Copyright © 1998, American Society for Microbiology
The Herpes Simplex Virus Type 1 Cleavage/Packaging Protein,
UL32, Is Involved in Efficient Localization of
Capsids to Replication Compartments
CARMELA LAMBERTI
ANDSANDRA K. WELLER*
Department of Microbiology, University of Connecticut Health Center,
Farmington, Connecticut 06030-3205
Received 25 September 1997/Accepted 10 December 1997
Six genes, including UL32, have been implicated in the cleavage and packaging of herpesvirus DNA into
preassembled capsids. We have isolated a UL32 insertion mutant which is capable of near-wild-type levels of
viral DNA synthesis; however, the mutant virus is unable to cleave and package viral DNA, consistent with the
phenotype of a previously isolated temperature-sensitive herpes simplex virus type 1 mutant, tsN20 (P. A.
Schaffer, G. M. Aron, N. Biswal, and M. Benyesh-Melnick, Virology 52:57–71, 1973). A polyclonal antibody
which recognizes UL32 was previously used by Chang et al. (Y. E. Chang, A. P. Poon, and B. Roizman, J. Virol.
70:3938–3946, 1996) to demonstrate that UL32 accumulates predominantly in the cytoplasm of infected cells.
In this report, a functional epitope-tagged version of UL32 showed that while UL32 is predominantly
cyto-plasmic, some nuclear staining which colocalizes with the major DNA binding protein (ICP8, UL29) in
replication compartments can be detected. We have also used a monoclonal antibody (5C) specific for the
hexon form of major capsid protein VP5 to study the distribution of capsids during infection. In cells infected
with wild-type KOS (6 and 8 h postinfection), 5C staining patterns indicate that capsids are present in nuclei
within replication compartments. These results suggest that cleavage and packaging occur in replication
compartments at least at 6 and 8 h postinfection. Cells infected with the UL32 mutant exhibit a hexon staining
pattern which is more diffusely distributed throughout the nucleus and which is not restricted to replication
compartments. We propose that UL32 may play a role in “bringing” preassembled capsids to the sites of DNA
packaging and that the failure to localize to replication compartments may explain the cleavage/packaging
defect exhibited by this mutant. These results suggest that the UL32 protein is required at a step distinct from
those at which other cleavage and packaging proteins are required and may be involved in the correct
localization of capsids within infected cells.
During infection of cells with herpes simplex virus type 1
(HSV-1), the large concatemeric products of DNA replication
are cleaved to unit length and packaged into preassembled
capsids. Capsids are icosahedral structures composed of 150
hexons and 12 pentons. Three types of capsids (A, B, and C)
can be isolated from infected cells by velocity centrifugation
(20). C capsids contain the viral DNA genome; B capsids
contain the scaffolding protein; and A capsids contain neither
DNA nor the scaffolding protein. Pulse chase experiments with
another alphaherpesvirus, equine herpesvirus 1, indicate that
at least some B capsids can package DNA and mature into
infectious virions, while A capsids cannot (46). By analogy with
the bacteriophages, these results suggest that B capsids
repre-sent procapsids which are intermediates in the packaging
pro-cess. However, a new intermediate in the assembly process has
recently been identified (41, 62). These newly identified capsid
forms observed in in vitro assembly extracts have the same
protein content as B capsids but are more spherical; these
capsids are unstable and adopt the more angular form
charac-teristic of B capsids after prolonged incubation in vitro. These
results suggest that the unstable spherical forms may represent
the true procapsid intermediate (41, 62).
In many bacteriophages, the procapsid contains at least
three essential components: an icosahedrally arranged protein
shell, an internal scaffold, and a dodecameric ring called the
portal vertex through or around which the phage DNA is taken
up (8, 11, 18). For HSV-1, the outer shell is composed of four
proteins: the major capsid protein, VP5; a small protein bound
to hexons, VP26; and a triplex structure made up of
heterotri-mers of VP19C and VP23 (reviewed in reference 56). VP24,
VP21, and VP22a are found in the interior of the capsid and
are encoded by overlapping genes UL26 and UL26.5; VP21
and VP22a are present in B but not A or C capsids and are
considered to make up the internal scaffold (reviewed in
ref-erence 56). Although bacteriophages contain a portal vertex,
no such structure has been observed in HSV-1 capsids.
Wheth-er the hWheth-erpesviruses have a unique portal vWheth-ertex through which
viral DNA is taken up is unclear; it is possible that this type of
unique vertex is only needed in viruses which have a tail.
Cap-sids indistinguishable from those isolated from HSV-1-infected
cells have been observed in extracts from insect cells infected
with recombinant baculoviruses bearing HSV-1 capsid genes
(42, 60). Therefore, it is clear that these proteins are sufficient
for capsid assembly in vitro; however, it is not known whether
capsids formed in vitro are competent for DNA uptake. It is
possible that minor components of capsids play important
roles in genome encapsidation.
In addition to the capsid proteins, at least six genes are
essential for the encapsidation of viral DNA: the UL6, UL15,
UL25, UL28, UL32, and UL33 genes. Temperature-sensitive
(ts) strains with mutations in these genes have similar
pheno-types, in that viral DNA can be replicated but not cleaved and
packaged (1, 2, 4, 6, 48, 51, 54, 55, 66). Strains with null
* Corresponding author. Mailing address: Department of
Microbi-ology, University of Connecticut Health Center, 263 Farmington Ave.,
Farmington, CT 06030-3205. Phone: (860) 2310. Fax: (860)
679-1239. E-mail: [email protected].
2463
on November 9, 2019 by guest
http://jvi.asm.org/
mutations in the UL6, UL15, UL25, UL28, and UL33 genes
have been isolated and characterized, thereby confirming the
roles of these genes in cleavage and packaging (5, 27, 37, 45,
59, 68). Despite the identification of these required genes, the
mechanism by which viral DNA is cleaved and packaged is not
understood, nor has the role of any of the gene products been
determined. The UL6 and UL25 proteins have been detected
in A, B, and C capsids as well as in virions (3, 28, 37, 44);
however, the precise role of these two proteins in capsids
remains to be determined.
A ts UL32 mutant, tsN20, defective in cleavage and
packag-ing, has been reported previously (51). Because mutants with
lesions resulting in temperature sensitivity are often prone to
problems associated with incomplete penetrance at the
non-permissive temperature, we isolated a UL32 insertion mutant,
hr64. Characterization of hr64 confirms that UL32 is essential
for cleavage and packaging. Previous studies demonstrated
that UL32 localizes to the cytoplasm of infected cells (13). We
have used a functional epitope-tagged version of UL32 to
confirm that in infected cells, this protein is mainly
cytoplas-mic, although some nuclear staining was observed.
HSV-1 DNA replication occurs in globular nuclear domains
termed “replication compartments” initially identified by ICP8
(UL29) staining patterns in an immunofluorescence assay (49).
All seven replication proteins have now been localized within
replication compartments (10, 24, 29–31, 43) as has regulatory
protein ICP4 (26, 50). Ward et al. have recently reported that
at late times after infection (18 h), capsids accumulate in the
nucleus in regions distinct from replication compartments (64).
These authors suggest that these regions represent assembly
stations in which DNA is packaged. We report herein,
how-ever, that at 6 and 8 h postinfection, capsids colocalize with
ICP8 in replication compartments. This suggests that at these
early times, cleavage and packaging occur within replication
compartments. Furthermore, we report that in cells infected
with the UL32 mutant virus, capsids are distributed throughout
the nucleus, accumulating in regions outside the replication
compartments. This suggests that UL32 may play a role in the
efficient localization of capsids in infected cells.
MATERIALS AND METHODS
Cells and viruses.African green monkey kidney cells (Vero; American Type Culture Collection, Rockville, Md.) were propagated and maintained as previ-ously described (65). HSV-1 strain KOS was used as the wild-type virus and the parental virus for the isolation of the UL32 host range mutant. tsN20, a ts UL32 mutant, was generously provided by Priscilla A. Shaffer (University of Pennsyl-vania, Philadelphia). hr74 containing a lacZ insertion in the UL6 gene was previously described (27). KUL25NS, a mutant with an in-frame stop codon in the UL25 gene, was generously provided by Fred Homa (Pharmacia and Upjohn, Kalamazoo, Mich.) (37).
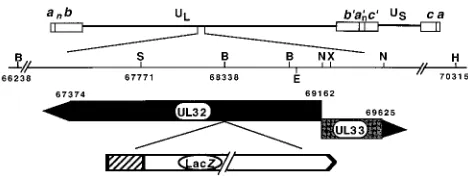
Plasmids.The pAPV vector contains the inducible HSV-1 promoter from the ICP6 gene and the simian virus 40 (SV40) polyadenylation sequence on either side of a BamHI site into which genes to be expressed can be cloned. pAPV was generated by ligating the HindIII-XhoI fragment containing the ICP6 promoter into pUC118 digested with HindIII-SalI. A BamHI-to-BclI fragment from SV40 containing the poly(A) site was then inserted at the BamHI site. pAPVUL6, containing the UL6 sequences from nucleotide (nt) 15103 to nt 17323, under the control of the ICP6 promoter, was generously provided by Fred Homa. p1BD1 contains the BglII D fragment of HSV-1 (14). p205, containing the UL31, UL32, and UL33 genes, was obtained by subcloning the EcoRV-to-HpaI fragment (nt 63987 to nt 70315) from p1BD1 into SmaI-digested pUC119. pAPVUL32, con-taining the UL32 gene under the control of the ICP6 promoter, was generated in several steps. A BamHI fragment from p205 containing the sequence of the UL32 gene (nt 68885 to nt 70168) encoding the N-terminal portion of UL32 was inserted at the BamHI site of pAPV to generate pAPVUL32 GP. To position the UL32 gene initiation codon (nt 69159) downstream from the ICP6 promoter, pAPVUL32 GP was digested with XbaI, which releases a fragment from nt 69247 to nt 70168, and religated to generate pAPVUL32 F. A BamHI fragment con-taining the UL32 gene sequences (nt 68338 to nt 66238) was inserted into the unique BamHI site of pAPVUL32 F to generate pAPVUL32 S. A BstEII-to-SalI fragment (nt 68938 to nt 67771) was subsequently inserted into pAPVUL32 S
digested with BstEII-SalI to obtain the final construct, pAPVUL32. In pAPVEEUL32 the coding sequence for the EE epitope was fused in frame to that for the N terminus of the UL32 protein in pAPV as follows. A SmaI-to-BamHI fragment containing UL32 gene sequences (nt 69144 to nt 68885) from pAPVUL32 was ligated with pUC119 digested with SmaI and BamHI to gener-ate p32 S/B. Two annealed synthetic complementary oligonucleotides containing coding sequences for the EE epitope (EEYMPME) and the four amino acids of UL32 upstream of the position corresponding to the SmaI site (nt 69144) were inserted in p32 S/B digested with EcoRI and SmaI. Oligonucleotide 59-GGGG GGCGAAGTTGCCATTTCCATTGGCATATA TTCTCCATGG-39 and its complement were designed to yield a blunt SmaI site and a cohesive EcoRI site on annealing. Nucleotides corresponding to the EE epitope are underlined. The resulting plasmid, pEE32S/B, was subsequently digested with NcoI and BstEII to obtain a fragment that contained the coding sequence for the EE epitope in frame with the N terminus of UL32; this fragment was subsequently inserted into pAPVUL32 digested with NcoI and BstEII to generate pAPVEEUL32. pUL32lacZ, containing the UL32 gene disrupted by the lacZ cassette at the codon for amino acid 274, was constructed in two steps. A KpnI-to-XbaI frag-ment containing the UL32 gene sequences (nt 66660 to nt 69247) was inserted into XbaI-KpnI sites of pUC119 to obtain pUC UL32. The ICP6::lacZ cassette was subsequently inserted in pUC UL32 at the BamHI site (nt 68338) (Fig. 1). The direction of transcription of the lacZ gene in pUL32lacZ is inverted with respect to the direction of transcription of the UL32 gene.
pGST-UL6, containing the coding sequence for the C-terminal region of UL6 (amino acid residues 381 to 676) fused with the glutathione S-transferase (GST) gene, was constructed as follows. The Asp718 fragment (nt 16273 to 17793) containing the sequence of the UL6 gene encoding the C-terminal portion of UL6 was inserted into Asp718-digested pUC119. This fragment was released by digestion with BamHI and EcoRI and cloned in frame with the GST gene sequence into plasmid pGEX-2T (Pharmacia) digested with BamHI-EcoRI. pAPVAU1-UL6 contains the UL6 gene, whose product has an epitope tag at its N terminus. Plasmid pNN9 containing two copies of the 5.9-kb BamHI K frag-ment from strain KOS cloned into pUC19 was generously provided by Mark Challberg (National Institutes of Health, Bethesda, Md.).
Isolation of UL32 cell lines.To generate cell lines containing the UL32 gene under the control of inducible promoter ICP6, Vero cells were cotransfected with pSV2neo and pAPVUL32 as described previously (21). In brief, pSV2neo (2mg) and pAPVUL32 (18mg) were coprecipitated in a final volume of 1 ml and mixed with freshly trypsinized Vero cells in suspension (25). Two weeks after transfection, G418-resistant colonies were collected and screened for their ability to support the growth of a ts UL32 mutant (tsN20) at the nonpermissive tem-perature of 39.5°C.
Marker transfer.Marker transfer experiments were performed as previously described (23, 71). Transfections were carried out in the UL32-permissive cell line with linearized pUL32lacZ and infectious wild-type (KOS) DNA at a molar ratio of 10:1. Cells (1.53106) were transfected with 0.4mg of the linearized plasmid and 0.8mg of infectious DNA.
Transient complementation test.Transient complementation was performed as previously described (35).
[image:2.612.310.544.68.156.2]DNA isolation, Southern blotting, and PFGE.Infectious viral DNA was pre-pared as previously described (72). Cellular DNA was isolated as previously described (67). For DNA analysis by Southern blot hybridization, virion or infected-cell DNA was digested with restriction endonucleases as directed by the manufacturer, subjected to agarose gel electrophoresis, and transferred to Gene Screen Plus nylon membranes (New England Nuclear Corp., Boston, Mass.) as recommended by the supplier. DNA fragments to be used as probes were labeled as described previously (19) by the random hexamer primed method (Boehringer Mannheim). Pulsed-field gel electrophoresis (PFGE) was performed as previ-ously described (34).
FIG. 1. Physical map of the region of the HSV genome containing the UL32 gene. The region of HSV-1 DNA spanning the UL32 and UL33 open reading frames has been expanded to show the cleavage sites for BamHI (B), SalI (S), XbaI (X), NcoI (N), HpaI (H), and BstEII (E) and the start (nt 69162) and the stop (nts 67374 and 69625) sites for UL32 and UL33, respectively. On the last line is a diagram of the insertion of the ICP6::lacZ cassette in UL32 at the BamHI site (nt 68338).^, ICP6 promoter. UL, unique long region; US, unique short region.
on November 9, 2019 by guest
http://jvi.asm.org/
DNase I protection.Vero cells on a 60-mm-diameter plate were infected at a multiplicity of infection (MOI) of 10 PFU/cell with wild-type KOS or mutant viruses. At the end of the 1-h adsorption period the cells were washed three times with medium and overlayed with complete medium. Time zero DNA is DNA from cellular extracts collected immediately after the 1-h adsorption period. The DNase protection experiment was performed as previously described (68).
Analysis of viral capsids and virions.HSV-1 viral particles were isolated as described by Sherman and Bachenheimer (55) with some modifications. At 18 h postinfection cells were scraped and collected by low-speed centrifugation (4°C, 10 min), resuspended in phosphate buffer (40 mM phosphate [pH 7.4], 0.15 M NaCl) with 1% Nonidet P-40 (NP-40), and treated with a probe sonicator. The total cellular lysate was layered onto 15-to-45% (wt/wt in phosphate buffer) sucrose gradients and centrifuged at 35,000 rpm for 30 min in an SW41 rotor. To prepare large quantities of capsids, 40 10-ml-diameter plates of 60 to 70% confluent monolayers of Vero cells were infected at an MOI of 6 PFU/cell for 24 h. Infected cells were lysed with 50 ml of phosphate buffer with 1% NP40. After low-speed centrifugation, the pellet was resuspended in 20 ml of TEN buffer (20 mM Tris-HCl [pH 7.4], 1 mM EDTA, 0.5 M NaCl) and frozen and thawed three times. A probe sonicator was used to clarify the extract (1 min) before it was layered on a 35% (wt/wt) sucrose cushion, which was centrifuged at 19,600 rpm in an SW41 rotor for 1 h. The pellet containing the capsids was carefully resuspended with 400ml of TEN–NP-40 containing 5 mM MgCl and 10 mM dithiothreitol and layered on a 15-to-45% sucrose gradient. Capsid bands were visualized by light scattering upon illumination with a halogen fiber optic lamp. Bands corresponding to A, B, and C capsids were removed from each gradient by side puncture with an 18-gauge needle on a 5-ml syringe. Each band was diluted fivefold with phosphate buffer and pelleted by centrifugation at 35,000 rpm in an SW41 rotor for 1 h. Alternatively, the collected band was diluted 1:3 in phosphate buffer and layered on a second sucrose gradient for further purification. The pellets were resuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and analyzed by Western blotting. Mature virions were prepared as previously described (58).
Antibodies.pGST-UL6 described above was used to transform UT481 cells. Expression of the fusion protein was induced by 0.4 M IPTG (isopropyl-b-D
-thiogalactopyranoside) when the culture reached an A600of 0.4 at 37°C. SDS-PAGE was used to visualize a protein of the expected size in the IPTG-induced bacterial culture which was not present in the uninduced culture. Cells were lysed with a French press apparatus. The vast majority (90%) of the expressed protein was present in the insoluble fraction of the lysed cells. This protein was purified by SDS-PAGE with a 10% preparative gel. After staining with Coomassie blue and destaining, gel slices containing the GST-UL6 protein were excised, washed in distilled water, and sent to HRP Inc. (Denver, Pa.) for antiserum production in rabbits (a-UL6). The UL32 polyclonal antibody was generously donated by Bernard Roizman (University of Chicago, Chicago, Ill.) (13). Anti-EE ascites was previously described (30). Anti-ICP8 was generously provided by William Ruyechan (State University of New York at Buffalo, Buffalo) (53). Monoclonal antibody 5C was generously provided by Jay Brown (University of Virginia Health Science Center, Charlottesville) (63).
Detection of proteins by immunoblotting.Vero cells were plated onto 60-mm-diameter tissue culture dishes. At 18 h postinfection, the cells were processed as previously described (35). Samples were subjected to SDS–10% PAGE, and the proteins were transferred for 2 h onto an Immobilon-P membrane (Millipore, Bedford, Mass.) in 25 mM Tris–192 mM glycine–20% methanol (61). The filters were incubated in TBST (10 mM Tris [pH 8.0], 150 mM NaCl, 0.1% Tween 20) containing 20% fetal calf serum and 1% bovine serum albumin for 1 h at room temperature and were incubated overnight at 4°C with thea-UL6 antiserum, diluted 1:1,500 in TBST. After five washes in TBST, the filters were incubated in TBST containing goat anti-rabbit immunoglobulin G–alkaline phosphatase con-jugate (Promega, Madison, Wis.). Alkaline phosphatase color development was carried out as previously described (72). For UL32 protein detection, the blot was incubated in 5% dry milk in phosphate-buffered saline (PBS) for 1 h at room temperature and incubated with a 1:500 dilution of the UL32 antibody for 1 h. After four washes with PBS-dry milk suspension the blot was incubated for 1 h at room temperature with secondary antibody and developed as described above. Alternatively, the enhanced chemiluminescence (ECL) detection system (Am-ersham, Arlington Heights, Ill.) was used as recommended by the supplier.
Indirect immunofluorescence.The ICP6 promoter can be induced with either of the two transactivators ICP0 and VP16 (17, 22, 57). Vero cells were cotrans-fected (21) with equal amounts of pAPVEEUL32 and pVP16 (33) and grown on coverslips for 24 h, or cells were transfected with pAPVEEUL32 and infected 18 h later for 6 h prior to fixation. Cells were fixed and processed for immuno-fluorescence as previously described (30). In these experiments the EE mono-clonal antibody was diluted 1:500 and anti-ICP8 was used at 1:400. Cells infected with the wild-type KOS strain or the mutant viruses were fixed and processed as described above. In these experiments monoclonal antibody 5C was used at 1:1,000. All double-labeled cells were checked for bleed-through staining and cross-reactivity by treatment with only a single primary antibody and both sec-ondary antibodies.
Imaging.Cells stained for immunofluorescence were imaged on a Zeiss Ax-iovert 135 laser scanning microscope (confocal) equipped with a ZeissX630 Plan Neofluar objective. Collected images were arranged and labeled with a Silicon Graphics workstation equipped with Adobe Photoshop version 3.0 software. To
obtain the three-dimensional images of replication compartment and capsid staining patterns, a series of two-dimensional slices (Z series) 0.3mthick were imaged on the confocal microscope. Collected Z series images were subsequently analyzed and displayed by the Voxel View 2.5 program.
RESULTS
To propagate a potentially lethal mutation in the UL32
gene, it was first necessary to isolate a complementing cell line.
Vero cells were transfected with pAPVUL32 and pSV2neo as
described under Materials and Methods. Ten G418-resistant
cell lines were isolated, and the one which could support the
growth of tsN20 at the nonpermissive temperature most
effi-ciently was designated 158. To isolate a viral mutant containing
a disrupted UL32 gene, a plasmid containing the ICP6::lacZ
cassette inserted at the codon for amino acid 274 within the
UL32 gene was used for marker transfer. The permissive cell
line, 158, was cotransfected with infectious KOS DNA and
pUL32lacZ as described under Materials and Methods. Three
blue plaques were purified and one, designated hr64, was
cho-sen for further study. We confirmed by Southern blot analysis
that DNA from hr64 contains the ICP6::lacZ cassette at the
expected position (data not shown).
Characterization of the UL32 mutant.
The UL32 mutant
was then examined for its ability to form plaques on permissive
and nonpermissive cells (Vero and 158). hr64 was unable to
form plaques on Vero cells, but it could efficiently form
plaques on 158 cells (titer on 158 cells, 5
3
10
7PFU/ml). The
ability to form plaques on 158 cells but not on Vero cells
indicates that hr64 carries a defective UL32 gene. This result
was further confirmed in a transient complementation assay;
Vero cells were transfected with pAPVUL32, which expresses
wild-type UL32, and superinfected with hr64. pAPVUL32, but
not a control plasmid containing the UL6 gene under the
control of the ICP6 promoter, was able to complement the
growth of hr64 (data not shown). In sum, these results indicate
that the mutation in hr64 lies within the UL32 gene.
In addition, hr64 was tested for expression of the UL32
protein during infection of permissive and nonpermissive cells.
The UL6 mutant hr74 was used as a control in these
experi-ments. Total cell extracts from infected cells were subjected to
Western blot analysis with the polyclonal UL32 antiserum as
described under Materials and Methods. Figure 2 clearly
shows that a band of 68 kDa corresponding to the UL32
protein is present in KOS- and in hr74-infected cells (Fig. 2,
lanes 2 and 4, respectively). On the other hand, the 68-kDa
protein was not detected in mock- or hr64-infected Vero cells
(Fig. 2, lanes 1 and 3, respectively). A smaller band of 30 kDa
is present in hr64-infected Vero cells (Fig. 2, lane 3). Since the
lacZ gene in hr64 was inserted at the codon for amino acid 274,
it is likely that this 30-kDa band represents the N-terminal
portion of UL32. This truncated UL32 protein is not sufficient
for viral growth, as demonstrated by the inability of hr64 to
form plaques on Vero cells (see above). Furthermore, the
behavior of hr64 on the complementing cell line (see below)
suggests that the truncated form does not exhibit any inhibitory
effect. When 158 cells were infected with the mutant hr64, two
bands corresponding to the full-length UL32 protein and to the
truncated version were observed (Fig. 2, lane 6). Thus, we
confirm that hr64 does not express the full-length UL32
pro-tein in nonpermissive Vero cells.
The UL32 mutant can synthesize viral DNA but is defective
for cleavage.
We tested the ability of hr64 to synthesize and
process viral DNA. DNA extracts from infected cells were
assayed for hybridization to an HSV DNA probe as previously
described (72); hr64 synthesizes viral DNA at near-wild-type
levels (data not shown). To measure genomic cleavage activity
on November 9, 2019 by guest
http://jvi.asm.org/
during viral infection, we tested for the presence of genomic
termini in DNA extracted from hr64- and UL6 mutant
hr74-infected cells at 18 h postinfection. BamHI digested-DNA
from hr64-, hr74-, and KOS-infected Vero and 158 cells was
subjected to Southern blot analysis. As shown in Fig. 3A, only
KOS-infected Vero and 158 cells (lanes 1 and 2, respectively)
and hr64-infected 158 cells (lane 6) contain genomic termini,
represented by the BamHI fragments S and Q. In contrast,
Vero and 158 cells infected with hr74 (lanes 3 and 4,
respec-tively) and Vero cells infected with hr64 (lane 5) contain only
DNA corresponding to the L-S junction fragment (designated
SQ), indicating that these viruses do not produce genomic
termini. The amounts of junction fragment are comparable in
all three lanes, confirming that viral DNA synthesis is
unaf-fected in mutants hr74 and hr64.
To confirm the inability of hr64 to carry out genomic
cleav-age, we analyzed the behavior of viral DNA from mutant and
wild-type viruses by PFGE. During replication, viral DNA
ac-cumulates as concatemeric complex intermediates that are
un-able to enter a pulsed-field gel (well DNA), while mature
monomeric viral DNA (152 kb) is well resolved (7, 34, 38, 52,
70). Figure 3B shows a Southern blot analysis of a pulsed-field
gel of viral DNA prepared from KOS-infected Vero cells (lane
1) or hr64-infected Vero or 158 cells (lanes 2 and 3,
respec-tively). Monomeric viral DNA (152 kb) is present in Vero cells
infected with KOS and not in Vero cells infected with hr64
virus. Monomeric viral DNA is present in 158 cells infected
with the hr64 virus. We conclude that the UL32 insertion
mutant synthesizes wild-type levels of viral DNA but fails to
process the DNA into monomeric units.
The UL32 mutant is defective for encapsidation.
Once
rep-licating DNA has been cleaved and packaged, a
DNase-resis-tant band representing a monomer unit of viral DNA can be
detected in extracts of infected cells. DNA extracts from cells
infected with KOS or hr64 for different times were treated with
DNase I as described under Materials and Methods. Figure 4
(lanes 1 to 6) shows that in KOS-infected Vero cells,
DNase-resistant monomeric DNA can be detected as early as 6 h
postinfection. On the other hand, no DNase-resistant DNA
was observed in cells infected with hr64 (Fig. 4, lanes 7 to 12).
The faint band observed in all the lanes (lanes 8 to 12) is
comparable to the band present at time zero (lane 7),
indicat-ing that these bands represent input viral DNA.
[image:4.612.93.247.70.302.2]By analogy with other cleavage/packaging mutants, cells
in-fected with the UL32 mutant would be expected to exhibit an
altered pattern of capsid formation compared to the wild type.
After lysis and sonication of infected cells in 1% NP40,
whole-cell extracts were analyzed by sucrose gradient sedimentation.
KOS-infected cells exhibit three types of capsids (A, B, and C),
while UL32 mutant virus-infected cells produce only B capsids
(data not shown). This result is consistent with a defect in
cleavage and packaging.
FIG. 2. Immunoblot of extracts from KOS- and hr64-infected cells. Mono-layers of Vero and 158 cells were infected with either KOS, hr74, or hr64 (5 PFU/cell) or were mock infected. Extracts were prepared at 18 h postinfec-tion as described under Materials and Methods and resolved by SDS-PAGE. Proteins were revealed by Western blotting with a UL32 polyclonal antibody and alkaline phosphatase-conjugated secondary antibodies. Lane 1, mock-infected Vero cell extracts; lane 2, KOS-infected Vero cell extracts; lane 3, hr64-infect-ed Vero cell extracts; lane 4, hr74-infecthr64-infect-ed Vero cell extracts; lane 5, mock-infected 158 cell extracts; lane 6, hr64-mock-infected 158 cell extracts.
FIG. 3. Southern blot analysis of total DNA from KOS-, hr74-, and hr64-infected Vero cells. (A) Vero cells were hr64-infected with the indicated virus at 5 PFU per cell. At 18 h postinfection, DNA was isolated as described under Materials and Methods and digested with BamHI. The blotted DNA was hy-bridized with a 32P-labeled probe containing the BamHI SQ fragment. SQ represents viral DNA junctions, and S and Q represent viral DNA termini. Lanes 1, 3, and 5, total DNA from KOS-, hr74-, and hr64-infected Vero cells, respec-tively; lanes 2, 4, and 6, total DNA from KOS-, hr74-, and hr64-infected 158 cells, respectively. (B) Vero cells were infected at 5 PFU per cell. At 18 h postinfection the cells were collected in low-melting-point agarose. Agarose blocks were in-troduced into the well of a 1.3% pulsed-field gel. The complex high-molecular-weight DNA which does not enter the gel is labeled “well,” and unit-length monomeric DNA is labeled “152Kb.” Lane 1, total DNA from KOS-infected Vero cells; lane 2, total DNA from hr64-infected Vero cells; lane 3, total DNA from hr64-infected 158 cells.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.353.504.305.600.2]UL32 is not detected in capsids or in virions.
Cleavage/
packaging proteins UL6 and UL25 are associated with A, B,
and C capsids found in infected cells (28, 37, 44); UL6 and
UL25 are also found in complete virions (3, 28, 44). In order to
determine whether UL32 is also associated with capsids and/or
virions, we prepared B capsids and mature virions and
ana-lyzed by Western blotting as described under Materials and
Methods. Figure 5 shows that UL32 is not detected in B
cap-sids or virions from KOS-infected cells (Fig. 5, lanes 1 and 5,
respectively). As expected, UL32 (marked by an arrow) can
easily be detected in KOS-infected cell extracts (Fig. 5, lane 3)
but not in mock- or hr64-infected cell extracts (Fig. 5, lanes 2
and 4). The band, just below the band marked by an arrow,
present in hr64-infected Vero cells is not specific since a similar
band is also present in mock-infected cells. Thus, it appears
that UL32 is not present in B capsids or in virions, although at
this point we cannot rule out the possibility that UL32 is
present at too low a level to be detectable with the antibody
used.
Expression and capsid localization of other
cleavage/pack-aging proteins in cells infected with the UL32 mutant virus.
Since mutations in the cleavage and packaging genes produce
similar phenotypes, we were interested in whether the
expres-sion of one viral gene affects the expresexpres-sion of the others.
Western blot analysis indicates that the levels of UL6, UL15,
UL25, and UL28 proteins, as well as levels of protease and
scaffold proteins, in Vero cells infected with a UL32 mutant
are similar to those seen in wild-type infections (data not
shown). This result indicates that the expression of these
pro-teins is not dependent on the presence of a functional UL32.
Likewise, we asked whether the presence of UL6 and UL25 in
capsids was dependent on the expression of UL32. Therefore,
hr64-infected Vero cells were collected at 20 h postinfection
and B capsids were prepared as described under Materials and
Methods. Figure 6A shows that the UL6 protein is present in
B capsids from both KOS- and hr64-infected cells (Fig. 6A,
lanes 1 and 2, respectively). As expected, the UL6 protein can
be detected in KOS-infected cell extracts and in virions from
KOS-infected cells (Fig. 6A, lanes 4 and 6) but not in mock- or
hr74-infected cell extracts (Fig. 6A, lanes 3 and 5, respectively).
Similar results were obtained with the UL25 polyclonal
anti-body, as shown in Fig. 6B. B capsids collected from KOS- and
hr64-infected cells contain UL25 protein (Fig. 6B, lanes 3 and
[image:5.612.328.530.68.267.2]4) as do KOS-infected cell extracts (Fig. 6B, lane 2) and virions
from KOS-infected cells (Fig. 6B, lane 6). On the other hand
UL25 is not detected in mock-infected cell extracts or in
ex-tracts from cells infected with virus lacking UL25 (Fig. 6B,
lanes 1 and 5, respectively). The presence of UL6 and UL25 in
FIG. 4. Southern blot analysis of DNase-treated DNA from KOS- and [image:5.612.95.245.68.189.2]hr64-infected Vero cells. Total DNA extracts from KOS or hr64 were treated with DNase I and subsequently digested with BamHI as described under Materials and Methods. The samples were collected at the end of a 1-h incubation time (time zero) and at 2, 4, 6, 8, and 16 h postinfection.
FIG. 5. UL32 can be detected in KOS-infected cell extracts but not in B capsid or virions. B capsids from KOS-infected Vero cells were collected from a sucrose gradient and purified on a second gradient as described under Materials and Methods. Monolayers of Vero cells were infected with either KOS or hr64 (5 PFU/cell) or were mock infected. Extracts were prepared at 18 h postinfection as described under Materials and Methods. Virions were prepared as described under Materials and Methods. Proteins were revealed by ECL Western blotting with UL32 polyclonal antibodies (13) and horseradish peroxidase-labeled sec-ondary antibodies. Lane 1, B capsids from KOS-infected Vero cells; lane 2, mock-infected Vero cell extracts; lane 3, KOS-infected Vero cell extracts; lane 4, hr64-infected Vero cell extracts; lane 5, virion from KOS-infected Vero cells.
FIG. 6. UL6 and UL25 are present in B capsids from wild-type- and infected cells. The presence or absence of UL6 and UL25 in KOS- and hr64-infected Vero cell extracts, in B capsids from KOS- and hr64-hr64-infected cells, and in virions from KOS-infected Vero cells was determined as described under Materials and Methods. Proteins were revealed by Western blotting witha-UL6 antibodies and alkaline phosphatase-conjugated secondary antibodies (A) or by ECL Western blotting with a UL25 polyclonal antibody and horseradish perox-idase-labeled secondary antibodies (B). Panel A lanes: 1, B capsids from KOS-infected Vero cells; 2, B capsids from hr64-KOS-infected Vero cells; 3, mock-KOS-infected Vero cell extracts; 4, KOS-infected Vero cell extracts; 5, hr74-infected Vero cell extracts; 6, virions from KOS-infected Vero cells (this lane was developed for a longer time to visualize the band). Panel B lanes: 1, mock-infected Vero cell extracts; 2, KOS-infected Vero cell extracts; 3, B capsids from KOS-infected Vero cells; 4, B capsids from hr64-infected Vero cells; 5, extracts from Vero cells infected with virus lacking UL25; 6, virions from KOS-infected Vero cells.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.95.250.451.635.2]B capsids in cells infected with viruses lacking UL32 indicates
that the accumulation of UL6 and UL25 in B capsids is not
dependent on the expression of full-length UL32.
Intracellular localization of UL32.
Previous studies have
shown that in infected cells, the UL32 protein was found
pre-dominantly in the cytoplasm (13). Although in that report a
small amount of nuclear staining was also observed at 18 h
postinfection, we were intrigued by the unexpected distribution
of a protein that has a role in cleavage and packaging. We
constructed an epitope-tagged version of UL32 containing the
EE epitope (EEUL32) in frame with the N-terminal portion
of the UL32 protein. The plasmid encoding this construct,
pAPVEEUL32, was found to be able to complement UL32
mutant virus hr64 as well as the wild type in a transient
comple-mentation assay (data not shown). This indicates that EEUL32
is functional. Vero cells were transfected with pAPVEEUL32
and a transactivator and processed for immunofluorescence
with the monoclonal EE antibody and the polyclonal UL32
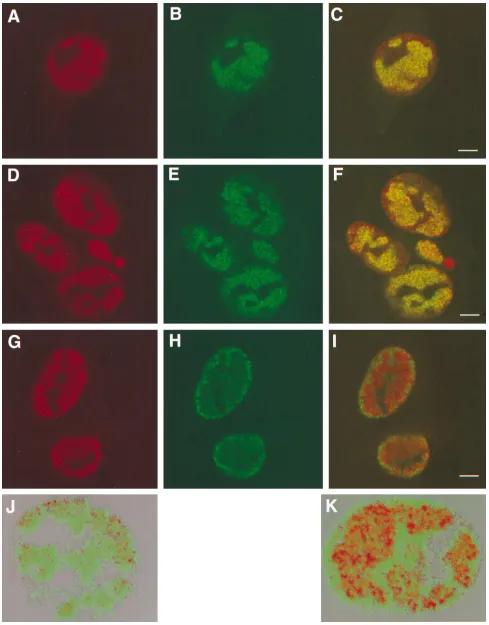
antibody as described under Materials and Methods (Fig. 7A
and B, respectively). Although both antibodies are presumably
detecting the UL32 gene product, the staining patterns differ
somewhat. The monoclonal antibody clearly detects UL32 in
the cytoplasm and in the nucleus (Fig. 7A); the polyclonal
antibody detects UL32 mainly in the cytoplasm with very faint
staining in the nucleus (Fig. 7B). In Fig. 7C, a merged image is
shown. Control experiments indicate that the EE antibody
does not react with untransfected cells (data not shown). It is
possible that the EE antibody reaction with its epitope is much
stronger than that of the polyclonal serum with UL32;
alter-natively, the polyclonal antibody may not recognize nuclear
UL32 as efficiently. In any case, we have demonstrated that
UL32, when expressed only with transactivator VP16,
accumu-lates in both the nucleus and the cytoplasm. In contrast, in cells
transfected with a plasmid bearing a tagged version of the UL6
gene, another cleavage/packaging gene, UL6 can efficiently
localize to the nucleus on its own (32).
In order to determine the localization of UL32 in infected
cells, Vero cells were transfected with pAPVEEUL32 and
su-perinfected with hr64 at an MOI of 10 PFU/cell (Fig. 7D to F).
Although most cells will be infected under these conditions,
only a fraction of cells are expected to be transfected. To
ensure that all the transfected cells were also infected, cells
were double stained with the EE antibody and with a
poly-clonal antibody directed against ICP8 (anti-ICP8), the major
DNA-binding protein of HSV-1. ICP8 is found in globular
nuclear domains termed replication compartments, in which
viral DNA synthesis is known to occur (49). In Fig. 7, panel D
shows the EE staining pattern, panel E shows the ICP8 staining
pattern, and panel F shows the merged image. ICP8 staining in
panel E indicates that most of the cells are infected;
charac-teristic replication compartments are observed. Figure 7D
shows that in cells that are transfected and infected, the EE
staining (UL32 protein) is predominantly in the cytoplasm.
Nevertheless, in these cells, some EE staining is seen in the
FIG. 7. Localization of UL32 in cells transfected with pAPVEEUL32. Cells shown in panels A to C were transfected with pAPVUL32 and pVP16 as described under Materials and Methods. Cells shown in panels D to F were transfected with pAPVEEUL32 and superinfected with hr64. In panels A and B, green represents staining with the EE monoclonal antibody and red represents staining with the UL32 polyclonal antibody, respectively. In panel D green represents staining with the EE monoclonal antibody, and in panel E red represents staining for ICP8. Panels C and F show the merged images of the staining patterns. Bars510mm.on November 9, 2019 by guest
http://jvi.asm.org/
nucleus, in a pattern that colocalizes with that of ICP8 (Fig.
7F). Cells that do not react with the EE antibody are cells
which are infected but not transfected (Fig. 7D and E); the lack
of reactivity in these cells indicates that the antibody does not
cross-react with other viral proteins. In sum, although in
in-fected cells most of the UL32 protein localizes to the
cyto-plasm, the UL32 protein in the nucleus appears to be present
in replication compartments.
Capsids localize to replication compartments.
Ward et al.
have proposed that at late times after infection, some capsid
and tegument proteins assemble in discrete nuclear structures
at the borders of replication compartments and that these sites
represent sites of capsid assembly and encapsidation (64).
Since we and others (70) have shown that encapsidation begins
as early as 6 h postinfection (Fig. 4), we decided to look at the
localization of capsids at earlier times. Furthermore, at 6 and
8 h postinfection, the cells are still well attached to the
cover-slips and cellular structures are more recognizable than at later
times. Three monoclonal antibodies (8F5, 5C, and 3B) which
recognize distinct epitopes of major capsid protein VP5 on the
capsid itself have been reported (63); however, they do not
recognize VP5 in a Western blot (40a, 63). These
conforma-tional epitopes have been used previously as probes for
assem-bled capsid structures (36); monoclonal antibodies 8F5 and 5C
recognize hexons, while 3B is specific for pentons.
In this study, assembled capsids were stained with
monoclo-nal antibody 5C, which recognizes hexons within preassembled
capsids. Vero cells were infected with KOS, the UL6 mutant
(hr74), or the UL32 mutant (hr64) for 6 h at an MOI of 10
PFU/cell and stained with monoclonal antibody 5C to label
hexon-containing capsids. Figure 8B and E show that in Vero
cells infected with KOS or hr74, hexon staining is restricted to
regions resembling replication compartments, well-defined
structures in which DNA synthesis occurs (49). Vero cells
infected with hr64 (Fig. 8H), on the other hand, exhibit an
altered pattern of hexon staining in which capsids are
distrib-uted throughout the nucleus, with some accumulation at the
periphery of the nucleus. In order to determine the localization
of capsids with respect to replication compartments, the same
Vero cells infected with KOS, the UL6 mutant (hr74), or the
UL32 mutant (hr64) were also labeled with the ICP8
poly-clonal antibody (Fig. 8A, D, and G, respectively). The
replica-tion compartments seen at 6 h postinfecreplica-tion are large globular
domains that take up a large portion of the nucleus; however,
as has been seen before, replication compartments do not
occupy the entire nucleus (Fig. 8A, D, and G). ICP8 staining is
not seen in the nucleolus, nor does it extend to the periphery
of the nucleus. These patterns are consistent with previous
reports by de Bruyn Kops and Knipe (16). When the staining
patterns of ICP8 and monoclonal antibody 5C are merged, it
becomes apparent that in KOS- and hr74-infected Vero cells,
hexon staining is indeed restricted to the replication
compart-ments (Fig. 8C and F, respectively). The demonstration that in
cells infected with wild-type virus for 6 h capsids colocalize
with replication compartments suggests that cleavage and
pack-aging occur in these structures.
On the other hand, hr64-infected cells exhibit a very
differ-ent staining pattern (Fig. 8G to I). As mdiffer-entioned above, these
cells contain replication compartments similar to those seen in
wild-type- or hr74-infected cells; however, the merged image
(Fig. 8I) indicates that the distribution of hexon-containing
capsids is not restricted to replication compartments. Hexon
staining is observed throughout the nucleus and is present in
the areas, especially around the periphery of the nucleus, in
which replication compartments are not seen (Fig. 8H and I).
To confirm that hexon staining is not restricted to the
replica-tion compartments, we used the Voxel View program to create
three-dimensional images of replication compartment and
cap-sid staining patterns. Figure 8J and K show Vero cells infected
with KOS and the UL32 mutant, hr64, respectively. The green
regions represent hexon-containing capsids, while the red
re-gions represent replication compartments. In KOS-infected
cells (Fig. 8J), the green and red regions colocalize. In
hr64-infected cells (Fig. 8K) some of the green color is observed
within replication compartments (red); however, a significant
portion of the green is visible outside the red regions. The
image reconstructions confirm our previous observations that
in Vero cells infected with the UL32 mutant, capsids are not
restricted to replication compartments. When hr64 was used to
infect 158 cells, the complementing cell line expressing the
UL32 protein, the hexon staining pattern was found to
resem-ble that of wild-type virus (data not shown), confirming that the
alteration in capsid staining in hr64-infected Vero cells is due
to the defect in the UL32 gene.
DISCUSSION
We report the construction and characterization of a UL32
insertion mutant, hr64, which fails to form plaques on Vero
cells and which is defective for cleavage and packaging. Both
the growth phenotype and the cleavage and packaging defects
are corrected when hr64 is grown on the complementing cell
line which expresses only the UL32 gene. These results confirm
previous results with ts mutant tsN20 (55). While two other
cleavage/packaging gene products (UL6 and UL25) appear to
be associated with A, B, and C capsids and with virions, in our
hands UL32 has not been detected in B capsids or in virions.
On the other hand, UL6 and UL25 are still present in B
capsids made in cells infected with the UL32 mutant,
indicat-ing that UL32 is not required for the correct localization of
UL6 and UL25 in the capsids.
Chang et al. (13) previously reported that UL32 can be
detected primarily in the cytoplasm of infected cells. Using an
epitope-tagged version of this protein, we were able to detect
some UL32 staining in the nuclei of infected cells in addition
to the cytoplasmic staining. The UL32 which is in the nucleus
appears to localize to replication compartments. Furthermore,
in cells transfected with the UL32 gene, both nuclear and
cytoplasmic staining was observed, indicating that no other
cleavage/packaging or capsid proteins are required for the
cytoplasmic and nuclear localization patterns. Finding UL32 in
the nucleus is consistent with its putative role in cleavage and
packaging. The reason for the presence of UL32 in the
cyto-plasm is not understood; however, it may indicate that UL32
carries out more than one function. It is thus possible that the
role of nuclear UL32 is in capsid localization and that the role
of cytoplasmic UL32 is performed at a later step in infection,
such as egress of the capsid once it buds from the nucleus.
Ward et al. previously reported that another
cleavage/packag-ing protein, UL15, also localizes to replication compartments
in infected cells, and we have recently confirmed this finding
(64, 69). Furthermore, UL6 has also been observed in
replica-tion compartments (47).
The presence of UL6, UL15, and UL32 in replication
com-partments prompted us to ask whether cleavage and packaging
occurs in replication compartments or at another adjacent site
as previously suggested by Ward et al. for late times after
infection. Using a hexon-specific antibody we observed capsid
staining which colocalizes completely with replication
com-partments at 6 and 8 h postinfection. Since DNase-protected
DNA can be detected as early as 6 h postinfection, we conclude
that at these early times, packaging is likely to occur within
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 8. Replication compartments and capsid localization. Vero cells were infected with KOS (A to C and J), hr74 (D to F), or hr64 (G to I and K) for 6 h and stained with anti-ICP8 polyclonal antibodies (A, D, and G) and 5C monoclonal antibodies (B, E, and H). Panels C, F, and I represent the merged images of staining patterns with ICP8 and 5C antibodies. Panels J and K represent three-dimensional reconstructions from Z series images of KOS- and hr64-infected Vero cells obtained with the Voxel View program as described under Materials and Methods. In a control experiment, mock-infected cells were treated with primary and secondary antibodies, and infected cells were treated with the secondary antibodies alone; in neither case was any cross-reactivity for anti-ICP8 or monoclonal antibody 5C observed (data not shown). Bars510mm.
2470
on November 9, 2019 by guest
replication compartments. In support of this conclusion, a
re-cent report by Phelan et al. (47) demonstrates that capsid
proteins and the HSV-1 DNA polymerase also colocalize at 8 h
postinfection. We also analyzed capsid distribution at 16 h
postinfection; at this time KOS-infected Vero cells exhibit
sev-eral different staining patterns (data not shown). In some cells,
hexon staining is present in aggregates that resemble the
“as-semblon” structures described by Ward et al. (64); however, in
other cells hexon staining was diffuse. Ward et al. have
pro-posed that the overlap of replication compartments with these
sites of capsid accumulation may represent sites at which
cleav-age and packaging occur (64). While we cannot rule out that
some packaging occurs outside replication compartments at
late times postinfection, we favor a model in which cleavage
and packaging occur within replication compartments. Our
model is based on our observations that (i) some cells do not
contain assemblon structures even at 16 h postinfection; (ii) at
6 h postinfection none of the cells contain assemblons in spite
of the fact that we can detect significant levels of cleavage and
packaging; and (iii) hexon-containing capsid staining occurs
within replication compartments at 6 h postinfection. It is
possible that assemblons which are only observed at late times
postinfection represent dead-end accumulations of capsids,
which are not destined for the cleavage and packaging
pro-cesses.
The most striking feature of the UL32 mutant phenotype is
the alteration of capsid distribution. Whereas in cells infected
with wild-type virus and a virus with a mutant version of
cleav-age and packaging gene UL6, capsids localize exclusively to
replication compartments, cells infected with the UL32 mutant
exhibit an unusual capsid-staining pattern in which capsids are
not restricted to replication compartments. Instead, many
cap-sids accumulate at the nuclear periphery in areas which are
devoid of ICP8. We thus conclude that UL32 may play a role
in the correct localization of capsids after their assembly, a role
in the packaging process which is distinct from that played by
UL6. The DNA bacteriophages provide useful models for the
process of genome encapsidation in the eukaryotic viruses. By
analogy with these better-studied viruses, we might expect
her-pesviruses to encode a terminase complex responsible for
translocation and cleavage of concatemeric DNA (reviewed in
reference 9). In lambda phage, the gpF1 protein influences the
efficiency of the terminase (12) and in combination with the
terminase may serve to facilitate capsid interactions with DNA
(40). We are intrigued by the possibility that UL32 may play a
role similar to that of gpF1 in facilitating the interaction
be-tween capsids and DNA by bringing capsids to replication
compartments. Our observations raise the possibility that
UL32 may be expected to interact with capsids. In this report,
however, we were not able to detect UL32 in purified B capsids
or in virions. It is possible that UL32 interacts only transiently
with capsids in a way that is not detected under the conditions
used in this study. In fact, if UL32 plays a role similar to that
played by the gpF1 protein of the lambda phage, one might
expect a transient interaction with capsids and with the
termi-nase. Although the HSV terminase has not been definitively
identified, it has been suggested that UL15 may form part of a
viral terminase complex (6, 15, 68). Recent work in our
labo-ratory suggests that UL15 may exhibit a transient association
with capsids; we can detect UL15 primarily in B capsids and in
much lesser amounts in C capsids (69). It is possible that UL32
also plays a transient role in interactions between capsids,
terminase, and viral genomes. Further experimentation will be
required to test these proposals.
The apparent colocalization of ICP8 and capsids in
replica-tion compartments deserves further comment. Replicareplica-tion
compartments are large globular domains in which several
viral processes are known to occur. In addition to the seven
viral replication proteins and several cellular replication
pro-teins which are now known to colocalize within replication
compartments, ICP4 and several cleavage and packaging
pro-teins are also observed within these structures. The
relation-ship between these various processes is not well understood. It
is worthwhile to note that there are viral processes which occur
in the nucleus outside the replication compartments. For
ex-ample, although we believe that encapsidation occurs within
replication compartments, our preliminary evidence suggests
that capsids themselves may assemble at discrete sites outside
replication compartments and subsequently move to
replica-tion compartments prior to packaging (28). One model, which
is consistent with all our data, is that UL32 facilitates the
interaction between capsids which may be assembled outside
replication compartments, viral genomes which are
presum-ably generated within the replication compartments, and the
cleavage/packaging machinery. In cells infected with UL32
mu-tant virus, some capsids are seen within replication
compart-ments, but a large number accumulate in spaces outside the
replication compartments. This may indicate that the
appro-priate interactions between capsids and the cleavage/packaging
machinery leading to the initiation of the encapsidation
pro-cess cannot occur in this mutant. In fact, the absence of A and
C capsids in UL32 mutant-infected cells indicates that
encap-sidation was not even attempted in these cells. In
bacterio-phage T4, packaging has been shown to compete with the
replication and recombination machinery (39). It is possible
that in HSV-1-infected cells, various competing reactions will
determine whether viral DNA undergoes replication,
recom-bination, or cleavage and packaging.
Two major conclusions can be made from the data presented
in this paper: (i) in contrast to the results of a recent study by
Ward et al. (64), we believe that cleavage and packaging are
likely to occur within replication compartments at least at early
times postinfection; (ii) the UL32 gene product acts at a
unique step in the cleavage/packaging process and likely plays
a role in the efficient localization of capsids to replication
compartments. We propose that the inability of capsids to
localize to replication compartments may be responsible for
the cleavage/packaging defect of the UL32 mutant.
ACKNOWLEDGMENTS
We thank the members of our laboratory and Fred Homa for
crit-ically reviewing the manuscript. We thank Kevin Nawotka for the
construction of pAPV plasmid and Fred Homa for pAPV UL6. We are
grateful to Bernard Roizman for providing the UL32 antiserum, Bill
Ruyechan for providing ICP8 antiserum (anti-ICP8), Jay Brown for
providing monoclonal antibody 5C, and Fred Homa for UL25
anti-serum and the KUL25NS virus. We especially thank Susan Krueger
and Frank Morgan for assistance with confocal imaging.
This study was supported by National Institutes of Health grant AI
37549.
REFERENCES
1. Addison, C., F. J. Rixon, J. W. Palfreyman, M. O’Hara, and V. G. Preston. 1984. Characterisation of a herpes simplex virus type 1 mutant which has a temperature-sensitive defect in penetration of cells and assembly of capsids. Virology 138:246–259.
2. Addison, C., F. J. Rixon, and V. G. Preston. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J. Gen. Virol. 71:2377–2384.
3. Ali, M. A., B. Forghani, and E. M. Cantin. 1996. Characterization of an essential HSV-1 protein encoded by the UL25 gene reported to be involved in virus penetration and capsid assembly. Virology 216:278–283.
4. Al-Kobaisi, M. F., F. J. Rixon, I. McDougall, and V. G. Preston. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388.
on November 9, 2019 by guest
http://jvi.asm.org/
5. Baines, J. D., C. Cunningham, D. Nalwanga, and A. Davison. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J. Virol. 71:2666–2673.
6. Baines, J. D., A. P. Poon, J. Rovnak, and B. Roizman. 1994. The herpes simplex virus 1 UL15 gene encodes two proteins and is required for cleavage of genomic viral DNA. J. Virol. 68:8118–8124.
7. Bataille, D., and A. Epstein. 1994. Herpes simplex virus replicative concate-mers contain L components in inverted orientation. Virology 203:384–388. 8. Bazinet, C., and J. King. 1985. The DNA translocating vertex of dsDNA
bacteriophage. Annu. Rev. Microbiol. 39:109–129.
9. Black, L. W. 1989. DNA packaging in ds DNA bacteriophages. Annu. Rev. Microbiol. 43:267–292.
10. Bush, M., D. R. Yager, M. Gao, K. Weisshart, A. I. Marcy, D. M. Coen, and D. M. Knipe.1991. Correct intranuclear localization of herpes simplex virus DNA polymerase requires the viral ICP8 DNA-binding protein. J. Virol. 65:1082–1089.
11. Casjens, S., and R. Hendrix. 1988. Control mechanism in dsDNA bacterio-phage assembly, p. 15–91. In R. Calendar (ed.), The bacteriobacterio-phages, vol. 1. Plenum Press, New York, N.Y.
12. Catalano, C. E., and M. A. Tomka. 1995. Role of gpFI protein in DNA packaging by bacteriophage lambda. Biochemistry 34:10036–10042. 13. Chang, Y. E., A. P. Poon, and B. Roizman. 1996. Properties of the protein
encoded by the UL32 open reading frame of herpes simplex virus type 1. J. Virol. 70:3938–3946.
14. Coen, D. M., D. P. Aschman, P. T. Gelep, M. J. Retondo, S. K. Weller, and P. A. Schaffer.1984. Fine mapping and molecular cloning of mutations in the herpes simplex virus DNA polymerase locus. J. Virol. 49:236–247. 15. Davison, A. J. 1992. Channel catfish virus: a new type of herpesvirus.
Virol-ogy 186:9–14.
16. de Bruyn Kops, A., and D. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857–868.
17. Desai, P., R. Ramakrishnan, Z. W. Lin, B. Osak, J. C. Glorioso, and M. Levine.1993. The RR1 gene of herpes simplex virus type 1 is uniquely trans activated by ICP0 during infection. J. Virol. 67:6125–6135.
18. Dube, P., P. Tavares, R. Lurz, and M. van Heel. 1993. The portal protein of bacteriophage Spp1: a DNA pump with 13-fold symmetry. EMBO J. 12: 1303–1309.
19. Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction fragments to high specific activity. Anal. Biochem. 132:6–13. 20. Gibson, W., and B. Roizman. 1972. Proteins specified by herpes simplex
virus. VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10:1044–1052.
21. Goldstein, D. J., and S. K. Weller. 1988. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166:41–51.
22. Goldstein, D. J., and S. K. Weller. 1988. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 62:196–205.
23. Goldstein, D. J., and S. K. Weller. 1988. An ICP6::lacZ insertional mutagen is used to demonstrate that the UL52 gene of herpes simplex virus type 1 is required for virus growth and DNA synthesis. J. Virol. 62:2970–2977. 24. Goodrich, L. D., P. A. Schaffer, D. I. Dorsky, C. S. Crumpacker, and D. S.
Parris.1990. Localization of the herpes simplex virus type 1 65-kilodalton DNA-binding protein and DNA polymerase in the presence and absence of viral DNA synthesis. J. Virol. 64:5738–5749.
25. Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467.
26. Knipe, D. M., D. Senechek, S. A. Rice, and J. L. Smith. 1987. Stages in the nuclear association of the herpes simplex virus transcriptional activator pro-tein ICP4. J. Virol. 61:276–284.
27. Lamberti, C., and S. K. Weller. 1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 226:403–407.
28. Lamberti, C., and S. K. Weller. Unpublished results.
29. Liptak, L. M., S. L. Uprichard, and D. M. Knipe. 1996. Functional order of assembly of herpes simplex virus DNA replication proteins into prereplica-tive structures. J. Virol. 70:1759–1767.
30. Lukonis, C. J., and S. K. Weller. 1996. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J. Virol. 70:1751–1758.
31. Lukonis, C. J., and S. K. Weller. 1997. Formation of herpes simplex virus 1 replication compartments by transfection: requirements and localization to nuclear domain 10. J. Virol. 71:2390–2399.
32. Lukonis, C. J., and S. K. Weller. 1996. The herpes simplex virus type 1 transactivator ICP0 mediates aberrant intracellular localization of the viral helicase/primase complex subunits. Virology 220:495–501.
33. Malik, A. K., L. Shao, J. D. Shanley, and S. K. Weller. 1996. Intracellular
localization of the herpes simplex virus type-1 origin binding protein, UL9. Virology 224:380–389.
34. Martinez, R., R. T. Sarisky, P. C. Weber, and S. K. Weller. 1996. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J. Virol. 70:2075–2085.
35. Martinez, R. M., L. Shao, and S. K. Weller. 1992. The conserved helicase motifs of the herpes simplex virus type 1 origin-binding protein UL9 are important for function. J. Virol. 66:6735–6746.
36. Matusick-Kumar, L., P. J. McCann III, B. J. Robertson, W. W. Newcomb, J. C. Brown, and M. Gao.1995. Release of the catalytic domain No from the herpes simplex virus type 1 protease is required for viral growth. J. Virol. 69:7113–7121.
37. McNab, A. R., P. Desai, S. Person, L. L. Roof, D. R. Thomsen, W. W. Newcomb, J. C. Brown, and F. L. Homa.1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 72:1060–1070.
38. McVoy, M. A., and S. P. Adler. 1994. Human cytomegalovirus DNA repli-cates after early circularization by concatemer formation, and inversion occurs within the concatemer. J. Virol. 68:1040–1051.
39. Mosig, G., D. Ghosal, and S. Bock. 1981. Interactions between the matura-tion protein gp17 and the single-stranded DNA binding protein gp32 initiate DNA packaging and compete with initiation of secondary DNA replication forks in phage T4. Prog. Clin. Biol. Res. 64:139–150.
40. Murialdo, H., and D. Tzamtzis. 1997. Mutations of the coat protein gene of bacteriophage lambda that overcome the necessity for the FI gene; the EFi domain. Mol. Microbiol. 24:341–353.
40a.Newcomb, W. W., and J. C. Brown. Personal communication.
41. Newcomb, W. W., F. L. Homa, D. R. Thomsen, F. P. Booy, B. L. Trus, A. C. Steven, J. V. Spencer, and J. C. Brown.1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free cap-sid formation. J. Mol. Biol. 263:432–446.
42. Newcomb, W. W., F. L. Homa, D. R. Thomsen, Z. Ye, and J. C. Brown. 1994. Cell-free assembly of the herpes simplex virus capsid. J. Virol. 68:6059–6063. 43. Olivo, P. D., N. J. Nelson, and M. D. Challberg. 1989. Herpes simplex virus type 1 gene products required for DNA replication: identification and over-expression. J. Virol. 63:196–204.
44. Patel, A. H., and J. B. Maclean. 1995. The product of UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206:465–478. 45. Patel, A. H., F. J. Rixon, C. Cunningham, and A. J. Davison. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6. Virology 217:111–123.
46. Perdue, M. L., J. C. Cohen, C. C. Randall, and D. J. O’Callaghan. 1976. Biochemical studies of the maturation of herpesvirus nucleocapsid species. Virology 74:194–208.
47. Phelan, A., J. Dunlop, A. H. Patel, N. D. Stow, and J. B. Clements. 1997. Nuclear sites of herpes simplex virus type 1 DNA replication and transcrip-tion colocalize at early times postinfectranscrip-tion and are largely distinct from RNA processing factors. J. Virol. 71:1124–1132.
48. Poon, A. P. W., and B. Roizman. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67:4497–4503.
49. Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857–868.
50. Randall, R. E. 1986. Intranuclear localization of herpes simplex virus imme-diate-early and delayed-early proteins: evidence that ICP4 is associated with progeny virus DNA. J. Gen. Virol. 67:2163–2177.
51. Schaffer, P. A., G. M. Aron, N. Biswal, and M. Benyesh-Melnick. 1973. Temperature-sensitive mutants of herpes simplex virus type 1: isolation, complementation and partial characterization. Virology 52:57–71. 52. Severini, A., A. R. Morgan, D. R. Tovell, and L. J. Tyrrel. 1994. Study of the
structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology 200:428–435.
53. Shelton, L. S. G., A. G. Albright, W. T. Ruyechan, and F. J. Jenkins. 1994. Retention of the herpes simplex virus type 1 (HSV-1) UL37 protein on single-stranded DNA columns requires the HSV-1 ICP8. J. Virol. 68:521– 525.
54. Sherman, G., and S. Bachenheimer. 1987. DNA processing in temperature-sensitive morphogenic mutants of HSV-1. Virology 158:427–430. 55. Sherman, G., and S. L. Bachenheimer. 1988. Characterization of
intranu-clear capsids made by ts morphogenic mutants of HSV-1. Virology 163:471– 480.
56. Steven, A. C., and P. G. Spear. 1996. Herpesvirus capsid assembly and envelopment. In R. Burnet, W. Chiu, and R. Garcea (ed.), Structural biology of viruses. Oxford University Press, New York, N.Y.
57. Sze, P., and R. C. Herman. 1992. The herpes simplex virus type 1 ICP6 gene is regulated by a leaky early promoter. Virus Res. 26:141–152.
58. Szilagyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72:661–668.
59. Tengelsen, L. A., N. E. Pederson, P. R. Shaver, M. W. Wathen, and F. L. Homa.1993. Herpes simplex virus type 1 DNA cleavage and encapsidation