Soluble Interleukin-6 Receptor-Mediated Innate Immune Response to DNA and RNA Viruses
Full text
Figure
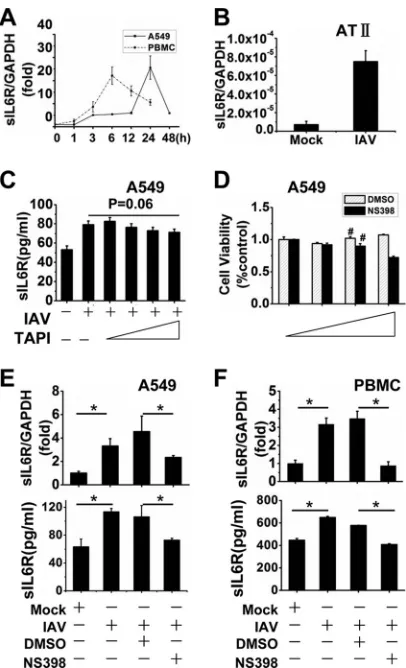
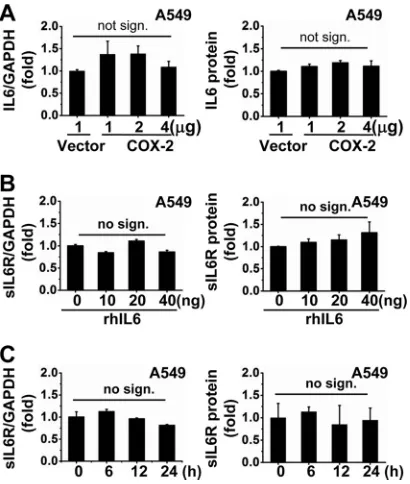
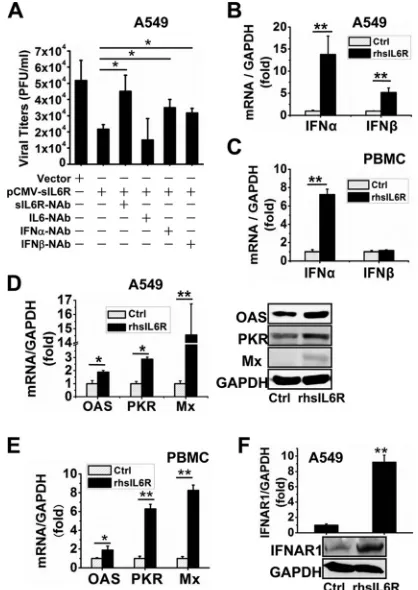
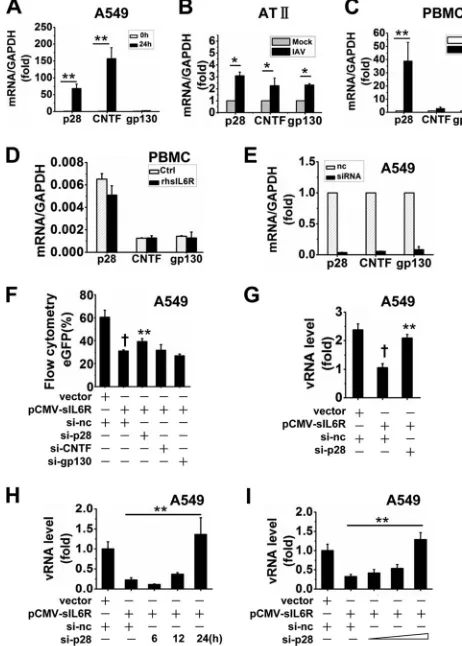
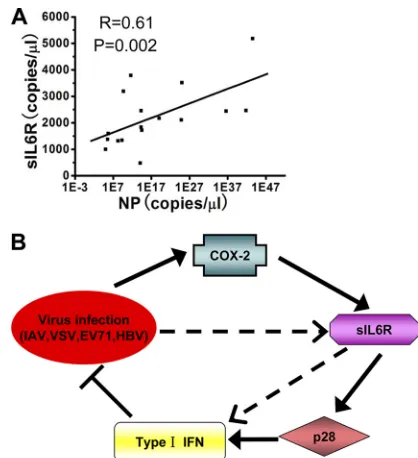
Related documents
Toll-like receptor 4 (TLR4) has been found to partici- pate in antiviral defense to RNA viruses in human cells by recog- nizing the fusion protein of respiratory syncytial virus
Measurement of the viral titer showed that overexpression of RIG-I Nter, as previously observed with MAVS, decreased the viral titer 10,000-fold compared to that in control
However, the overex- pression of miR-212 significantly decreased ZEB2 protein levels in A549 cells, while inhibi- tion of miR-212 significantly increased ZEB2 protein
After steroid treatment, the mean plasma levels of TPO showed a significant decrease, but platelet count was significantly increased as compared to ITP group before the treatment,
Inhibition of either tissue factor or thrombin in WT mice also significantly increased CVB3 levels in the heart and cardiac injury compared with controls.. BM
IL-18R α protein levels in RA-FLS remained unaffected by IFN- γ stimulation in western blotting experiments ( n = 3, data not shown), and IL-18R α mRNA levels in U937 cells were
The levels of IFN- α produced by SLE peripheral blood mononuclear cells (PBMCs) induced by SLE serum that contained an endogenous IFN- α -inducing factor, herpes simplex virus type
Mice deficient in IL-1RI signaling showed reduced expression of IL-10 and type I IFN and increased susceptibility to dextran sulphate sodium–induced colitis and failed to mount
