JOURNAL OF VIROLOGY, June 1994, p.3896-3907 Vol. 68,No.6 0022-538X/94/$04.00+0
Copyright C 1994, American Society for Microbiology
Substrate Features Important for Recognition and Catalysis by
Human
Immunodeficiency Virus Type 1 Integrase
Identified by Using Novel DNA Substrates
SAMSON A.CHOW't
AND PATRICK0. BROWN'23*Department of
Pediatrics,'
Department
of Biochemistry,2 and Howard Hughes Medical Institute,3
Stanford University
MedicalCenter,
Stanford, California 94305-5428
Received 17 December 1993/Accepted 17 March 1994
Theintegrase encoded by human immunodeficiency virus type 1 (HIV-1) is required forintegration of viral DNA into the host cell chromosome. Invitro,
integrase
mediates a concerted cleavage-ligation reaction (strand transfer) that results in covalent attachment of viral DNA to target DNA.Witha substratethat mimics the strand transfer product, integrase carries out disintegration, the reverse of the strand transferreaction,
resolving this integration intermediate into its viral and target DNA parts. We used a set ofdisintegration
substratestostudy thecatalytic
mechanism of HIV-1integrase
and theinteraction between theprotein
and the viral and target DNA sequence. One substrate termed dumbbell consists ofa singleoligonucleotide thatcan fold to form a structure that mimics theintegration
intermediate. Kinetic analysisusing
the dumbbell substrate showed thatintegrase turned over,establishing
thatHIV-1 integrase isanenzyme.Analysis
of thedisintegration activity
onthe dumbbell substrate andits derivatives showed that both the viral and target DNA partsof themolecule wererequired
forintegrase
recognition.
Integraserecognized
targetDNAasymmetrically:
the target DNA upstream of the viral DNA joining siteplayed
a much moreimportant
role than the downstream targetDNA inprotein-DNA
interaction. The site oftransesterification
wasdeterminedby
both the DNA sequence of the viral DNA end and the structure of the branched substrate.Using
a series ofdisintegration
substrates with various basemodifications,
we found thatintegrase
had relaxed structuralspecificity
for thehydroxyl group used in transesterification and could tolerate distortion of the double-helical structure of these DNA substrates.Integration of
adouble-stranded
DNA copyof the
retrovirus
genome into a chromosomeof the
host cellis essential for
efficient viral replication
(9, 24, 27,
30). Genetic studies
have shown thatretrovirus
integration requires
twoviral
compo-nents:integrase (encoded by the 3' end of the
pol gene) and
DNA sequences atthe
endsof the
virallong terminal
repeats(for reviews
onretrovirus
DNAintegration,
seereferences 3,
17,
and
35). Analysis of the integration reaction in vivo and
invitro has demonstrated that it
proceeds
inthe
following three
steps:(i) 3' processing, in which integrase
removes 2nucleo-tides
fromthe
3'end
ofeach
strandof linear
viral DNAsothat the viral 3' endsterminate
with the CAdinucleotide (5,
14, 22,28);
(ii)
strandtransfer,
aconcertedcleavage-ligation reaction
during which integrase makes
astaggered
cut inthe
target DNAand ligates
therecessed 3' ends of
theviral
DNA tothe
5'ends of the target DNAatthecleavage site
(5, 10, 13, 21),
producing
agapped
intermediate; and (iii)
gaprepair,
inwhich
the
integration
processis
completed by removal of the
2unpaired nucleotides
atthe
5' endsof the viral
DNAand
repair
of the gaps between the viral and target DNA se-quences,thereby generating the
shortdirect
repeatsthatflank
the
provirus.
Theprotein factors involved
in the last stepof
thereaction
remain to becharacterized.
The
first
two steps ofthe reaction, which
areintegrase
dependent,
do not require the presence of a nucleotide*Correspondingauthor.Mailing address: Howard Hughes Medical
Institute,B255 Beckman Center, StanfordUniversity Medical Center, Stanford, CA 94305-5428. Phone: (415) 725-7569. Fax: (415) 723-1399. Electronic mail address:
pbrownC@)CMGM.stanford.edu.
tPresentaddress:
Department
of Molecular and Medical Pharma-cology,UCLA School ofMedicine, LosAngeles,CA 90024.cofactor
as anenergysource(4,
10),
andeach
stepproceeds by
a one-step
transesterification mechanism
(12).
Using
asub-strate
that mimics the initial
integration intermediate,
we showedpreviously
that humanimmunodeficiency virus
type 1(HIV-1) integrase
cancatalyze
the strand transfer reaction
in reverseand resolve the
intermediate into its viral and
target DNA parts. The reversereaction is called
disintegration (8).
Integrase
canalso
mediate
DNAcleavage-ligation
on analtered
disintegration
substrate in
which the entire
viralpor-tion is
single stranded,
anactivity
wetermed
DNAsplicing
(7,
8, 31, 36,
37). The discovery
ofthis
DNAsplicing
activity
ledus to proposethat
integrase
mayhave other
biological roles
during the retrovirus life cycle, such
asviral 5'-end
joining
and
viral
recombination.
Asexemplified by the
DNAsplicing
activity, the
sequence and structurerequirements
for
disinte-gration
are lessstringent
than thosefor
3'processing and
strand transfer
(8). This characteristic allows
manygenetic
variants of
integrase that lack detectable
activity
in
3'
process-ing
andstrand
transfer toretain
disintegration
activity (7, 11,
26, 34, 36,
37). Thus, the disintegration
assayhas
played
anessential role
inlocating
thecatalytic domain of
integrase and
will be useful inmapping other functional domains
of theprotein.
Compared with the substrates
usedin
other invitro
assays forintegrase activity, the disintegration substrate
has theunique
propertythat
thesite of
integration
ofviral
DNAinto targetDNAis
predetermined
and
canbe
manipulated.
Inthis
study, this
propertywasexploited by using novel
DNA sub-strates toidentify features of
viral and target DNA structureimportant
forintegrase
recognition, the structural
specificity
of
the
nucleophile,
and the
sequence and structureinvolved
indetermining
the site oftransesterification
during
disintegra-3896
on November 9, 2019 by guest
http://jvi.asm.org/
SUBSTRATE RECOGNITION AND CATALYSIS BY HIV-1 INTEGRASE 3897
tion. Our
results show
that
integrase recognizes
target DNAasymmetrically
and thatseveral
features of the substrates do
notrequire
double-helical
structurefor
recognition by
the
protein.
The
reversibility
of the strand transfer reaction
suggested
thatintegrase
mayfunction
as anenzyme.The
presentstudy
provides
kinetic
evidence
proving
that
HIV-1integrase
is
an enzyme.MATERIALS
AND METHODSEnzymes.
HIV-1integrase
wasexpressed
inEscherichia coli
using
the
T7polymerase-promoter
system(32)
andpurified
asdescribed
previously (36).
T4polynucleotide kinase,
T4 DNAligase,
andrestriction endonucleases
werepurchased
from
NewEngland
Biolabs. Exonuclease-free
Klenowfragment
of
E.coli
DNApolymerase
Iwasfrom United States Biochemical
Corp.
E.coli
exonuclease
III wasfrom
Stratagene
and NewEngland
Biolabs. Terminal
deoxyribonucleotidyl
transferase
wasobtained from
Boehringer
Mannheim.Nucleotides and
oligonucleotides. Deoxyribonucleotides
were
purchased
from Pharmacia
LKB.[ct-32P]dATP,
[a
_32p]
dCTP,
and[_-32P]ATP
withspecific
activities of
3,000
to4,500
Ci/mmol
were obtainedfrom ICN.
[_-32P]3'-dATP
(cordyce-pin 5'-triphosphate)
with aspecific activity
of5,000
Ci/mmol
was
from
NewEngland
Nuclear.
[cx-32P]ddATP
with
aspecific
activity
of3,000
Ci/mmol
wasfrom Amersham.
Acontrolled-pore
glass
column
containing 2',3'-dideoxyadenosine
for
oli-gonucleotide synthesis
wasobtained from Midland Certified
Reagent Company. Oligonucleotides
werepurchased
from
Operon Technologies,
Inc.,
and
werepurified by
electrophore-sis
through
a15%
denaturing polyacrylamide gel
before
use.Disintegration
assayforintegrase
activity.
Oligonucleotides
were
labeled
eitheratthe
5' endwith
T4polynucleotide
kinase
and
[_-32P]ATP
oratthe 3' end with
exonuclease-free Klenow
fragment
of
E.coli
DNApolymerase
Iand[ct-32P]dCTP
(28a).
Unincorporated
radioactive nucleotides
were removedfrom
the labeled
oligonucleotide by centrifugation
through
small
columns
(0.9-ml
bed
volume)
of
Sephadex
G-15
(Sigma).
Preparation
of the
Yoligomer (Fig.
IB)
and its
structurally
related substrates for the
disintegration
assay wasdone
asdescribed
previously
(8).
The novel
DNAsubstrate,
the
dumb-bell
(Fig. IC),
and its
derivatives
wereprepared by heating
the
oligonucleotide
inasolution
containing
10 mMTris-HCl, (pH
7.5),
1 mMEDTA,
and 0.1 M NaClfor
3 minat80°C,
then
slowly cooling
to roomtemperature,
andfinally chilling
to4°C.
The reaction
conditions for the
disintegration
assay wereessentially
the
same aspreviously
described
(8). Typically
ina20-LI
volume,
the
DNAsubstrate
wasincubated with
HIV-1integrase
for
60 minat37°C
in areaction buffer
containing
final concentrations
of 20 mMN-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
(HEPES) (pH 7.5),
10 mMMnCl,,
10mM
dithiothreitol,
0.1 mM3-[(3-cholamidopropyl)-dimethyl-ammonio]-1-propanesulfonate
(CHAPS),
and0.05%
Nonidet
P-40.
The concentrations
ofintegrase
usedthroughout
this
report refer
toprotomers and
notnecessarily
active
enzymemolecules.
Thereaction
wasstopped
by
theaddition of
18 mM EDTA and0.1%
sodium
dodecyl
sulfate.
Thereaction
prod-uctswere heatedat
90°C
for 3 minbefore
analysis by
electro-phoresis
on15%
polyacrylamide
gels
with 7 M urea inTris-borate-EDTA
buffer.
Quantitation
of
theproducts
wascarried outwith
aMolecular
Dynamics
Densitometer
or aMolecular
Dynamics
Phosphorlmager.
Ethylation
interference. Theconditions
ofethylation
wereessentially
the same as those ofSiebenlist
andGilbert
(31a).
Oligonucleotides,
labeledatthe
5'termini,
weresuspended
in50 mM sodium cacodylate (pH 8.0). An equal volume
of
ethanol, saturated with N-nitroso-N-ethylurea (Sigma) wasadded,
and the solution was incubated at 50°C for 30 min. Afterincubation,
the solution was spun through aSephadex
G-15 column (0.9-ml bed volume). The DNA was further purified by electrophoresis on a 15% denaturing polyacryl-amide gel. The labeled,ethylated DNA was then annealed with otheroligonucleotides
toformthe Yoligomer.Disintegration
of theethylated
substrate was carried out asdescribedabove,
and the labeled products were isolated on a 15% native ordenaturing polyacrylamide
gel. The isolateddisintegration
products were resuspended in 0.15 M NaOH and cleaved atethyl phosphates
by heating at 90°C for 30min.
Anequal
volume of formamide
buffer
was added, and the cleavedproducts
were analyzed on a 15% denaturingpolyacrylamide
gel.RESULTS
The dumbbell substrate
undergoes
disintegration mediated by HIV-1 integrase. The dumbbell substrate is made up of asingle oligonucleotide
38nucleotides
long. The folded
struc-ture of the dumbbell substratedepicted
inFig.
IC was based onprevious results obtained
fromstudies
examining
the for-mation ofhairpins
using similar DNA sequences (1, 2). We have not determined the structure of the dumbbell substrateexperimentally
orthe influence of otherfactors,
such as helicalstacking,
on the final structure ofthe
substrate. However,
we believethat most, ifnotall, of the structural features shown
are correctbecause ofthefollowing results. (i)
Thesubstrate
wasrecognized by integrase
andformed
expected products
(seebelow). (ii)
The substratemigrated
at a faster rate on adenaturing (7
Murea) polyacrylamide gel
than oligonucleo-tides ofanidenticallength
butcomposed
ofrandom
sequences(data
notshown). The rapid mobility is
acharacteristic
sharedby
otheroligonucleotides
that form
stablehairpins
(18, 19).
(iii)
Morethan95% of the substrate
migrated
as asingle
band with a sizecorresponding
to that of a monomolecule on anative
polyacrylamide
gel, indicating that species resulting
from
annealing
of
two or moremolecules
were not formed(data
notshown).
The foldedsubstrate
has 5bp of
viral sequence and 10bp
of nonviral
sequence.The nonviral
se-quences,which
arearbitrary,
wereused
astarget DNAand arereferred
to assuch
inthis
report.Disintegration, the
reverseof the strand transferreaction,
is a concerted strandcleavage-ligation
reaction
(8).
The strand
cleavage
occursprecisely
at thejunction
between
the viral and target DNA sequences and iscoupled
to therejoining
of theinterrupted
target DNA sequences.Disintegration
of the dumbbell substratewas there-foreexpected
to form twoproducts:
a14-nucleotide
hairpin
anda24-nucleotide circular molecule.
Thereaction
wasmon-itored
by labeling
either
the5'
or3'
endof
theoligonucleotide.
Inboth5'-
and3-labeled
substrates,
wedetected
the appear-anceof
products
with the
expected
sizes(Fig.
2).
Similar
tothedisintegration
of the
Yoligomer,
the reaction
required
inte-grase andMn2+ (8).
The24-nucleotide
molecule,
after itsisolation from the
gel,
showed theexpected cleavage
patternsafter
digestion
with
HpaII
and HaeIII(data
notshown).
Weverified
that theisolated
product
was a closedloop
by
itsinability
toundergo
chain
elongation
by terminal
deoxyribo-nucleotidyl
transferase
and itsresistance
todigestion by
E.coli exonuclease III under conditions where control substratescorresponding
totheunligated
target DNA partof the dumb-bell molecule were acted onby
these enzymes(data
notshown).
Demonstration of
disintegration activity
with theVOL. 68? 1994
on November 9, 2019 by guest
http://jvi.asm.org/
3898 CHOW AND BROWN
3' EndLabel
A
B
-+ +
-+-_ +
ICGCAGCACTGCGCGC
X
CGTTGCGTTCGAAC CCGACGTCCAGCTGI
13 (3U)
c
G G C C Ce CG G
CC
G GGFIG. 1. Novel syntheticDNA substratesthat mimic the recombi-nation intermediate of retrovirus integration. (A) Diagram of the integrationintermediate. Thick linesrepresentthe U5 and U3 ends of the long terminal repeats of provirus DNA. Thin horizontal lines represent targetDNA.Vertical linesrepresentthe 5bp oftarget DNA between thestaggered breaks. Half-arrows denote the locations of the 3'-OH ends ofDNAstrands. Theregion of the intermediate mimicked by the syntheticDNAsubstrates(see panelsBandC) ishighlighted by the dashed circle. (B) Y oligomer. The substrate is formed by annealing fourseparateoligonucleotides asdescribed previously (8). The DNA sequences of the U5 end of the HIV-1 (HXB2) long terminal repeat (in boldface type) and the target DNA sequences derived from plasmid wAN13 (not in boldface type)are shown. The namesand lengths (in nucleotides) of the oligonucleotides are indi-cated. (C) Dumbbell substrate. The substrate consists of a single oligonucleotide 38 nucleotides long. The twounpaired5' nucleotides ofthe viral DNA, whichare notessential for disintegration(8),are not included in the dumbbellsubstrate. The predictedsecondary structure shown and the numbers of nucleotides in the hairpin loopsarebased on the results of Blommersetal. (2) and Antaoetal.(1). The DNA sequencesof theHIV-1 U5end(in boldface type) in thehairpinstem and the arbitrary DNAsequences representing target DNA (not in boldface type)are shown.
j5
(38)3'
5z
5'EndLabel
_ + - + IN
+ + - Mn2+
(14)
+ U
[image:3.612.318.548.70.255.2]I-A{<
(24)FIG. 2. Disintegration of the dumbbell substrate by HIV-1 inte-grase.Correct strandcleavage andligationof the dumbbell substrate (38-mer)isexpectedtoformtwoproducts:a14-nucleotidehairpinand a 24-nucleotide closed circular DNA. The dumbbell substrate was
labeledatthe 5' end(closedcircle)with T4polynucleotidekinase and [y-_32P]ATP. The DNAsequence ofthe starting oligonucleotide (37-mer) for 3'-end labeling isidenticaltothe first 37 nucleotides of the 38-merdumbbell substrate(Fig.IC),and theterminal nucleotidewas
filled in with exonuclease-free Klenow fragment of E. coli DNA polymeraseI and
[o-32P]dCTP
(closed square).The labeled oligonu-cleotide 38 nuoligonu-cleotideslongwaspurified by electrophoresisthrougha15% denaturing polyacrylamide gel. The oligonucleotide, labeled eitheratthe 5'or3'end,wasincubated with(+)orwithout(-) HIV-l integrase(IN) andMn2+asindicated above thegels. The reactionwas carried out as described in Materials and Methods. The reaction productswereanalyzedon a15% denaturing polyacrylamide gel.As sizemarkers,oligonucleotideswithsequencesidenticaltothose of the twoexpecteddisintegration productsweresynthesized.The endsof the synthetic 24-nucleotide molecule were then ligated with T4 DNA ligaseand usedasamarker for the circularproduct (markersare not
shown in thefigure).
dumbbell substrate
indicated that smallfragments
of viral DNAand target DNA weresufficient for
disintegration.
As
previously observed
with the Yoligomer (8),
the viral DNA sequenceinfluenced
thedisintegration
reaction
cata-lyzed by
integrase. Substitution
of the conserved CAdinucle-otide
attheviral
3'end with TC reduced
disintegration
fivefold(data
notshown).
HIV-1 integrase is
capable
ofturning
over. Weuseddisin-tegration
of thedumbbell
substrate as an assay for kineticanalysis of
HIV-1integrase.
By
determining
the initial
velocity
of the reaction
atdifferent
substrate
concentrations,
wecalcu-lated
theK1,,
for thissubstrate
tobe
140 nM(Fig.
3A).
Todetermine whether
integrase
is
capable
of
turning
over, we incubated theprotein with
a30-fold
molar excess of the dumbbell substrate andmonitored
theformation of
product
over
time
(Fig.
3B).
The concentration of
the dumbbell substrate used in theturnoverexperiment
wassevenfold
higher
than the Km to ensure that the substrate was
saturating
theintegrase.
After 12 h ofincubation,
areaction mixturecontain-ing
33 nMintegrase generated
morethan
600 nMproduct.
This result indicates that
integrase
wascapable of
turning
over, with an apparentkcat (catalytic constant) of
1.8h-'
(Fig. 3B).
The demonstration thatintegrase
couldturn overcatalytically
provesthat HIV-1integrase
is an enzyme.Recently,
Jonesand coworkers(20)
showedthatintegrase
of Roussarcomavirus
is
J. VIROL.
IOn'
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.75.270.84.498.2]SUBSTRATE RECOGNITION AND CATALYSIS BY HIV-1 INTEGRASE 3899
A
.c
E
C',
0
0
I-,
B
C
I.-I
-60
0
0'
2
2
1.5 1 0.5
2 4 6 8 10
V/S( 103,min-1) 12
B0O I
600
400
-200 kcat= 1.8
h1
I I I I
A , ,
-8 12 16 20 24
Time
(h)
A
a. db-v18 (0)
b. db-v36 (A)
C. db-t24(*)
d. db-t38 (-)
B
100
c
0
co
.E
0
U--o
0
FIG. 3. Kinetic experiments using the dumbbell substrate. (A) Determination ofKm.Variousconcentrations(50to1,500 nM) of the dumbbell substrate labeled atthe5' endwereincubated with 50 nM
HIV-1 integrase under standard reaction conditions. Aliquots of the reactionmixturewereremovedat2, 5,and10min after thestartofthe reaction, and the formation of the labeled 14-mer hairpinwas
deter-mined. Therateof the reaction ateach substrate concentrationwas
calculated,andthe Km value of the dumbbell substratewasdetermined withanEadie-Hofsteeplot.v,reactionrate;s,substrate concentration. (B) Turnover of HIV-1 integrase. The dumbbell substrate (1 mM) labeledatthe 5'endwasincubatedwith33nM HIV-1integrase. The formation of the labeled 14-merhairpinproductwasdeterminedat0, 2, 4, 8, 10, 12, 14, 16, 20,and 24 h after thestartofthe reaction.The apparent kcat wasdetermined by calculating the slope of the curve
duringthe first 10 h of the reaction. The apparentkcatobtained islikely
an underestimation, since the calculation method assumes that all integrasein thereaction isenzymatically activeandthat the activeunit ofintegraseisa monomer.
capable
ofturnover in the 3'processing
and strand transfer reactions.Both the viral and target DNA parts
of
thedumbbell
substrate arerequired
forintegrase recognition.
Since the dumbbell substrate contains both viral and target DNAse-quences,itwasnotknown whether its
recognition by integrase
wasmediated
mainly
via the viralortargetDNApartorboth. Thisquestion
was addressedby carrying
out acompetition
experiment
inwhichintegrase
wasincubated withanequimo-larconcentration of labeled dumbbell substrate
(Fig. 4).
Inthepositive control,
a 1- to 10-fold molar excess of unlabeleddumbbell substratewasadded
simultaneously
with thelabeledmolecule and the formation of
product
wasmeasured 25min
after the start of the reaction. As shown in
Fig. 4B,
when unlabeled dumbbell substrate was added so that the overall substrate concentrationranged
from0.7to4times theK,,,,
the unlabeled substratecompeted
with the labeled molecule for the fixedconcentration ofintegrase
and resulted inapropor-80
60
40
20
0
2
4 6 8 10 [image:4.612.101.274.77.366.2]DNA Ratio
(Unlabeled/Labeled)FIG. 4. Neither the viral nor target DNA part of the dumbbell substratecompeteswith the dumbbellsubstrate for HIV-1 integrase. (A) Sequences and predicted secondarystructuresofDNAsubstrates used in the competition experiment. Thepredicted secondary
struc-turesand thenumbers of nucleotidesinthehairpin loopsarebasedon
the results ofBlommersetal.(2) and Antaoetal.(1). Analysis of these DNA substrateson a15% nativepolyacrylamidegel showed that they didnotformbimolecularorhigher-molecular-weight species under the
describedreactionconditions(datanotshown).Insubstratesaandb, the DNAsequencescorrespondtotheU5 end ofHIV-1. Insubstrates
aandc,theDNAsequencescorrespondtothe viral andtarget partsof
the dumbbellsubstrate, respectively. The DNAsequenceinsubstrate d isarbitrary andrepresents target DNA. The 3'-OH endsof DNA strandsaredenotedbyhalf-arrows.The lengths of substratesa,b,c,
and dare18, 36, 24, and 38 nucleotides, respectively. (B) Competition
experiment.Thedumbbell substrate(50 nM) labeledatthe 5'endwas
mixedwith0, 50, 125, 250,or500 nMof unlabeled dumbbell substrate (O)oroneofthe four substrates shown inpanel A. The mixturewas
incubated with 50nM HIV-1 integrase under the standard reaction conditions for25minat37°C.Thereactionproductswereseparated by
electrophoresison a15%denaturing polyacrylamide gel.The forma-tion of the labeled 14-mer hairpin productwas quantitatedwith a
MolecularDynamics PhosphorImager.
tional decrease in
product
formation. When we divided the dumbbell into its viral(db-v18
[Fig. 4A])
and target(db-t24
[Fig. 4A])
DNAparts,neitherfragment
wasefficient incom-peting
with the labeled dumbbell substrate(Fig. 4B).
We attributed this to the small sizeof the dumbbelloligonucleo-tide;
loss ofstabilizing
interactionsprovided by larger
viral and target DNA substrates leadsto an absoluterequirement
for interactions with both viral andtargetDNAsequencesof the dumbbell substrate. When the dumbbell wasseparated
into individual parts, each part was too small toprovide
stable interaction.Indeed,
whenweincreased thelength
of thetarget part of the dumbbell from 24 to 38 nucleotides(db-t38 [Fig.
4A])
ortheviralpart from 18to 36 nucleotides(db-v36 [Fig.
Km= l4OnM
I
VOL.68, 1994
0
4
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.329.563.82.365.2]3900 CHOW AND BROWN
4A]),
weobserved effective competition against the labeled
dumbbell molecule
(Fig.
4B). The observed competition
was not anartifact
causedby
thehairpin
structureof the
compet-itors: similar
results wereobtained with
double-stranded,
blunt-ended
oligonucleotides containing viral
or target DNA sequences(data
notshown). The inefficient binding of db-v18
to
integrase
wasconsistent with its
inability
toundergo 3'
processing andstrand
transfer
reactions, whereas db-v36,
which
was anefficient competitor, showed normal levels of 3'
processing and strand transfer under identical reaction
condi-tions
(data
notshown). The effectiveness of
nonspecific
target DNA as acompetitor is consistent with
previous
reportsshowing
thatoligonucleotides containing
nonspecific
se-quences cancompeteeffectively
against oligonucleotides
con-taining
viral end sequences forbinding
toHIV-1integrase (29,
33,
39).
Asymmetric recognition
of target DNA by HIV-1integrase.
Previous
results from
ourlaboratory suggested that integrase
mayrecognize the
target DNAasymmetrically, i.e., the 5' and
3' sides of the
targetDNA,
inreference
tothe site
ofintegration
of viral DNA,
arerecognized unequally
by
inte-grase.When the
lengths of the
products formed from the
strand
transfer reaction
wereanalyzed, products
with sizes
corresponding
tointegration
nearthe 3'
end
wereabundant,
but
products of integration
events nearthe
5' end were notdetected
(Fig.
SB).
To testthe
possibility
that the
asymmetric
pattern
of
integration
wasdue
toasequenceeffect
imposed by
the
HIV-1U5
DNA(C220/120
[Fig. 5A]),
weexamined the
sites
ofintegration
by using
amodified strand transfer
assaysimilar
tothat described
by Leavitt
etal.
(25).
The unlabeled
U5
DNA wasused
as adonor molecule.
Oligonucleotides,
which
weremade
upof
arbitrary
sequencesandhave
nooverthomology
amongthemselves
or tothe
U5
orU3
region
of
HIVlong terminal
repeat, weresingly labeled
atthe 3' end and
wereused
astarget DNA(Fig.
SA). Integration
of the
processed
U5
DNA(19
nucleotides)
atthe
furthest 3'
position
of
any3'-labeled
target DNA generates a20-nucleotide
product,
whereas
integration
atthe furthest 5'
position
generates a40-nucleotide
product for S21/22, TI 1/12, and T227/228, and
a39-nucleotide
product
for
T45C/45.
Similar
tothe results
obtained with U5
DNA,
using
the described random
oligonu-cleotides
astargetDNA,
weobserved
integration
nearthe 3'
end andthe internal
region of the
DNAmolecules,
but nointegration
occurrednearthe 5'end
(Fig.
5C).
The absence of
an
integration product longer
than 36
nucleotides indicates
thatthe terminal
4nucleotides
atthe 5' end
were notused
as targetsites. This
led us tohypothesize
that
recognition
of
target DNAby integrase
is
asymmetric.
We
studied further the
asymmetric requirement
of
target DNAwith three modified dumbbell substrates
(Fig. 6).
Inthe
standard dumbbellsubstrate
(Fig.
IC),
there
are5bp
of
target sequencesoneach side of the viral
DNA-target
DNAjunction.
In the
modified
substrates, the
5' deleted(Fig.
6, substrate
A)
and3'-deleted
(Fig.
6, substrates
BandC)
substrates, the right
(corresponding
tothe 5'
end)
and left
(corresponding
tothe
3'
end) sides,
respectively,
of thedisrupted
target DNAstrand
wereunchanged, while
theremaining sides
werereduced
to 2bp.
The5'-deleted
substrate showed anactivity
that wasalmost
indistinguishable
from the
standarddumbbell, whereas both of
the
3'-deleted molecules
hadonly
8 to18%
ofthe
standardactivity.
Itispossible
that the poor activitiesof the
3'-deleted
substrates were not due to the deletion of the target DNAbut
to
instability
of the shorthairpin
stem. We did notfavor this
possibility
because substrate C
(Fig. 6)
contains
a sequence(5'-GCGAAAGC)
that forms anunusually
stableminihairpin
with
amelting
temperatureof
76.5°C
(19).
The result obtainedA
HIV-1 U5
C220/120 - -::
RANDOM
T45C/45
S21/22
--T11/12
T227/228 .. .. ....
B C
+ M M
-44
3 -6
::'
30
-U5
+ - + +
21
U5 T227/228 T45/45C T11/12 S21/22
FIG. 5. Absence ofintegrationnearthe 5' end oftargetDNA.(A) Oligonucleotides used in determining sites of integration. C220/120 DNAcontainsDNA sequencesof the U5 end of the HIV-1 (HXB2) long terminalrepeat,andthe conserved CA dinucleotide is indicated inboldfacetype.The DNAsequencesofrandomoligonucleotidesare arbitraryand haveno overthomologyamongthemselvesortotheU5 orU3 regionofHIV-1 longterminal repeat. Synthetic oligonucleo-tides were purified as described in Materials and Methods. The purified oligonucleotides were labeled at the 5' or 3' end, and unincorporated radioactive nucleotides wereremoved by centrifuga-tion through spin columns or electrophoresis in 15% denaturing polyacrylamide gels. The labeled DNAs were annealed with their complementarystrandsbyheatingto80°C,followedbyslowcoolingto room temperature. The numbers inparenthesesindicate the lengths (in nucleotides)of theoligonucleotides.(B)Strand transferassayusing HIV-1 U5 DNA.C220/120 DNAwaslabeledatthe 5' end of the C220 strand using T4 polynucleotide kinase and [_y-32P]ATP. The labeled substrate(0.1 pmol)wasincubated with 1.5pmolof HIV-1integrase, and the reaction conditions were as described in Materials and Methods. (C)Strand transferassayusingrandomoligonucleotides as targetDNA.Oligonucleotides S22, T12, T228,and T45werelabeledat
the 3' end using terminal deoxyribonucleotidyl transferase and
[a-32P]ddATP
beforeannealingwiththeircorresponding complemen-tary strands (panel 5A). Unless otherwise indicated, unlabeled U5 DNA(0.1 pmol)wasincubated with 1.5pmolof HIV-1 integraseinatotal volume of 20p.lfor1minat37°Cbefore the addition of 0.1pmol oflabeled target DNA.The reaction mixturewasthen further incu-bated for 60min at37'C. InpanelsB andC, thesize markerswere
prepared by digesting a5'
32P-labeled
44-nucleotide DNA(38 FLM) with 2p.g
of P1 nucleasepermlfor 5 minat37°C.The absence(-)or presence (+) ofC220/120 DNA (U5) and HIV-1 integrase (IN) is indicated above thegels.Thenumbers between thetwopanelsdenote the lengths(in nucleotides)of the size markers(M).with
the5'-deleted
substrateindicated
that for fullactivity
the 5' side of thetarget
DNAwasdispensable.
Thelarge
difference inactivity
between the 5'- and 3'-deleted substrates is consis-tentwith thehypothesis
that therecognition
oftargetDNA isasymmetric.
Mismatches
in the target DNA part of adisintegration
J. VIROL.+ - + +
- + +
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.312.549.76.344.2]SUBSTRATE RECOGNITION AND CATALYSIS BY HIV-1 INTEGRASE 3901
5'-Deleted
Activity
-
+
(% Control)
A i; T
GGCN
3-Deleted
B
c G
T A!
GC
~~~~~8
TOGc GOCGG
c
'AT
':G C:.GC 18G
[image:6.612.68.307.71.427.2]39
A T 5'FIG. 6. Differential effectondisintegrationofdeleting the 5'or3' side of thetargetDNAof the dumbbell substrate.SubstratesA, B,and Cweremodified from the standard dumbbell molecule(Fig. 1C).The viral DNA sequences (in boldface type) and target DNAsequences
(not inboldface type) are shown. The 5'- or3-deleted molecule (5 nM)waslabeledatthe 5' end andincubated with(+)orwithout(-)
75 nM HIV-1 integrase under the standard reaction conditions.The formation of the labeled 14-mer hairpin was determined, and the activities of the modifiedsubstrateswereexpressedaspercentagesof that obtained usingstandard dumbbell as acontrol. Under identical
conditions,85% of the labeled standard dumbbellwasconvertedtothe labeled 14-mer hairpin molecule. For each set of reactions, the positionsof the substrate(open arrowhead)anddisintegration product (solid arrowhead) are shown.
substrate showed analogous asymmetric effects on
disintegra-tion. The mismatched substrates were modified from the Y oligomer, rather than the dumbbell substrate,tominimize the effect of a mismatch on the overall helical structure of the DNA molecule. We founddrastic differences indisintegration activities, depending on whether the mismatch was located
upstreamordownstream of the viral DNAjoining site(Fig. 7).
Theactivity obtained with the substrate containingamismatch
downstream of the joining site was 10-fold higher than that
obtained with the substratecontainingamismatchupstreamof
thejoiningsite.
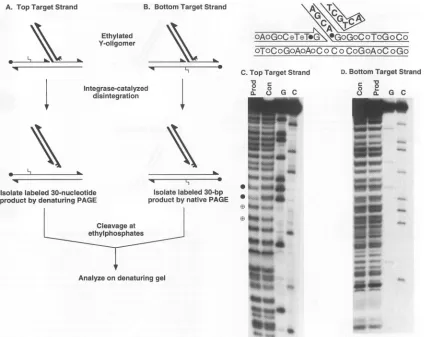
Asymmetric recognition of target DNA at the integration site was further corroborated by an ethylation interference
experiment (Fig. 8). Ethylation interference has been used recentlytoprobe the positionsonviral DNAthatarerequired
5'
Target Mismatch
A
Activity
(%Control)
- +
-.4
50
3 Target Mismatch
IK
*-5
[image:6.612.332.556.72.291.2]S04 FIG. 7. Differential effect on disintegration of a mismatch located upstream ordownstream of the viral DNA-target DNAjunction site. The substrates were modified from the Y oligomer (Fig. 1B) and contained a mismatch in the target DNA downstream (5' target mismatch) or upstream (3' target mismatch) of the viral DNA joining site. Except for the mismatched nucleotides shown, the remaining DNAsequencesand the lengths of the strands are identical to those of theYoligomer. The labeled, modified substrateswereprepared in the same manner asthatof theYoligomer (8). Thick lines represent viral DNA, andthin lines represent target DNA. A solid circle denotes the location of the
5'
32p
label, and ahalf-arrow denotes the3-OHend of the labeled strand. The formation of the disintegration productswas determined, and the activities of the modified substrates were ex-pressedaspercentagesof that obtained using the standardYoligomer (Fig. 1B) as a control. Under identical conditions, 40% of the labeled standard Yoligomerwasconverted to the labeled 30-mer product. The absence(-) and presence (+) of integraseareindicated. For eachset of reactions, the positions of the substrate (open arrowhead) and disintegration product (solid arrowhead) areshown.for
integrase-mediated
strandtransfer
in vitro(6).
Treatmentof
DNAwith
N-nitroso-N-ethylurea
attachesethyl
groups to thesugar-phosphate backbone (31a).
Inthis study,
weethyl-ated the
target partof the
Yoligomer
andmapped
thesites
atwhich
ethylation affected
theability of integrase
torecognize
the
modified
DNA as asubstrate
(Fig.
8A andB).
DNAethylation
at twopositions
inhibiteddisintegration, while
ethy-lation
at twootherpositions enhanced the reaction
(Fig. 8C).
These
positions
wereclustered at andimmediately
upstreamof
the
junction between
viraland
target DNA. Wedid
notdetermine, however,
whether DNAethylation affected
disin-tegration directly by interfering with integrase
binding
orindirectly by altering the
structureof the substrate. The
earlier
experiment
carried outby
Bushman andCraigie (6)
wasdesigned
todetermine functional
contacts onviral DNA. The substratethey
used did notpermit
detection ofpossible
contacts between
integrase
and the upstream target DNA.However,
they
found thatethylation
of the target DNAat theposition
immediately
downstream of the viraljoining
site inhibited strandtransfer.
We do not believe that the down-stream site represents an essential contact withintegrase
becausewe didnotdetect suchan interference for
disintegra-tion and because strand transfer events can occur one base VOL.68,1994
on November 9, 2019 by guest
http://jvi.asm.org/
3902 CHOW AND BROWN
B. BottomTarget Strand
Ethylated Y-oligomer
Ix.._L
grase-catalyzed
isintegrationIsolatelabeled30-bp
product by native PAGE
Cleavage at ethylphosphates
Analyzeondenaturing gel
oAoGOC¶Te
GoGoCoToGoCo
OTOCoGoAoAoCoCo
CoGoAoC
oGoC.Top Target Strand
*0
U- 0
CD hqi0,
A__L
;.
D.Bottom Target Strand
0 C
on.'6 G C
_ ._ _,,i:
*|'.'
. ....
__
*: _
._. _
._. i_
_: _
_: _E
*.
_
.
*i_twg
_ _.
...
#-S8 :4iidr: *_ U
_t: 'SlgtH, _ _i
t_
_ S.2:L::: ::,
aW
FIG. 8. Probingintegrase-DNA interactions by ethylation interference. The oligonucleotides usedtopreparethe DNA substrateswereidenticalto thoseof theYoligomer (Fig. 1B).Thezigzaglines representethylgroups.For theothersymbols,seethelegendtoFig.6. The top targetstrandrefers tothe targetstrand thatisjoinedtothe viralDNA;the bottom target strand referstothecomplementof the top strand.(A) Ethylatedsubstrate for detection ofinterferenceonthetop targetstrand. TheTi oligonucleotidewaslabeledatthe 5' end. The labeled Ti and unlabeledV1/T2DNAwere
ethylated by using N-nitroso-N-ethylureaasdescribedinMaterials and Methods. The ethylated strandswereannealedwith untreatedV2andT3DNA toformthe Yoligomeraspreviously described (8).Each Yoligomer had,onaverage,lessthanoneethylgrouprandomly placedonthesugar-phosphate backboneof the Ti orV1iT2 strand.Disintegrationwasthencarriedoutunder thestandardreactionconditions.Alabeled30-nucleotideDNA,formed afterintegrase-catalyzed disintegration,wasisolatedby electrophoresison a15%denaturingpolyacrylamide gel. (B) Ethylated substratefor detection ofinterferenceonthe bottomtargetstrand. TheT3 oligonucleotidewaslabeledatthe5' end and ethylatedasdescribedinMaterialsandMethods. The
labeled, ethylated strandwasthenannealed witholigonucleotides Ti, V2, and V1iT2toform theYoligomer. The disintegration product,a30-bpDNA fragment,wasisolatedbyelectrophoresisona15% native polyacrylamide gel. In both panels A and B, the isolated disintegration productswerecleaved
atthepositions of ethylphosphates by heating in 0.15MNaOH. The NaOH cleavage productswereanalyzedona15% denaturingpolyacrylamidegel.
PAGE,polyacrylamide gel electrophoresis. (C) Integrase contactsonthe toptargetstrand. Lane Prod containsproducts of NaOH cleavageof the disintegration product made with the ethylated Y oligomer shown in Fig. 8A. Lane Con contains products of NaOH cleavage ofanunreacted, ethylated
30-nucleotideDNA(Tl-2)that isidenticalin DNAsequencetothat of thetoptargetstrand formed afterdisintegration. Therefore, theDNAsequence
ofTl-2 strand is complementarytothat ofT3. Lanes G and C contain products of the Maxam-Gilbert sequencing reactions performedontheunethylated Tl-2 DNA (26a). (D) Integrasecontactsonthe bottomtargetstrand. Lane Prodcontains products of the NaOH cleavage of the disintegration product madewiththeethylatedYoligomer shown in Fig. 8B. Lane Con contains products of NaOH cleavage ofanunreacted, ethylatedT3 DNA. Lanes G and
Ccontainproducts of theMaxam-Gilbert sequencing reactions performedonthe unethylated T3 DNA. NaOH treatmentofapopulation of the unreacted, randomlyethylated, and end-labeled DNA yieldsaladderof bands after separationon adenaturinggel. A position atwhichethylation interfereswith substrate recognition by integrase is recognizedas achange in band intensity by comparing the NaOH cleavagepatternsobtained with thedisintegration product and theunreacted substrate DNA. Positions where ethylation inhibited product formation (0), where ethylation enhanced product formation(G3),andwhereethylation didnotappreciably alter productformation (0)areshown. Only the relevantpartof theYoligomer is shown.
from
the 3'
end of the joining
strand oftargetDNA (Fig.5),indicating that integrase does
notabsolutely
require contacts withthephosphodiester bond immediately
downstream from thesite ofintegration.
Relaxed structural specificity for the nucleophilic group
during
disintegration. The
mechanism of 3' processing
and strand transfermediated
by integrase is
a one-stepin-line
transesterification
(12).
Theattacking nucleophile
in strand transfer is the 3'-OH ofDNA;
in 3'processing,
anumber ofnucleophiles,
such as water,glycerol,
and certainaliphatic
alcohols,
canbeused(12, 38). Disintegration
normally involves anucleophilic
attackby
the 3'-OH of target DNA on thephosphodiester
bond between the viraland
target DNA se-quences(8).
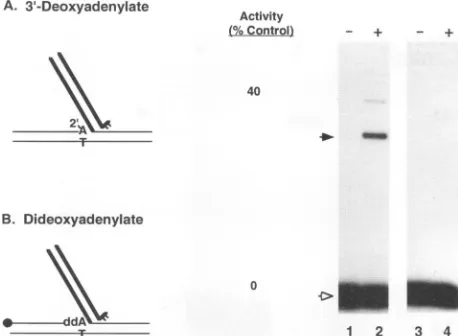
A 2'-OH group,however,
couldreplace
theA. Top Target Strand
'~~~~~~~~~~~~~~~~~~~
Integ
di
Isolatelabeled30-nucleotide
productby denaturingPAGE
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.92.517.69.406.2]SUBSTRATE RECOGNITION AND CATALYSIS BY HIV-1 INTEGRASE 3903 A.
3'-Deoxyadenylate
Activity (%Control)
40
B. Dideoxyadenylate
0
--d
[image:8.612.68.297.76.244.2]FIG. 9. Nucleophile usage during disintegratio
strates were modified from the Y oligomer. For
symbols,seethe legendtoFig. 7. In panelAand la end of the TI oligonucleotide was labeled with th
nucleotidyl transferase and
[r-32P]3'-dATP
(corcphate[A-2']).The labeledoligonucleotidetherefori
instead ofa3-OH endof normal DNA. The banc
the product and those between the product and s
products from reintegration ofaviral DNA end lil
gration. In panel B and lanes 3 and4,theterminal r
32P-labeled targetstrandwas2',3'-dideoxyadenylat
3'-OH
group,butwitha 2.5-folddecreaseinaThe target DNA product after disintegratioi
strate presumably contained an unusual 2'-5
linkage. The essential role of the -OH grol
confirmed in Fig. 9B in which substitution nucleotide with a 2',3'-dideoxy analog comj
the disintegration activity that rejoined the ir strand. Integrase, however, could mediate an age reaction at the viral DNA joining site containing substrate usingwaterorvarious
ale
philes (data notshown).
Reaction site determinants and effects of double-helical structureon disintegration. A
ously thatintegrasecanmediatedisintegratior
in which the viral sequences are single stran
disintegration also occurs, albeit with a sixf
activity, using a substrate in which the highl
dinucleotide at the viral 3' end is replacedv
results indicate that integrase can recognize
nothave a B-form structure and that substra thanthe CA dinucleotide playa rolein
deterr
transesterification. We introducedbasedeleti
nearthe viral DNA-target DNAjunction site
DNA substrate and examined their effects o
Substrates containing a 1- or 2-nucleotide d
end of the interrupted target strand could st
tegration (Fig. IOA).The disintegration prod deletion substrate was isolated from the gel.
wasconfirmed by DNA sequencing. Sequence
that the cleavagestill occurred precisely 3' to
otide and that cleavage was coupled to th( interruptedtargetstrand (datanotshown). Sii ligation reaction is mediated by a nucleophi
3'-OH on the phosphodiester bond3' tothe (12), the activity observed with these deletic plies that there must be a considerable degre
the catalytic site or that the DNA in this
region
can be + _ +distorted
toallow
correcttransesterification
tooccur.In the other group of modifications, wefound that
insertion
of a base pair beyond the CA dinucleotide at the viral end decreased the activity only twofold;insertion of2 bpdecreased
the activity
to around10% of
that seenwith
the standard Y oligomer (Fig.10B).
Analysis of the disintegrationproducts
from these insertionsubstrates provided
someinteresting
observations(Fig.
lOB). When the insertion was I bp, a majority of the cleavage-ligation events took place at the phosphodiester bond immediately 3' to the CAdinucleotide,
i.e., between the terminal Anucleotide
ofthe viralsequence
and the inserted nucleotide, while asmallfraction occurred
atthe
branchpoint,
i.e., the phosphodiester bond between the _ _ inserted nucleotide and its 3' neighbor in the target DNA (Fig.B10B).
When the
insertion was
2bp, the amount of activity
at 1 2 3 4the branch
point
remained
the same as that of1-bp
insertion,
but the frequency of cleavage-ligation
eventsoccurring at thene
The DNA sub- bondimmediately
3' to the CAdinucleotide decreased,
sothat
ines
xand
2,
the 3 theactivities
at these twosites
becamecomparable.
However, erminal deoxyribo- no product was detected with a length that corresponded tolycepin
5-triphos-
disintegration
atthe
phosphodiester
bond between
the 2*e
had a 2'-OH end insertedbp.
Theability
ofthe modified substrates toundergo
is appearing above correct disintegration againdemonstrated the tolerance of
the,ubstrate
represent catalytic site to structural distortion. This result shows that berated by disinte- during disintegration the viral DNAsequence
is themajor
tucleotide
of the 5' factor indetermining
the site oftransesterification. When
thete
(ddA). position of the viral sequence isshifted
by baseinsertions,
there is residual activity that is dependent onthe
structure, namely, the branch point.Lctivity
(Fig. 9A).n of such a sub-
DISCUSSION
phosphodiester
up of DNA was During the retrovirus life cycle,
integrase
isrequired
for
of the terminal integration of retrovirus DNA into the host cell genome. Inpletely
abolished vitro, integrase alone issufficient
to carry out the major stepsiterrupted
target of retrovirusintegration, namely,
3'processing
and strand inefficient cleav- transfer. The strand transfer reaction that results in the of the dideoxy- formation of an integrationintermediate
isreversible.
Using
acohols
as nucleo- modelsubstrate termed
a Yoligomer, prepared
byannealing
four separateoligonucleotides
to form a structure thatmimics
distortion of the the integrationintermediate,
we showedpreviously
thatHIV-1
{e
showed previ- integrase efficiently and precisely resolves the intermediate using substrates into its viral and target parts, a process we termed disintegra-ded (8). Correct tion (8). In the present study, we devised a novel DNA old reduction in substrate called adumbbell.
The substrate consistsof
asingle
ly conserved CA oligonucleotide containingsequences
that can form a second-vith TC (8). The ary structureresembling
the integrationintermediate.
Al-DNA that does though thestructure
of thedumbbell substrate
has notbeen
tefeatures otherdetermined experimentally,
for reasonsdescribed
earlier (seemining
the site of Results), we believe that the structure shown in Fig.IC
islikely
ions or insertions correct. We showed that with such asubstrate,
integrase
can in a Y-oligomer mediatecorrect
andefficient
disintegration.
Experiments
using
ndisintegration.
thedumbbell
substrate,
the Yoligomer,
andtheir
structural
eletion at the 3'variants provided
information
on thefunctional
andenzymatic
:ill
support disin- properties of HIV-1 integrase.uct ofthe
1-base
Compared
with the Yoligomer,
the dumbbell substrate has and itsidentity
theadvantages
ofbeing small,simple,
and easy to prepare. The analysis showed dumbbell substrate also offers a meansof
studying
aspects of the CAdinucle-
integrase-catalyzed disintegration
that cannot beeasily
ad-e joining of thedressed
byusing the Yoligomer.
Asshownby
thecompetition
nce
thecleavage-
experiment, because
of its small size,recognition
of theilic
attack of the dumbbell substrate required interactions ofintegrase
with both CAdinucleotide
the viral andtarget
DNA partsof
the substrate. This is in nsubstrates
im-contrast
with the larger Yoligomer
wherein either the viral
or eofflexibility
at target DNA part of thesubstrate
contributes
substantially
to Vot,.68? 1994on November 9, 2019 by guest
http://jvi.asm.org/
3904 CHOW AND BROWN
A.
Deletion
1-Base
Gap
Activity
(%
Control)
41
_ + - +
J.
13
-~~~~~~~0
1 2 3 4
B.
Insertion
_ + _
\\
1-bp
2-T
to .
2-bp 2
D 0.
Site1:
5
Site 2:
35
Sitel:
5
Site 2:
5
1 1 2 3 4
FIG. 10. Effects of nucleotide deletion or insertionon disintegration. All of the DNA substrates are modified from the Y oligomer. For
explanation of thesymbols,seethelegendtoFig.7.(A)Deletion. The modified substrateswereprepared usinga5'-end-labeledtargetstrand that is1 (lanes 1 and2)or2nucleotides(lanes3 and4)shorterthan that of the standard Yoligomer (Fig. 1B).Theresulting unpairednucleotides
aredenotedbyvertical lines.(B)Insertion.The location of the conserved CA dinucleotideintheviral DNA strand and the 1-(lanes1 and2)or
2-bpinsertion(lanes3 and4)after the CA dinucleotideareindicated. Thetwosites of transesterificationaredenotedbyshadedarrows.For each setofreactions,thetop and bottomdisintegration products correspondtotransesterificationoccurringatsites 2 and 1, respectively.
integrase recognition (36). Several
recentstudies
on mutantintegrases
indicatethat
certainintegrase
variantsareunabletocatalyze
3'processing
and strand transfer but arecapable
ofcarrying
outdisintegration (7, 11, 26, 34, 36, 37).
Weinterpret
this observation as an indicationthatinteraction
ofintegrase
with eitherthe viral ortarget DNApartofthe Y
oligomer
is sufficient for substratebinding.
Therefore,
thedifferential
abilities ofmutantintegrases
touseYoligomer
anddumbbell asdisintegration substrates
can serve as anindicator
for variantproteins
that aredefective in recognizing either
theviral
ortargetDNA
(36).
Another
distinguishing
feature between the dumbbellandYoligomer
wasthat HIV-1integrase
showedturnoverusing
the dumbbell substrate(see
below)
but not with theYoligomer.
Toexplain
thedifference,
wehypothesize
that indisintegra-tion,
release of reactionproducts
isordinarily
therate-limiting
step. Stable association of
disintegration products
withinte-grase occludes a DNA
binding
site and prevents turnover.There
isevidence that HIV-1integrase
forms stable protein-DNAcomplexes
during
3'processing,
strandtransfer,
anddisintegration reactions;
the formation ofstablecomplexes
isdependent
onthepresenceofthetwounpaired
5'nucleotides 2-BaseGap
I I
J. VIROL.
... ...ft.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.131.485.72.531.2]SUBSTRATE RECOGNITION AND CATALYSIS BY HIV-1 INTEGRASE 3905
of the viral
DNA(lOa).
However, the disintegration products
of the dumbbell
substrate, perhaps
because of their small sizes
and the
absence of the
twoterminal
unpaired 5' nucleotides,
do
notform stable
complexes with the enzyme and thus allow
the
protein
to turn over.We
tested the
hypothesis
that
integrase
recognizes target
DNAasymmetrically. Depending on the position of target
DNArelative
tothe viral sequences, dumbbell substrates of
identical
lengths had
a5-
to10-fold difference in activity. A
similar
difference in
activity
wasobserved when
wecompared
the effect of locations of mismatches
using Y oligomers. The
cleavage pattern obtained from the ethylation interference
experiment also pointed
toessential
interactions between
integrase and target
DNAupstream,
but not downstream, of
the viral
DNA-target
DNAjunction site. The results are
consistent with the
hypothesis
that the target
DNAupstream
of the viral
joining
site
plays
amoreimportant
role than the
downstream target
DNAin
interacting
with
integrase.
Incorroboration with this observation is the result obtained from
strand transfer reaction
showing
that
the 5'-terminal
region of
target
DNA wasrarely
used
as anintegration
site.
Experimen-tal
evidence is consistent with the notion that the
natureof
target
recognition
is similar in the forward and
reversedirec-tions of the
integration
reaction. Our result therefore suggests
that
in
mediating integration, integrase
interacts strongly with
about
4bp
of target
DNAupstream
of the site of
integration,
but
contactswith target
DNAdownstream
areof
less
impor-tance.Inthe in vivo reaction where
twoviral ends
arejoined
atstaggered
sites
onopposite strands,
these
key
contactswith
target
DNAwould flank the
twojoining sites,
such
that contactswith target
DNAwould span
atleast 13
bp (Fig. 11).
Data
obtained from
sequencing integration
targets
used
by
murine leukemia virus
(27a)
and avian
myeloblastosis
virus
(16) provide
consistent evidence of sequence bias in target
DNAimmediately flanking
the
twoviral
DNA-target
DNAjunction sites,
while
nosequence
bias is observed in the
staggered
region
between
thejunction
sites.
Therefore, the
positions
of the nucleotides that show sequence bias
arein
good
agreement
with the
protein-DNA
contactsrevealed
by
the
ethylation
interference
experiment.
The
findings
that
integrase
could
catalyze disintegration
onsubstrates that contained base
substitutions, deletions,
inser-tions,
ormismatches
suggested
that the
catalytic
site of
inte-grase
waspromiscuous
and
could tolerate helical distortion of
DNAsubstrates.
Integrase
normally
usesthe 3'-OH group of
DNA as anucleophile
for
disintegration,
but
a2'-OH group
could substitute for the 3'-OH group of
DNA asthe
nucleo-phile.
Moreover, the
position
of the
attacking
-OH group
relative
tothe
junctional phosphodiester
bond
was nothighly
restricted.
Wealso found
that,
in addition
tothe
DNAsequence
atthe viral
end,
the
structureof
the branched
substrate
played
arole in
determining
the
site of
transesteri-fication. Site-directed
mutagenesis
studies of HIV-1
integrase
showed that substitutions of conserved amino acids in the
central
region,
the
D,D(35)E domain,
render the
protein
inactive in
carrying
out3'
processing,
strand
transfer,
and
disintegration (11, 23, 26, 37).
This result suggests
that theD,D(35)E
motif defines
asingle catalytic
domain that is
responsible
for all the
catalytic
activities of
integrase.
Ifthis is
indeed
thecase, the
samecatalytic
domain that mediates
ahydrolytic cleavage during
3'
processing
is also involved
inmediating
transesterification
during
strandtransfer
anddisin-tegration.
Since the substrates in these different reactions
arelikely
topose
different structural constraints
onthe
nucleo-phile
and the
phosphodiester
bond
involved in
breakage
andjoining,
wepropose that
accommodation of these differences
syn -
Configuration
5'
trans
-
Configuration
5'1
FIG. 11. Amodelshowing key sites of HIV-1 integrase contact with target DNA. Dashed lines represent thepositions ofintegrase pro-tomers. Open and filled symbols denote the positions at which ethylationinfluencesdisintegration.Filledsymbolsdenote the sites of viral DNAjoining, whicharestaggered by 5 bp. If we assume thatjoint integrationoftwoviralends in vivo involves twosymmetrically placed integrase protomers, the interaction between an integraseprotomer and target DNA can have two possible configurations. In the syn configuration, the integrase protomer that catalyzes the cleavage-ligationreaction on atarget strand makescontactswith target DNA immediatelyupstreamonthesamestrand. Inthetransconfiguration, the integrase protomer that catalyzes cleavage-ligation on a target strand makescontactwith thecomplementarystrandacrossthemajor groove.Filled and open circles represent thejoiningandcontactsites, respectively, of one protomer; whereas filled and open rectangles represent the joining and contact sites, respectively, of the other protomer. Eventhough the contact sitesare located onboth target strands acrossthe majorgroove,webelieve that for each viral DNA end, in both syn and transconfigurations, thecontact siteson target DNA thatare mostcritical for integraseinteraction are those
posi-tionedimmediatelyupstreamof the viralDNA-targetDNAjunction. This is basedonthe observations that the strand transfer reaction in vitrocan occur onebase from the 3' endof the target DNA and that disintegrationofaYoligomerwas notaffectedbyethylationof target DNAatpositionsdownstreamof the viralDNA-targetDNA
junction
site.
may be best
accomplished by
locally distorting
thedouble-helical
structureof both
viral and target DNA sequences, sothat the
structureof the substrate
in the active site neednotbedetermined
by
the
secondary
structureof the free substrate.
Thus, the
ability
of
integrase
totolerate various modifications
on
the
DNAsubstrates may be
areflection of
asingle catalytic
site
being required
tocatalyze
twoor moredistinct reactions.
Ithas
beenestimated
that there
areabout 100
copies
ofintegrase
pervirion
(35),
and
presumably
in vivointegrase
VOL.68, 1994


![FIG. 5.withcomplementarypurifiedorunincorporatedpolyacrylamidelongroomstrandsubstratetidespresenceintionofarbitrarybatedpreparedOligonucleotidesDNAHIV-1andtheMethods.targetthetarytotal(inindicatedDNA[a-32P]ddATP boldface U3 labeled Absence of integration n](https://thumb-us.123doks.com/thumbv2/123dok_us/1293472.82296/5.612.312.549.76.344/withcomplementarypurifiedorunincorporatedpolyacrylamidelongroomstrandsubstratetidespresenceintionofarbitrarybatedpreparedoligonucleotidesdnahiv-andthemethods-targetthetarytotal-inindicateddna-boldface-labeled-absence-integration.webp)