Vol.46,No. 1 JOURNALOFVIROLOGY,Apr. 1983,p.83-93
0022-538X/83/040083-11$02.00/0
CopyrightC1983,AmericanSociety for Microbiology
Genetic
Variability of Herpes Simplex Virus: Development of
a
Pathogenic
Variant During Passaging of a Nonpathogenic
Herpes
Simplex Virus Type 1 Virus Strain in Mouse Brain
H.C. KAERNER,* C. H.SCHRODER,A. OTT-HARTMANN, G. KUMEL,AND H. KIRCHNER
Institute ofVirusResearch,German Cancer Research Center, 6900 Heidelberg,Federal Republic of
Germany
Received 22 July1982/Accepted2December 1982
Herpes simplex virus type 1 ANG(HSV-1 ANG) is originally nonpathogenic for inbred mice upon intraperitoneal intravenous, or intravaginal inoculation. In contrast, micediedof encephalitiswithin 4 to 5 daysafter intracerebral
inocula-tionwiththis strain. HSV-1 ANG wasserially passaged in mousebrains. In two independent series, peripherally pathogenic virus variants had developed and accumulated in the virus progeny after 12 to 15 intracerebral passages. In mixed
infections both nonpathogenic and pathogenic viruses replicated at the primary
site of infection and spread to various organs. However, only the pathogenic
phenotype could be recovered from the spinal cord and the brain. Comparison of therestriction enzymecleavagepatterns of pathogenic ANG andnonpathogenic
ANG virus DNAs revealeddistinctalterations in the S-segment(Us) sequences
bounded by coordinates 0.953 and 0.958 in the prototype orientation and by
coordinates0.862to0.867 in theIs orientationofthe viral genome.However,itis
notknown whether these alterations arephysiologicallyrelevant to the observed
changes in pathogenicity. When coinjected intraperitoneally at 50 to 100-fold
excess, thenonpathogenicHSV-1ANGprotectedmiceagainstitsown pathogen-ic variant as well as against otherpathogenic HSV-1 strains. PathogenicHSV-1 ANG proved to be genetically and phenotypically stable for at least 25 serial passagesin tissue culture at either high or low multiplicityof infection.
It is known from the workofLopez (15-17) and otherinvestigators (11,12) thatanumberof
inbred mice strains differ with respectto their
susceptibilitytoindividual strains ofherpes
sim-plex virus (HSV) upon intraperitoneal (i.p.) in-fection. Conversely, different virus strains
ex-hibit variable pathogenicity in the same inbred
mousehost. Forexample, HSV type1(HSV-1)
WALishighlypathogenicfor6-week-oldDBA/2
mice wheninjected i.p., whereas HSV-1 ANG does not kill the animals under these circum-stances,even atveryhighdosesofvirus(above 107PFUpermouse) (11, 12, 25).The latterfact
has been documented already for very early passages of this strain (passage 3; G. H. von
Mittelstaedt,Ph.D.thesis, Universityof
Heidel-berg,Heidelberg, WestGermany,1972). On the
other hand, intracerebral (i.c.) infection with eitherofthesetwovirus strainsresultsin lethal
encephalitis of mice(7, 23). Therefore, in
addi-tion to thegenetically determinedhost defense mechanisms involved in resistance of mice
againstHSVinfections,virus-encodedfunctions
appear to play a role in different patterns of pathogenicity.
Inthe present paper wedescribethe
develop-mentandproperties ofahighlypathogenic
vari-ant ofa plaque-purified clone of the originally
non-pathogenic HSV-1 strain ANG standard
(21). This variant(HSV-1 ANGpath)originated
in the courseofserial passagesofHSV-1 ANG standard in mouse brain and, in contrast toits parent, kills adultDBA/2miceeven atvery low doses of virus upon i.p. infection. Restriction enzyme analyses revealed that HSV-1 ANG path shows distinct alterations in certainregions
of its genomecomparedwith theparental
apath-ogenicstrain. It alsowill be demonstrated that
both HSV-1 ANG standard and its pathogenic
derivativereplicate similarlyattheprimarysite of infection for some time. However, whereas infectious HSV-1 ANGpathparticlesappear in
thebrain, thenonpathogenicvirus couldnotbe
recovered from thattissue,and thei.p.infected
animals survive.
An additional pathogenic variant of HSV-1
83
on November 10, 2019 by guest
http://jvi.asm.org/
84 KAERNER ET AL.
ANG was isolated from a second independent
series of viruspassages inmousebrainandwas
found by restriction enzymes analyses to be
indistinguishable from the first ANG path iso-late.
MATERIALSAND METHODS
Virus and cells.Plaque-purified HSV-1strains ANG
standard (21) and WAL (20) were propagated on Africangreenmonkey kidney cells, RC 37 Rita (Ital-diagnostics, Rome, Italy),asdescribed earlier(21).
Mice.DBA/2Jmicewereobtained from G.
Bomholt-gard (Rye, Denmark). Males of 4to 6 weeks ofage
wereusedthroughoutthis study.
Extraction andpurificationofHSV DNA. Extraction
and purification of HSV DNA were performed as describedearlier(21).
Restrictionendonucleases. BamHIwas aproductof
BethesdaResearchLaboratories, Inc.,Rockville,Md.
All other restriction enzymes were purchased from
NewEngland Biolabs Inc., Beverly,Mass.Digestions wereperformed by followingthemanufacturers
proto-cols.
Gel electrophoreses of HSV DNA restriction
frag-ments.Electrophoreseswerecarriedoutin0.5to0.8% verticalagarose(Seakem) slab gelsat40 Vfor 18 hat
room temperature. The gels were stained with
ethi-diumbromide andphotographed under UVlight.
Extraction ofDNA restrictionfragments from agar-osegels.Extraction of DNA restrictionfragmentswas
performed according to Langridge et al. (13). The fragmentswereseparatedinlow-melting pointagarose
gels (BethesdaResearchLaboratories).
Passaging of HSV-1 ANG inmouse brain. Plaque-purifiedHSV-1 ANG standard wasused. About3 x 102PFUin0.05mlof 0.15MNaClwereinoculatedi.c. peranimal. Tenmiceper passagewereinfected. The mice died after1week withinanintervalof2to3days.
The brains of the dead animals were recovered and
homogenizedin a10-fold volumeofsaline. The sus-pensionwas centrifuged at 10,000 x g for10 minat
4°C. The supernatant, which was designated as the
firsti.c.passage,wasused forthe similarpreparation
of subsequent i.c. passages. The virus titers, which weredeterminedonRC 37 Rita cells(19),wereabout5
x 105 to 1 x 106 PFU per brain in all of the i.c.
passages. Two independent series of i.c. passages
wereperformed.
Individual i.c.passages weretestedfor
pathogenici-tyini.p. infectionsofDBA/2mice. About2x103PFU wereinjectedperanimal.Thebrains of micethat had
died within 6 and 10 days after i.p. infection were
recovered andassayedfor thepresence ofinfectious
virus.
Determination of virus titers in differentorgansof i.p.
HSV-infected mice. The progress of i.p. HSV
infec-tions in micewasfollowedby titrating infectious virus particles in the peritoneum, the spleen, the spinal cord, andthe brain. The organswereresected atthe
indicatedtimespostinfection, homogenized, and sus-pended in saline. The suspensions were frozen (-70°C)andthawed threetimes, ultrasonicated for5s (position 4, Branson Sonifier), and centrifuged at
10,000x gfor10min.at4°C.From theperitoneumthe viruswasextensivelywashedoutwith1 mlof saline.
All virustitersweredeterminedonRC37cells(19).
RESULTS
Isolation of an i.p. pathogenic HSV-1 ANG
variant from i.c. virus passages in mice. Two
independent series of i.c. virus passages in DBA/2 mice were performed as described above, starting withplague-purified, peripheral-ly (i.p.) apathogenic HSV-1 ANG standard. In-dividual i.c.passages wereassayed for
peripher-alpathogenicityby i.p. infectionof mice. Inone
of these seriesperipheral pathogenicitywasfirst detectedin the 11th i.c.passage,which killed 30
to 40% of the mice upon i.p. infection with approximately 103 PFUper mouse.
Allearlier i.c.passagesprovedtobe peripher-ally (i.p.) nonpathogenic. In the following i.c.
passages pathogenicity increased rapidly: the
15th i.c. passage and all subsequent serial i.c.
passageskilled100% ofi.p.infected mice.Inthe
second series of i.c.passages, peripheral patho-genicitywas first observed in the 10th i.c.
pas-sage, and afteri.p. infection 100% mortality of
micewas achieved with the 13th i.c. passage.
Virusconstituting the 15th i.c. passageof the first series was plaque purified onRC 37 cells. Of 15 picked clones, 3 proved to be highly
pathogenic in that they killed 100% of mice between 6 and 8dayspost-i.p.infection with 300 PFUperanimal. Theremaining cloneswerei.p. nonpathogenic, like their parental clone HSV-1 ANG standard. Thisfinding suggested that the
15th i.c. passage of HSV-1 ANG in mice still
contained a considerable fraction of i.p.
non-pathogenic virus. Analogous results were ob-tained with the 13th i.c. passage of the second series. The above suggestionwas supported by
restriction enzyme analyses of the viral DNA isolated from the progeny of the 15th i.c.
pas-sage,which will bepresented below.Asjudged
by physical markers, this DNAis amixture of
HSV-1 ANG standard andANGpathgenomes.
From these resultsit becomesmoreevident that both thepathogenic and the nonpathogenic
vari-ants replicate when they are simultaneously
inoculated intomousebrain.
Selective appearance of HSV-1 ANG path in
mousebrainafterperipheral (i.p.)infection with
i.c. virus passages. Groups of 10 DBA/2 mice
were inoculated i.p. with (i) HSV-1 ANG
stan-dard, (ii) HSV-1 ANG path (i.e., one of the pathogenic clones isolated from the 15th i.c.
passage as described above), (iii) the 15th i.c.
ANG passage, and(iv) a1:1 mixture of HSV-1
ANG standard and HSV-1 WAL, as acontrol. Within6 to8days
100lo
of the animals died after infection with thepathogenic ANGvariant,the15th i.c. passage, and theANG standard-WAL
mixture.
Thebrains ofthemice killedby virus infection
andofthesurvivingmiceinoculated withHSV-1
ANG standard were resected and assayed for
on November 10, 2019 by guest
http://jvi.asm.org/
GENETIC VARIABILITY OF HSV 85
TABLE 1. Peripheral (i.p.) infection of mice withdifferentHSV-1 stocks
Infectious Genotype'/
Infectingviu PFU/ Mice killed virus per peoye fic
mouse per group mouse brain henotypeo
(PFU) Virus
HSV-1 ANG standard 300 0/10 <2
HSV-1 ANG standard 104 0/10 <2
HSV-1 ANG standard 106 0/10 <2
15thi.c. passage 300 10/10 1.2 x 104 HSV-1 ANGpath
HSV-1 ANG path 300 10/10 1 x 104 HSV-1 ANGpath
1:1 mixture, ANG 105each 10/10 1.4 x 104 HSV-1 WAL
standard/WAL
a Asjudged byrestriction enzyme analyses of viral DNAs (see legends, Fig. 2, 3, and 4).
bHSV-1 ANG standard and ANG pathareSyn; strain WAL is Syn+.
the presenceof infectious virus and its respec-tive phenotypes and genotypes. HSV-1 ANG standard and ANGpatharephenotypically Syn, whereas strainWALisSyn+.Aswillbe shown
below, ANG path DNA could be distinguished from ANG standard DNA by restrictionenzyme analyses. The data obtainedaregiven in Table1. The results showthat, after mixed i.p. inocula-tions with either the "homologous" variants HSV-1 ANG standard and ANG path (repre-sented by the 15th i.c. passage)orthe heterolo-gousstrains ANG andWAL, only the pathogen-ic virustypes ANG pathorWAL,respectively, appeared in the brain of mice. Identical results wereobtained with mixtures of ANG standard and ANGpath for i.p. infection.
Thefollowing experimentswerecarriedoutto test whether the inability to recover the non-pathogenic ANG standard virus from mouse
brains reflected the lack ofvirus replication at
the site of inoculationorthe lack of virus spread
toother tissuesorboth.Groups of 10 micewere inoculated i.p. with 103 PFU of HSV-1 ANG
standard and ANGpath, respectively.At
appro-priate times postinoculation, various organs of
the mice were resected and assayed for the presence of infectious virus. The virus titers
found are listed inTable 2. The data suggested
that both HSV-1 ANG standard and its patho-genic variant replicated similarly atthe site of primary infection and mainlyinthespleen of the
animals.Inagreementwith theprevious results,
only the ANG path virus could be recovered
from the mousebrains.
Finally, HSV-1 ANG pathwaspurified by i.p. infection of mice with i.p. pathogenic i.c. ANG standardpassagesandby isolatingthe pathogen-ic virus from the brain of the animals. All 15
clones pickedfrom intracerebral viruswere
like-wise i.p. pathogenic.
A similar selection ofapathogenic variantof HSV-1 ANGwasachievedusingthe 13th of i.c. passages ofthe second series.
Thecloned HSV-1ANGpathwasalsoplaque purified by transfection of BSC-1 cells with
purified viral DNA. The resulting virus stock
proved to be pathogenic, suggesting that the
acquired pathogenicitywasencodedbytheviral
genome.
The i.p. 50% lethal dose in 4- to6-week-old
DBAI2 mice oftheclonedHSV-1 ANGpathwas determinedtobe about 30 PFUpermouse.
Protection of miceagainst lethali.p. infection
withHSV-1pathby coinfectionorpriorinfection
with HSV-1 ANG. As pointed out above, the
15th i.c. passage of HSV-1 ANG (first series), which killed 100% of i.p. infected mice, con-tained about 80%o(12 of 15 subclones) i.p. non-pathogenicvirus. Precedingserial i.c.passages,
being only partially i.p. pathogenic, proved to
containdecreasing fractions of ANGpath.
From this fact itwas suspectedthat the
non-TABLE 2. Spreadofi.p.infection of mice with HSV-1 ANG standard and ANGpath
Titersof infectious virus(PFU/mouse)'
Organ Timei.p. (days) HSV-1 ANG HSV-1 ANGpath
standard
Peritoneal exudate 1 3 x 103 5 x 103
Spleen 1-4 5 x 103 (max) 1 x 104(max)
Liver 1-7 <2 <2
Kidney 1-7 <2 <2
Spinal cord 6 <2 5 x 104
Brain 6 <2 1-3 x 104
a Titersweredeterminedas
described
in thetextand represent averagevaluesof10mice in each case. VOL.46,1983on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.490.47.439.573.666.2]TABLE 3. Simultaneous i.p. infection with HSV-1 ANG standard and ANG path at different ratios
Infecting virus Total PFU/mouse Mice killedper group
HSV-1 ANG stan- 104
dard
ANGstandard/ANG path mixture
1/1 2 x 104 10/10
2/1 3 x104 10/10
5/1 6 x 104 7/10
10/1 1.1 x 105 2/10
50/1 5.1 x 105 0/10
100/1 10.1 X 105 0/10
HSV-1 ANG path 104 10/10
pathogenic virus type could interfere with the
fatal infection ofits pathogenic variant. It has
been shown earlier(20) thati.p. infection with
an approximately50- to 100-fold excess of the
nonpathogenic strain ANG standard protected
mice against simultaneous lethal i.p. infection
with the heterologous pathogenic strain WAL.
The mechanisms involved in this phenomenon
are not understoodatthemoment.
Inanalogous experiments micewere injected
i.p. with different mixtures of HSV-1 ANG standard and ANG path. The results are
com-piled in Table 3 andsuggestthat ANG standard protected theanimals when injected inat least
10- to 50-fold excess over the pathogenic
vari-Hind III
Eco RI
KpnI
BamHi
ant. Regarding the i.p. inoculation with i.c.
passages that resulted in 100% mortality, this
findingsuggeststhatthefraction of ANGpathin these virus passages exceeded 10%of the total infectious virus.
Kinetics of accumulation of HSV-1 ANG path
inthe progenyduring serial i.c. viral passages of
ANG standard: doesANGpath originate
sponta-neously from ANG standard or is it originally
present in the parental clone? There are two
possible explanations for the accumulation of ANG path inmousebrain. Either ANG pathwas
present at a low copy number in the original
ANG standard, due to its spontaneous
genera-tionduringplaquecloning in tissue culture (i.e.,
representingaminor fraction of theprogenyina
single plaque), oritdevelopedduringi.c. repli-cation of ANG standard.
One argument supporting the latter
assump-tion is the fact that a considerable number of individual HSV-1 ANG subclones isolated in
this laboratory in the course of several years
from infected cells proved to be peripherally
nonpathogenic for mice. Further evidence to
this point could be provided by the following
experiments. Five mice were i.c. coinfected
with1 PFUof ANG path and200 PFU ofANG
standard per mouse. As expected, all of the animals diedwithin 3to 4days. Thebrains ofthe
deadanimalswererecovered andmixed
togeth-er.Thetiter ofprogenyviruswasdeterminedto
beabout 1.5 x 105PFUper mouse.The homog-enized brains were used for subsequent serial
H 0-I J A k L D M N, G
J D G ,N F MPLi A I E Hk ,k
G Y E H
ElI$SSNIP
V B L C A ,,T,OI J Ql I F kIAl Z
E C IIA IMITI 1P,VI1 G IRII DI H __I F WilB ;k'SN, I
D W IY
L
0 0,1 0,3 Q4
Map Units L-S Joint
FIG. 1. Scale maps ofHindIll, EcoRI, KpnI, and BamHI endonuclease cleavage sites on the prototype isomerofHSV-1 ANGDNA.Thejointof theL-andS-segmentsis markedbyaverticalline.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.490.75.440.431.653.2]GENETIC VARIABILITY OF HSV 87
i.c. passages, injecting 200 PFU per mouse in all cases.The individual i.c. passages were assayed forperipheral pathogenicity by i.p. infection of mice with 3 x 102PFU per mouse. In each of two completely independent series of virus pas-sages thethird i.c. passage displayed about 50% mortality in groups of 10 mice, and the fourth
i.c. passages killed 100% of the animals (i.e.,
contained about 10 to 20% pathogenic virus).
BamH 1 1.8/ AG AROSE
-p.o
.... a
-Ts
---_S
-~u
-W.X.w. .Z
zB
A
-Y
-A -B
When compared with the results of serial i.c. passaging of the pure ANG standard described above, thesefindingssuggested that (i) the vari-ant clone ANG path is not contained in the
parentalvirus population ofHSV-1 ANG stan-dard but originated spontaneously during i.c.
replication; and (ii) ANG path has a consider-able growth advantage over ANG standard n
mouse brain tissue. Furthermore, it appears
BamH 0,.6/ AGAROSE
[image:5.490.97.382.188.606.2]... ...
K
-N.
-N
--a
_---A
-B
-. DC
-F
H G
-Q.,P. __
v--Y
I'
ci
Qi
nL
ci
Ci,
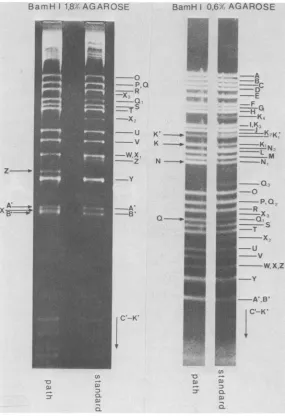
a-FIG. 2. Ethidium bromide-stainedBamHI restriction fragmentpatternsof HSV-1 ANG standard DNA and
HSV-1 ANG pathDNA. Toachieve betterresolution, the fragments were run through 1.8 and0.6%agarosegels.
The nomenclature of the fragments refers to the physical map of BamHI cleavage sites in Fig. 1. Subscript
numbers indicate steps of "band ladders" originating by stepwise amplification of500-bpsequences inthe
inverted S-repeats TRS andIRS(10). The fragmentsconcerned(Fig.1)intheseamplificationsareBamHI-X, -Q, -K and-N (Fig. 1).Arrows markalterations of fragment mobilities in ANG path DNA compared with ANG
standardDNA. VOL.46,1983
on November 10, 2019 by guest
http://jvi.asm.org/
KAERNER ET AL.
likely from the kinetics of the accumulation of
ANG path that its formation is a multiple-step rather than asingle-step event.
Restriction enzyme analyses and mapping of
sequence alterations of HSV-1 ANG path DNA.
Figure1showsvariousrestriction endonuclease
cleavage maps of HSV-1 ANG standard DNA.
Aswas reported earlier (8, 9),theS-segment of
the HSV-1 ANG genome displaysadiscretesize
heterogeneity due to multipleamplification of a
500-base pair (bp)sequencemappingnearthe S-terminus in the S repeatTRs and at the
corre-sponding regioninIRS.Asa consequenceofthis
amplification allDNArestriction fragments
con-E, ..
..-_ %!..i
t::
...Lt
C;g8,~
_. _
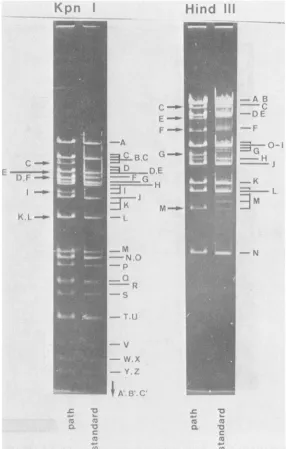
FIG. 3. Ethidiumbromide-stainedKpnIandHindIII restriction fragments of HSV-1 ANG standard and ANG path DNAs electrophoretically separated in 0.8 and0.6%agarose gels, respectively. Arrows indicate alterations offragments in ANG pathDNA.As a consequence of theamplification of500-bpsequences inTRs andIRS,the following fragmentsarestepladders of minor bands:KpnI-K, -I,-C, -D; HindIII-B, -C, -E, -F, -G, and -M. In the
casesofhigh-molecular-weight fragments, the ladders are not resolved in these gels and appear as broad smears.
'r- 1,
)
-1-4w,
:",, X,
.,i %..
I
i
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.490.111.397.166.615.2]VOL.46,1983
taining this sequence, i.e., S-terminal and L-S
joint fragments, appear as "stepladders" of
DNAbands differing in size by500bp.Recently, A. Podbielski (Ph.D. thesis, University of
Hei-delberg, Heidelberg, West Germany, 1982) in ourlaboratory hasfound that anothersequence
of similar size is amplified in the
TRs-Us
andIRS-Us boundaries of HSV-1 ANG DNA. All
restriction fragments containingthese
amplifica-tions also form stepladders in agarose gels.
Individualfragments that display this
phenome-non are listed in thelegendsof Fig.2, 3, and 4.
The BamHI, KpnI, HindIII, and EcoRI
re-striction patterns of ANG path DNA are
com-pared with the corresponding ANG standard DNA patterns in Fig. 2, 3, and 4. The
corre-sponding patterns of the ANG path variant
iso-lated during the second series of i.c. passages are identical to those in Fig. 2 to 4 (data not shown). The identity of the altered ANG path
DNA fragments was further verified by blot
hybridizationof KpnIfragmentsFandKand the
totalof BamHIfragmentsof HSV-1 ANG
stan-dard DNA to BamHI restriction patterns of ANG standard andpath(Fig. 5). Thefollowing
alterations wereobserved. (i) ANG path DNA
displays no amplification of the 500-bp
se-quences at both ends of
TRs
andIRS;
i.e.,BamHIfragments Q, X, N,and K, EcoRI
frag-ments K, B,C,andH,HindIIIfragmentsB,C,
E, F,G, and M, andKpnIfragmentsK, I,C,and
D are nolonger stepladdersof DNA bands but
form singlebands in therespective gels (Fig. 2,
3,and4). (ii)Thereisadeletion of about 500bp
in ANGpath DNAmappinginBamHI-X(Fig.2
and 5). This deletionapparently does not con-cern EcoRI-K, KpnI-C and -I, BamHI-N, and
HindIII-M, but it does concern HindIII-G and
KpnI-D and -K. It therefore must map exclu-sively in
Us,
spanningthe left end ofBamHI-X,and doesnotcompriseTRSorIRSsequences.(iii)
Another deletionofapproximately150bp
appar-entlymapsin BamHI-ZofANGpathDNA(Fig.
2 and 5). (iv) Fragment KpnI-F of ANG path DNA is about 230 bp smaller than the
corre-sponding ANG DNA fragment (Fig. 3 and 5). Since KpnI-F overlaps BamHI-Z completely,
one might assume that the 150-bp deletion in
BamHI-Zispartof the230-bpdeletionmapping
inKpnI-F.Theremaining80bpdeleted in
KpnI-F could not be unequivocally localized. Since
neither BamHI-JnorBamHI-N displays altered mobilities, it could concern the small BamHI
fragment of200bp recentlydetectedbyWatson
andVande Woude(26)tomap between
BamHI-Xand -Zandwhichwecouldnotidentifyin our
gels.
Of interest was how far the physical
alter-ations found tobeassociated withthe DNA of
ANGpathcorrelates specificallywiththe
prop-GENETIC VARIABILITY OF HSV 89
erty of peripheral pathogenicity. As shown
above, the 15th i.c. passages ofHSV-1 ANG
proved to be a mixture of about 80o ANG
standard and 20% ANG path phenotypes. In
Fig.6 the KpnI restrictionpatternof the parental
ANG standardDNA is compared with those of
ANG path DNA and the DNA isolated from
virions of the 15thANG standard i.c. passage.
-A.B
C.DE..F.G
H.
_~
I
K,L--.K..K
----KK
;j
... L
K.
---M
--N
-Oo
cn
::s CL
Q6)
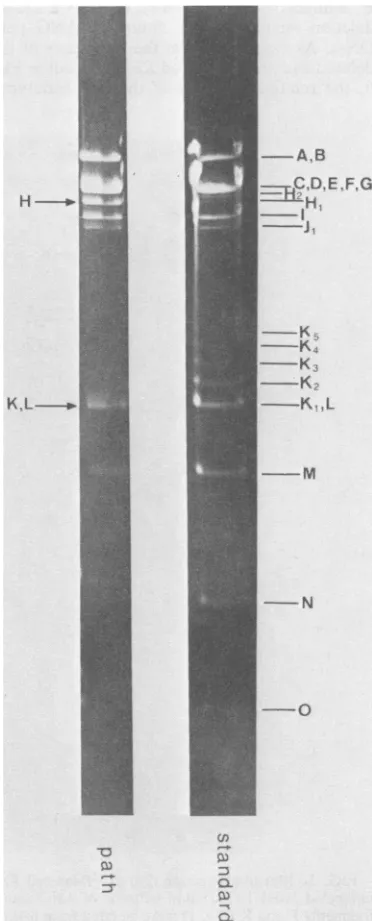
FIG. 4. EcoRI restriction fragment patterns of HSV-1ANG standard and path DNAs in0.8%agarose
gels. Arrows mark fragments altered in ANG path
DNAcompared with ANG standard DNA.
i'liv..110
H 10
I
.:-41.on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.490.252.438.167.628.2]90
The latter pattern shows that the total popula-tion of DNA molecules has lost the 500-bp amplifications at both ends of
TRs
andIRS
found in the parental ANG standard DNA. Fur-thermore, 15thi.c. passage DNA appears homo-geneous with respect to the 500-bp deletion located in KpnI-K. The only detectable evidence that this DNA would represent a mixture of different genotypescomesfromfragment KpnI-F, a minor fraction of which displays a 230-bp deletion similar to that found in ANG pathDNA. As estimated from the intensities of the
deleted and the nondeleted KpnI-F bands in Fig. 6, the relative amounts of the two genotypes
N_* |
K1-n
N- W
''X
z o..$;.... AW.I<<f.:
."
-.. xl
a b c
correlate approximately to the amounts of the corresponding phenotypes ANG path and ANG standard present in the mixture.
The results indicate that the amplification of the500-bp sequenceslacking in
TRs
andIRS
of theparental ANG genome as well as the 500-bp deletion in KpnI-K are notspecifically associat-ed with the observed peripheral pathogenicity. The only physical marker which can betenta-tively correlated to the pathogenic phenotype
from the present results is the 230-bp deletion mapping in KpnI fragment F (Fig. 3 and 5). This assumptionwasfurther supported by restriction analyses of viral DNA extracted from a number
-
K1
0
-Y
-Xi
d e f
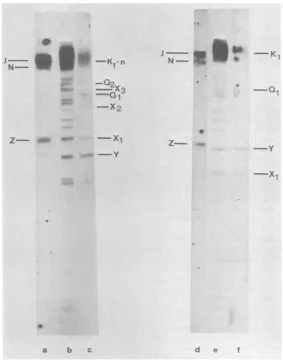
FIG. 5. Blot hybridization (23) of32P-labeledKpnI restrictionfragmentsof HSV-1 ANGstandard DNAto
unlabeledBamHI restrictionpatternsof ANG standardandpath DNA, monitored byautoradiography. KpnI
fragmentsFand K(Fig.1)wereisolated fromlow-melting-pointagarosegels (13)andwere32P-labeledbynick
translation (18). Lanesaand c, BlothybridizationofKpnI-Fand-K,respectively,to BamHIrestrictionpatterns
ofANG standard DNA. Lanes d andf,BlothybridizationofKpnI-Fand-K,respectively,toBamHI restriction
patternsof ANG path DNA. Thecomplete BamHI restrictionpatternsof ANG standard andpath DNAs(lanesb
and e, respectively) are visualized by blot hybridization of the total of KpnI restriction fragments of the respectiveDNAs.
J. VIROL.
A,:-j x VW
N %
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.490.113.396.233.593.2]GENETIC VARIABILITY OF HSV 91
C-del. F L
K L _
-A
t .>
~~~B.C
D.
7=
-Q-I _-E ;_] i~~
-T. U
v
-W.x
-Y.z
- A.
i B',CI
C-'
I.
-r.l
FIG. 6. Comparison ofKpnI restriction fragment
patternsofANGpathDNA and DNA extractedfrom virionsofthe 15th of serialpassagesof HSV-1 ANG
path. Arrowsmark ANGpathDNAfragmentsaltered ascompared with the corresponding ANG standard
DNAfragments.
ofplaque-purified nonpathogenicclones isolated
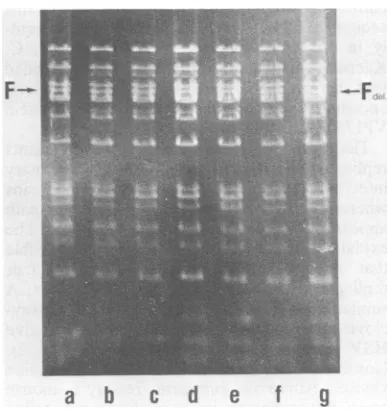
from the 15th ANG i.c.passage. Figure7shows
the KpnI restriction patterns offive
nonpatho-genicclones. None ofthemdisplaysthe 230-bp
deletion in KpnI-F. However, there is no
evi-denceatthemomentthatthe deletion inKpnI-F
wouldinanyrespectphysiologicallycorrelateto
the alteredpathogenicitypattern of HSV-1 ANG
path.
It should be pointed out that HSV-1 ANG path was found to begenetically and
phenotypi-cally stableduringatleast 25 passages in RC 37 Rita cells in tissue culture performed so far at eitherhigh (10 PFU per cell) or low(0.01 PFU
percell)multiplicity.
DISCUSSION
Infection i.p. of inbred mice with HSV is widely used as amodel to study antiviral defense mechanisms (11, 15, 24, 28). The mice die from encephalitis about 5 to 10 days after i.p. infec-tion with HSV-1. It is well established that there
isconsiderablevariability regarding the i.p.
sus-ceptibility of different mouse strains to
individ-ual HSV-1 strains and vice versa. Similar obser-vations have been reported for other routes of infection byseveral authors (4, 6). In addition to the HSV-1 strain HFEM, which was
demon-stratedby Lopez (15) to be completely avirulent
in i.p. infections for aconsiderable number of mouse strains, it was found in our laboratory thatHSV-1 ANG standard is peripherally
non-pathogenic for mice(12, 20). It should be
empha-sized here that HSV-1 ANG standard is also
nonpathogenicfor mice in intravenous and
intra-vaginal infections (Kumel, unpublished data). We have focused on the possible role of viral
geneticsin thephenomenon of different
suscep-F
-
F
a
b
cd
efg
FIG. 7. KpnIrestrictionfragment
patterns
ofindi-vidualnonpathogenicclones,plaquepurifiedfrom the 15th i.c. passageof HSV-1 ANG standard. The
pat-ters are compared with ANG standard DNA and ANG path DNA as controls. The fragments were
electrophoretically separated on a0.8% agarose gel.
Individualtracks show thepattersof: a, ANG
stan-dard;b-f, nonpathogenicclones 1 to5;g,ANGpath.
VOL.46,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.490.51.231.61.505.2] [image:9.490.250.444.387.593.2]KAERNER ET
tibilities ofmicetoperipheral(i.p.) HSV-1 infec-tions. The development ofahighly pathogenic varient, ANG path, from HSV-1 ANGstandard promisedtobeavaluabletool in such studies. It is of interest that HSV-1 ANG standard was originally avirulent for mice (von Mittelstaedt, Ph.D. thesis). Most recently, 2 of 20 tested HSV-1 strains freshly isolated from patients have beendemonstratedinourlaboratorytobe avirulent for mice in i.p. infections (M.
Batta-Buchle and H. C. Kaerner, unpublished data). Furthermore, we were able to develop i.p. pathogenic variants of both of these strains, which surprisingly originated following similar kinetics asANG path in thecourse of 13 to 15 consecutive i.c. virus passages in mice. From these findings it appears that the property of peripheral virulence is genetically acquired de novo in a multiple-step process by unknown mechanisms involved in the interaction ofthe
viruses with braintargetcells.
Nothing is currently known concerning the
HSV-1viral functions involvedinpathogenicity.
Thephysical alterationsfoundonthegenomeof
ANG path partially mapinregions which have been foundtobe "hotspots"ofgenetic
variabil-ity of HSV-1byanumber ofinvestigators (5, 14, 22)and as yetare physiologicallyundefined. It shouldbe mentioned that the amplification ofa 500-bpsequencelackingatthe S-terminus(TRS)
of the ANG standardgenomeobsereved in ANG
path DNA does not mean the total loss of this
sequence: DNAsequencedataobtained recent-ly in our laboratory (C. P. Gray and H. C. Kaerner, manuscript in preparation) revealed
that this sequence specifies part of the mRNA
encoding for the nonstructural virus protein VP175 (3).
That both the avirulent and virulent variants
replicatetoasimilarextent atthe siteofprimary infection and readily spread to several organs
whereas only infectious virions of ANG path appearin the brainofi.p.infectedmice could be explained in several ways. First, it is possible
that the avirulent strain is unable to infect or
replicate in the cells of the nervous system. A
similarexplanationhas beensuggestedfor
Acy-clovir-resistant and thymidine kinase-negative
HSV mutants by Field and co-workers (1, 2). However, HSV-1 ANG standard is thymidine
kinase positive and replicates readily in mouse
brain,if itis directly injected.Anotherpossible explanation is that strain ANG induces an
ex-traordinary early actinghost defense mechanism
upon peripheral infection which prevents virus
replication in the brain and resultant lethal en-cephalitis, ashas been suggestedby Kastrukoff
et al. (10). One argument against the latter
hypothesis is our finding that 4 days after i.p. infectionwith ANG standardnoviralantigensor
J.VIROL.
antibodies could be detected in the brains of the micebeyond theblood-brain barrier, whereas in the case of ANG path infection the brains of the animals contain significantamounts of both viral antigens and antibodies (H. Fischer and H. C. Kaerner,unpublisheddata).
Ithas also been found that ANG standard and ANG path do not differ with respect to the stimulation of NK cells (Kirchner, unpublished data).
Wewould prefer at the moment the hypothe-sis thatHSV-1 ANG standard can neither repli-cate in the cells of the central nervous system nor betransported axonally to the brain or pass theblood-brain barrier.
Experiments are now under way to introduce the property of peripheral pathogenicity by marker transfer techniques from HSV-1 ANG path and other virulent HSV-1 strains into the genome of ANG and otheravirulent strains to identify genomeregions andfunctions involved in thephenomenonof neurovirulence.
LITERATURE CITED
1. Field, H. J., J. R. Anderson, and P. Wildy. 1982. Atypical patterns of neuralinfectionproduced in mice, by drug-resistant strainsofherpes simplexvirus. J. Gen. Virol. 59:91-99.
2. Fleld, H. J., and P. Wildy. 1978. The pathogenicity of thymidine-kinase deficient mutants of herpes simplex virus in mice. J. Hyg. 81:267-277.
3. Haillburton,I. W.1980.Intertypic recombinantsofherpes simplex viruses. J. Gen. Virol.48:1-23.
4. Harbour, D. A., T. J. Hill, and W. A.Blyth.1981. Acute andrecurrentherpes simplexin several strainsof mice. J. Gen.Virol.SS:31-40.
5. Hayward, G. S., N. Frenkel, and B. Roizman. 1975. Anatomyof herpes simplex virusDNA: straindifferences and heterogeneity ofrestrictionendonuclease cleavage sites. Proc. Natl. Acad.Sci.U.S.A. 72:1768-1772. 6. HIll,T.J., H.J. Field, and W. A. Blyth. 1975. Acute and
recurrent infection with herpes simplex virus in the
mouse: amodelforstudyinglatencyand recurrent dis-ease.J.Gen. Virol. 28:341-353.
7. Johnson, R. T. 1964. Thepathogenesis of herpessimplex virus encephalitis. I. Virus pathways to the nervous system of suckling mice demonstrated by fluorescent antibody staining.J. Exp.Med. 119:343-356.
8. Kaerner, H.C., J. B. Maichle, A. Ott, and C. H. Schr6der. 1979.Origin oftwodifferent classesofdefective HSV-1 AngelottiDNA. NucleicAcidsRes. 6:1467-1477.
9. Kaerner, H. C., A. Ott-Hartnann, R. Schatten, C. H. Schr6der,andC. P.Gray.1981.Amplificationofashort nucleotidesequencein the repeat units of defectiveherpes simplexvirus type 1AngelottiDNA.J.Virol. 39:75-81.
10. Kastrukoff, L. F., C. Long, and H. Koprowskli. 1981. Herpes Simplex virus immunosystem interaction in a
murine model, p. 320-325. In A. J. Nahmias, W. R. Dowdle, and R.F. Schinazi (ed.), The human herpesvi-ruses.ElsevierPublishingCo., New York.
11. KirchnerH.,M.Kochen, H. M.Hirt, and K. Munk. 1978. Immunological studies of HSV infection of resistant and susceptibleinbredstrains of mice.Z. Immunitaesforsch. 154:147-154.
12. KirchnerH., M. Kochen, K. Munk, H. M. Hirt, S. E. Mergenhagen, and D. L.Rosenstrelch. 1978. Differences in susceptibilitytoherpes simplexvirusinfection of inbred strainsof mice. IARC (Int. Agency Res. Cancer)Sci. Publ. 24:783-788.
on November 10, 2019 by guest
http://jvi.asm.org/
GENETIC VARIABILITY OF HSV 93
13. age, J., P. ngge,and P. L. Berquist. 1980.
Extraction of nucleic acids from agarose gels. Anal. Biochem. 103:264-271.
14.Lonsdale,D. M.,S.M.Brown, J.Lang,and J. H.
Subak-Sharpe. 1980. Variations in herpessimplex virus isolated from human ganglia and a study of clonal variation in HSV-1. Ann. N.Y. Acad. Sci.354:291-308.
15. Lopez, C. 1975.Genetics of natural resistanceto
herpesvi-rusinfections in mice. Nature(London) 253:152-153.
16. Lopez, C. 1978. Oncogenesis and herpesviruses. IARC (Int. AgencyRes. Cancer) Sci. Publ. 24:775-781.
17. Lopez,C. 1981. Resistancetoherpessimplex virus-type1
(HSV-1). Curr.Top. Microbiol. Immunol. 92:15-24. 18.Rigby, P. W., J. M. Dleckmann, C. Rhodes,andP. Berg.
1977. Labeling deoxy-ribonucleic acid to high specific activity in vitroby nick translation with DNA polymerase I. J. Mol.Biol. 113:237-251.
19. Russel, W. C. 1962. A sensitive and precise plaqueassay
forherpes virus. Nature (London) 195:1028-1029. 20. Schr0der, C. H., H. Engler, and H. Kirchner. 1981.
Protection of mice byan apathogenic strain of HSV-1 against lethalinfection byapathogenic strain of HSV-1. J. Gen. Virol.52:159-161.
21. Schr6der, C. H., B.Stegnan,H. F. Lauppe,andH. C. Kaerner.1975176. An unusual defectivegenotypederived fromHerpesSimplex Virus1ANG.Intervirology
6:270-284.
22. Skare, J., W. P. Summers, and W. C. Summers. 1975. Structure and function of herpes virusgenomes.I.
Com-parison of five HSV-1 andtwoHSV-2 strainsby cleavage of their DNA with EcoRI restriction endonuclease. J. Virol.15:726-732.
23. Southern, E. M. 1975. Detection of specific sequences amongDNA fragments separated by gelelectrophoresis. J. Mol. Biol. 98:503-517.
24. Stevens, J. C., and M. L. Cook. 1971. Restriction of herpes simplex virusby macrophages. An analysis of cell-virus interactions. J.Exp. Med. 133:19-38.
25. Wildy, P.1967. The progression of herpessimplextothe centralnervoussystemof themouse.J. Hyg. 65:173-192. 26. Watson, R. J., andE. F. Van de Woude. 1982. DNA sequence of an immediae-early gene (IE mRNA5) of herpes simplex virustypeI.Nucleic Acids Res.
10:979-991.
27. Zawatzky, R., J. Hilfenhaus,F.Marcucci,andH.
Kirch-ner. 1981. Experimental infection of inbred mice with
herpes simplex virus. I. Investigation of humoral and
cellular immunity and of interferon induction. J. Gen. Virol. 53:31-38.
28. Zisman, B., M. S. Hirsch, and A. C. Alison. 1970. Selective effects ofanti-macrophage serum, silica and
anti-lymphocyteserum onpathogenesis ofherpes simplex
virus infection ofyoung and adult mice. J. Immunol.
104:1155-1159. VOL. 46, 1983
on November 10, 2019 by guest
http://jvi.asm.org/