfrey WR. 2010. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol 28: 1099 –1105.https://doi.org/10.1200/JCO.2009.25.0597.
42. Sebastian S, Gilbert SC. 2016. Recombinant modified vaccinia virus Ankara-based malaria vaccines. Expert Rev Vaccines 15:91–103.https:// doi.org/10.1586/14760584.2016.1106319.
43. Grandpre LE, Duke-Cohan JS, Ewald BA, Devoy C, Barouch DH, Letvin NL, Reinherz EL, Baden LR, Dolin R, Seaman MS. 2009. Immunogenicity of recombinant modified vaccinia Ankara following a single or multi-dose vaccine regimen in rhesus monkeys. Vaccine 27:1549 –1556.https://doi .org/10.1016/j.vaccine.2009.01.010.
44. Ikonen N, Strengell M, Kinnunen L, Osterlund P, Pirhonen J, Broman M, Davidkin I, Ziegler T, Julkunen I. 2010. High frequency of cross-reacting antibodies against 2009 pandemic influenza A(H1N1) virus among the elderly in Finland. Euro Surveill 15(5):pii⫽19478.https://doi.org/10.2807/ ese.15.05.19478-en.
45. Amanna IJ, Slifka MK, Crotty S. 2006. Immunity and immunological memory following smallpox vaccination. Immunol Rev 211:320 –337.
https://doi.org/10.1111/j.0105-2896.2006.00392.x.
46. Amanna IJ, Carlson NE, Slifka MK. 2007. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med 357:1903–1915.
https://doi.org/10.1056/NEJMoa066092.
47. Dorner T, Radbruch A. 2007. Antibodies and B cell memory in viral immunity. Immunity 27:384 –392.https://doi.org/10.1016/j.immuni.2007 .09.002.
48. Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, Flano E, Mejias A, Albrecht RA, Blankenship D, Xu H, Pascual V, Banchereau J, Garcia-Sastre A, Palucka AK, Ramilo O, Ueno H. 2013. Induction of ICOS⫹CXCR3⫹CXCR5⫹TH cells correlates with antibody
responses to influenza vaccination. Sci Transl Med 5:176ra132. 49. Koutsakos M, Wheatley AK, Loh L, Clemens EB, Sant S, Nussing S, Fox A,
Chung AW, Laurie KL, Hurt AC, Rockman S, Lappas M, Loudovaris T, Mannering SI, Westall GP, Elliot M, Tangye SG, Wakim LM, Kent SJ, Nguyen THO, Kedzierska K. 2018. Circulating TFHcells, serological
mem-ory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci Transl Med 10:eaan8405. https://doi.org/10.1126/ scitranslmed.aan8405.
50. Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romão VC, Pires AR, da Silva SL, Fonseca JE, Sousa AE, Linterman MA, Graca L. 2017. Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol 2:eaan1487.
https://doi.org/10.1126/sciimmunol.aan1487.
51. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, Sun X, Vandenberg K, Rockman S, Ding Y, Zhu L, Wei W, Wang C, Karnowski A, Belz GT, Ghali JR, Cook MC, Riminton DS, Veillette A, Schwartzberg PL, Mackay F, Brink R, Tangye SG, Vinuesa CG, Mackay CR, Li Z, Yu D. 2013. Circulating precursor CCR7loPD-1hiCXCR5⫹CD4⫹T cells indicate Tfh cell activity
and promote antibody responses upon antigen reexposure. Immunity 39:770 –781.https://doi.org/10.1016/j.immuni.2013.09.007.
52. Heit A, Schmitz F, Gerdts S, Flach B, Moore MS, Perkins JA, Robins HS, Aderem A, Spearman P, Tomaras GD, De Rosa SC, McElrath MJ. 2017.
Eldi et al. Journal of Virology
March 2019 Volume 93 Issue 6 e01795-18 jvi.asm.org 16