Vol.64, No.6 JOURNAL OFVIROLOGY, June1990,p.2588-2593
0022-538X/90/062588-06$02.00/0
Copyright© 1990,American Society for Microbiology
Specific Effect of Interferon
on
the
Herpes Simplex
Virus
Type 1
Transactivation Event
P. RANIERO DE STASIO ANDMILTON W. TAYLOR*
PrograminMicrobiology, Department ofBiology, Indiana University, Bloomington, Indiana47405 Received6 December1989/Accepted 21 February 1990
Human recombinantgammainterferonandtoalesser extentalphainterferonwereshown to inhibitherpes simplex virus type 1 replication in the human cell lines WISH and HEp-2. Dot blot analysis of viral DNA synthesis and viralRNAtranscription indicated that the inhibition occurredatanearlystep in infection. A study ofthe earlyevents after herpes simplex virus type 1 infection indicated thatadsorption, penetration, uncoating,and transportof viralDNAwerenotaffectedbyinterferon. Northern(RNA)blotanalysisrevealed that bothimmediate-early and delayed-earlygenetranscriptionwasinhibitedbyinterferon.Transactivationof theimmediate-early responsive element linkedtoareportergene(CATortk)wasspecificallyinhibitedbyboth classes of interferon. Our data would indicate that either the transactivating protein VP16 or thecomplex formed between VP16 andahostprotein(s)isattenuated byinterferon.
The molecular basis of interferon (IFN) antiviralactivity hasbeenonly partly elucidated. Thedouble-stranded RNA-dependent 68-kilodalton protein kinase complex and the
2'-5'-oligoadenylatesynthetaseinducedby IFN treatmentof responsivecells have been directly correlated with the onset of the antiviral state forencephalomyocarditis and mengo virus (2). The mechanism whereby the IFN-induced genes
discriminate between viral and cellular mRNAs has only
been partially determined (7, 14). Both groups have pro-posedanintracellular localizationof the2'-5'-oligoadenylate synthetaseand theprotein kinaseactivities in thevicinityof viral double-stranded RNA molecules. Many DNA viruses are sensitive to IFN action by mechanisms still unknown (13).
Herpes simplex virus type 1 (HSV-1) replicates via the
expression ofat least three groups of genes regulated in a
temporal cascade:immediate-early (IE), delayed-early(DE), and late (L) genes (6). Two IE genes, ICPO andICP4, are
strictly requiredfor thetranscriptionof theDEgenes(6, 22). Noexpressionof DE and L genesoccursif IE genesarenot properly expressed (since at least one of the DE genes is a
transactivator ofLgenes). L genes areexpressedonly after
the startofDNAsynthesis,and HSV DNA is synthesized by aviralDNApolymerasethatbelongstothe DE gene group. All five IE genes share a common characteristic: an up-streamenhancerlikeregulatory sequencecontainingthe
mo-tif TAATGARAT(R =purine) (18, 20, 26).This sequenceis
involvedin theregulation of IE genes through a
transactiva-tion event mediated by the virion (tegument) protein VP16 (also called Vmw65 or trans-inducing factor). Preston et al.
(20)andO'Hare et al. (17, 18) have shown by gel-shift assays that the IE gene transactivation by VP16 occurs via the
formation ofa protein complex (IE complex [IEC]) rather than by direct interaction of the protein with HSV-1 DNA sequences. This complex is made up of the cellular octamer
bindingfactor, TAATGARAT recognition factor (5), oraH
(8) and possibly other cellular factors. The cellular
compo-nent(s) ofthis complex are responsible for the interaction with DNA, while VP16 is required for interaction with the RNApolymerase 11 (21, 23).
The model described for the IE transactivation suggests
*
Corresponding
author.thatVP16 isrequired for efficient infection with HSV-1. This concept has been confirmedbyTriezenberg and co-workers (4), whodemonstrated that cellsexpressing a truncated form of VP16 (lacking the activating C-terminal domain) are restricted in HSV-1 replication.
Obermann and Panet (15, 16) have studied the effect of alpha IFN(IFN-a) on theearly stages of HSV replication. They report inhibition of IE gene transcription but no effect of IFN on any stepsprior to transactivation. Viraluncoating appears to be normal in IFN-treated cells. These authors proposethat theeffectof IFN-amustbe on somefacet ofthe VP16-complex interaction(16).
In agreement with theabovefindings,wereportan effect ofIFN--y on the early stages of HSV-1 replication. Viral DNAsynthesis,aswell as IE and DE genetranscription, is inhibited in IFN-treated cells. Similar results were found whenchimeric plasmids were used, involving activationof IE genesby VP16. These results indicate that inhibition of HSV-1replication by IFN involvesa stepinthe interaction of VP16 with the IEC complexor actiononVP16 itself.
MATERIALS ANDMETHODS
Cell lines and viruses. Cell lines and viruses wereobtained from the American TypeCulture Collection(ATCC, Rock-ville, Md.). African green monkey kidney (Vero), human
larynx carcinoma (HEp-2), amniotic (WISH), and murine L-929 cellswere grown in Dulbecco minimal essential me-dium (GIBCO Laboratories, Grand Island, N.Y.)
supple-mented with 100 ,ug of streptomycin per ml, 100 U of
penicillin per ml, 2 mM glutamine, and 5% fetal bovine serum (Hyclone,Logan, Utah).
HSV-1(strainMP,providedbyB.Roizman, Chicago,Ill.) wasplaque purified and was grown andtitered on Vero cells. Aliquots of theoriginalstock obtainedafter plaque
purifica-tionwerestoredat-80°C, and each vial was used onlyonce. Encephalomyocarditis virus was purchased from ATCC (strain VR 129-B), plaque purified, and grown and titered on L-929orWISH cells.
Viralinfections.Viralinfections wereperformed in 60-or 100-mmplastictissue culture dishes(CorningGlass Works,
Corning,N.Y.). Immediately priortoinfection, the medium was removed and the monolayer was rinsed twice with
phosphate-buffered saline. One of the cultures was
2588
on November 10, 2019 by guest
http://jvi.asm.org/
trypsinizedtocalculatethe total cellnumberperdish. A 0.2-to 0.4-ml portion ofviral lysate (diluted to the appropriate concentration of PFUpermilliliter) wasadded tothe mono-layer at a multiplicity of infection (MOI) of 1. Virus was allowedtoadsorb at37°Cfor 60 min,after which 3or 10 ml of medium was added. Viral incubation was terminated at appropriate times,and cellswereharvested eitherby usinga cell scraperorby trypsinization.
IFN pretreatment of cells. WISH, HEp-2, or Vero cells were treated 14 to 16 h prior to infection with human recombinant IFN-a or --y (Genentech Inc., San Francisco, Calif.) at the concentration shown in each experiment. Following theIFN treatment, viruswas added as described above. Fresh IFN was added to the medium during viral incubationin someexperiments. AllIFNpreparationswere assayed routinelyonWISHorHEp-2cellsby using enceph-alomyocarditis virus, and they were titered with National Institutes of Health standard IFN preparations.
Plaque assays. HSV-1 lysates were titered on Vero cells. Plaque assays wereperformed on monolayers of Vero cells overlaid with 1.3% methylcelluloseand 2% calfserum. The
monolayers were stained with crystal violet for plaque counting36 to48 h postinfection.
Purification and labeling of herpes viral particles. HSV-1 wasgrown onVero orWISH cellsasdescribed above. Viral particleswerepurified bythemethod ofSpearand Roizman (25), withsomemodifications. After clarification of the viral supernatants, the virus was pelleted by centrifugation in a
rotor(model SW27; Beckman Instruments, Inc., Fullerton, Calif.)at15,000rpmfor2 h at 4°C.The viralpellet wasthen
suspended in 50 mM Tris hydrochloride (pH 7.5)-10 mM EDTA and layered onto a continuous 5 to 40% sucrose
gradientmade in the samebuffer. Theviruswascentrifuged throughthe sucrosegradientinarotor(model SW27;
Beck-man) at 15,000 rpm for 2 h at 4°C. Purified virions were collected as a light-scattering band about halfway through the gradient by piercing the tube with an 18-gauge needle. Labeled virions were obtained by adding 5 ,uCi of
[3H]thymidine per ml to the incubation medium after viral
adsorption.
RNA isolation. TotalRNA frominfected, transfected, and uninfected cells was isolatedby the single-step method (1). RNA concentrations were determined by optical density readingsat 260 and 280 nm.
Plasmids and molecular probes. HSV-1 recombinant plas-mids pRB201 (containing IE genes on HindIII restriction
fragmentsH and M in pBR322) andpRB123 (containing DE and L genes on BamHI restriction fragment J in pBR322 [24]) were kindly provided by B. Roizman, Chicago, Ill.
Plasmid pIEllOCAT (5) containing the IE regulatory
se-quencelinked toabacterial CATgene inpUC9wasprovided by P. O'Hare, Oxted, United Kingdom, and plasmid pS12TKU containing the IE regulatory sequence linked to
the HSV tkgenewasprovided by C. M. Preston, Glasgow, UnitedKingdom. PlasmidpSAM-I containing a mouseaprt
genewasprovided byJ. Tischfield (Indianapolis, Ind.). When plasmids were used as probes, they were
radiola-beledby using theoligonucleotide random-priming method
(3), with some modifications. The DNA (0.2 ,ug) was
dena-turedby boilingandwaslabeled inareactionbuffer
contain-ing50 mMTrishydrochloride (pH 7.2); 10 mMMgSO4; 0.1 mMdithiothreitol;50,ugof bovine serumalbuminper ml; 1
nM (each) dCTP, dGTP, dTTP; 100 pM [a-32P]dATP (Am-ersham Corp., Arlington Heights, Ill.); 1 ,ug of
hexaoli-gonucleotide random primer; and 2 U of Escherichia coli DNApolymeraseI Klenowfragment (BoehringerMannheim
Biochemicals,
Indianapolis,
Ind.)
in atotal volume of30pAl.
Thereactionwas incubatedat
10°C
for 12h,
after which the labeled DNA waspurified by
ethanolprecipitation.
This methodroutinely yields
probes
withspecific
activities of1 x 108cpm/,g.
Theprobe
wasdenaturedby boiling
beforeuse. Molecularhybridizations.
Thehybridization
ofDNAfilterswas
by
the method of Maniatis et al.(11).
Somemodifica-tions were introduced for RNA filters. The nitrocellulose
(NC)
membranes were first wetted in 3x SSC(lx
SSC is 0.15 M NaClplus
0.015 M sodiumcitrate)
and underwentaprehybridization
step
for 2 h at65°C
in 3x SSC-35%formamide-1x Denhardtsolution-0.5%sodium
dodecyl
sul-fate-100 ,ug of sonicated salmon sperm DNA per ml. Theprehybridization
bufferwasthenremoved,
and the filterwashybridized
for 12 h with the denaturedprobe
in theprehy-bridizationbuffer
adjusted
to 10 mM EDTA. Thefilterwas washedsequentially
at roomtemperature
with2x
SSC-0.5% sodiumdodecyl
sulfate for 15 min, lxSSC-0.5%
sodium
dodecyl
sulfate for 15 min, and 0.1xSSC-0.5%
sodium
dodecyl
sulfate at55°C
until the radioactiveback-ground
was low.Autoradiography
was doneovernight
at-70°C
withanintensifying
screen. DotblotassaysforDNA and RNAwereperformed
asdescribedpreviously
(19).
DNAtransfections. DNAwastransfectedintocells
by
the DEAE-dextran(Mw
500,000;
Pharmacia,
Uppsala,
Sweden)
method
(10),
with some modifications. TheDNA-DEAE-dextran mixture was
prepared
inprewarmed
TBS buffer(25
mMTris
hydrochloride
[pH
7.4],
137 mMNaCl,
5 mMKCl,
0.6 mM
Na2HPO4,
0.7 mMCaCl2,
0.5 mMMgCl2)
at37°C.
The mixture was then mixed with anequal
volume ofprewarmed
2x Dulbecco modifiedEagle
medium andlay-ered onto washed
monolayers.
The final concentration ofDEAE-dextran
applied
to the cells was500,ug/ml,
and the finalconcentration ofDNA was 1,ug/ml.
The volumeusedwas0.3 mlon35-mm
dishes,
0.8 mlon60-mmdishes,
and 2 ml on 100-mm dishes. Cells were incubated for 4h,
after which the mediumwasremovedand10%
dimethyl
sulfoxidein
phosphate-buffered
saline was added for 2 to 5 min atroom
temperature.
Cellswerethenwashedtwicewithphos-phate-buffered
saline andincubatedingrowth
medium. Afterovernight
incubation,
themediumwasreplaced
and IFNwas addedasneeded.Samples
wereincubatedafurther 48 h andharvested.
RESULTS
Quantitation
ofHSV-1
viralyield
in the presenceof IFNsin differentcelllines. Inordertodeterminethein vivo effectofIFNsonHSV-1
replication,
threedifferent celllines,
HEp-2
(human
epithelial
fibroblasts),
Vero(African
greenmonkey
kidney),
and WISH(human
amnionfibroblasts),
were pre-treated for 16 to 18 h with different amounts of humanrecombinant IFN-a or --y and infected with HSV-1 at a
calculated
MOI
of0.1. The viralyield
was determinedby
plaque
assay(in
duplicate
samples)
of thelysates
48 h later. The viralyield
wascompared
withthatof untreated controls(Table
1).
(Previous
experiments
had shownthatatahigher
MOI,
protection
by
100 U ofIFN(a
or-y)
per mlwas notcomplete.)
The virus titer decreasedover10,000-fold
when WISH andHEp-2
cells weretreated with atleast 100 U ofIFN--y
per ml. In Verocells,
however,
large
amounts of infectious virus were scored in the presence ofIFNs,
indi-cating
that Verocellsarerefractiletoinhibition of HSV-1by
human IFN at this MOI. At lowerMOIs,
there was someinhibition. IFN-a appears to be
considerably
less effectivethan
IFN--y
ininhibiting
HSV-1replication
in thesecelllines.on November 10, 2019 by guest
http://jvi.asm.org/
2590 DE STASIO AND TAYLOR
TABLE 1. Inhibition of HSV-1replication by interferonsa
IFNand
Yield
in Yield with following concn of IFNcells used untreated 12UM
controls 102 U/ml 103 U/ml 104U/ml
IFN-a
Vero 6 x 107 5x 107 2 x 107 1.2x 107
WISH 4 x 105 2 x 105 5x 104 4 x 104 HEp-2 2 x 105 1.3 x 104 3x 103 2x 103
IFN--y
Vero 6 x 107 4.9 x 107 1.9 X 107 1.1 X 107
WISH 4 x 105 <102 <102 <102
HEp-2 2 x 105 <102 <102 <102
a Effectof human IFN-a and IFN-yonHSV-1yield in Vero,WISH, and
HEp-2cells.Infectionwasata MOIof0.1. Values weredetermined by plaque
assay ofthe viral lysates producedinthepresenceorabsence of IFN.Plaque
assays wereperformedon Verocells.TheresultsareexpressedinPFU per
milliliter.
Inorder to study the IFN effect underconditions of
approx-imately total inhibition of virus replication, WISH cells treated with 1,000 U of IFN--y and infected at a MOI of1 wereused in subsequent experiments. In some experiments, IFN-awas alsoused.
IFN-induced inhibition of HSV-1 DNA synthesis. A time course ofthe effect of 1,000 U ofIFN--y per ml on
HSV-1-infected WISH cells was performed. Infected cells were
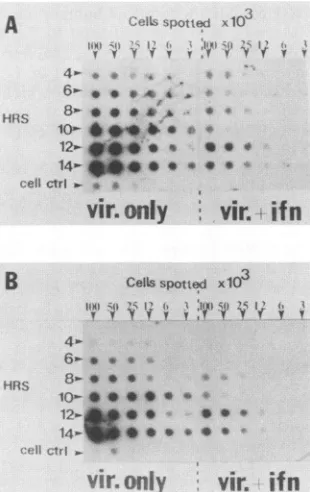
harvested atdifferenttimes afterinfection andimmediately applied onto NC membranes. The samples were processed for DNA analysis as describedpreviously (19). The results (Fig. 1)indicated a markedinhibition (and delay) of HSV-1 DNA synthesis in the presence ofIFN-y at an early time afterinfection(6 to10 h). There was a recoveryofviral DNA
synthesis at 18 to 24 h postinfection, probably reflecting escapesynthesis. That this delayed synthesis might be due to decay of the IFN effect was ruled out by adding high
concentrationsof IFN- y to theincubationmedium after viral
infection. Since HSV DNA synthesis requires a specific DNA polymerase, a viral DE gene product, we examined
transcription ofIEand DE genes.
IFN-induced inhibition of HSV-1 transcription. (i) RNA dot-blots. WISHcellswere treatedwithIFN-yandinfected
Cellsspotted x103
HRS
cell ci
,* ...::.-.W...:.
120s 14
vir.
only
vir.
if
n
FIG. 1. HSV DNA replication in IFN--y-treated WISH cells.
Cellspretreated for 12 h with IFN--ywereinfected with HSV-1ata
MOIof 1 inthepresenceof103 U of IFN--yperml.Sampleswere
collectedat2-hintervals. Twofold serial dilutionswereblottedonto
NCmembranes, denatured, and immobilized forDNAhybridization
asdescribedpreviously (19). Plasmid pRB201wasusedas aprobe.
A
(A.fCLSpoteAJ Xlo " V'f ji :yY1 Y4- * * , .
6- * 9 o .* i !.#
HRS 8-* ** *
10- .}
12c ** *
14> 0 'o * 0 6 .,
c:elletrl w_ .* *.
vir.
only
vir.-
if
nB
Celtsspottedxlo3
v
Vi
'V
V
r X
1!v
4-HRS 8 4&0
10'- 41 * * *
12-- w*
14- 46 * 4
c(tri
vir. only
vir.
ifn
FIG. 2. Detection of HSV-1 RNA transcripts inIFN--y-treated WISH cells. CellsweretreatedwithIFN--y andinfected, andcells wereharvestedasdescribed inthelegendtoFig.1.NCmembranes werefixed in1% glutaraldehyde andprobed asdescribedpreviously (19).(A)IEgenetranscription(pRB201);(B)DE genetranscription (pRB123).
with HSV-1 as described in Materials and Methods. Cell samples were harvested and counted, and twofolddilutions wereblotted ontoNC paper (19). After fixation and
proteo-lytic digestion, the filter paper was probed with labeled plasmids containing different HSV-1 DNA coding se-quences. Figure 2 shows that the inhibition of thetwotypes of viral messenger RNAs tested followed the pattern ob-served for DNAsynthesis (cf. Fig. 1 and 2); HSV-1 IE gene transcripts appeared in the untreated control 4 h after
infection (Fig. 2A), while glycoprotein D transcripts fol-lowedat6 h(Fig. 2B). DNA synthesiswasfirst detected at about 8 h. Thus, all steps of this cascade are inhibited by
IFN--y treatment. Inorderto confirm that specific mRNAs wereinhibited by IFN--y and that the results were not dueto
degradation of viral mRNA, the newly synthesized RNA was analyzed by Northern (RNA) blots.
(ii) Northernblot analysis. The steady-state level of
tran-scription ofIE and DE geneswasdetermined by Northern blot analysis following IFN--y treatment of WISH cells. Infectedcellsamples (treated and untreated)wereharvested at10hpostinfection, and RNAwasresolvedby denaturing agarosegel electrophoresis.The blotswerehybridizedwith different HSV-1probes,and theresultsobtainedareshown in Fig. 3. A remarkable decrease in both the IE and DE
signalsindicates that
IFN-y
specificallyinhibited IE mRNAexpression. DE genetranscription is probably inhibitedas a consequenceof the inhibitionofIEexpression, although we cannot eliminate a multitargeteffect ofIFN. Additionally, overexposureof theautoradiograms (Fig. 3Band D) showed that the samepatternof transcription foundinthe untreated control could be detected in the IFN-treated sample. This result suggests that the IFN--y action was due to inhibition
(ordelayof theonset)oftranscription rather than degrada-J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.57.300.90.206.2] [image:3.612.63.296.514.667.2]EFFECT OF IFN ON HSV-1 TRANSACTIVATION 2591
A
v
28S-18S- Po
B
BC;,&
j $C,:t
28SX
18S' _
'B
C
28
-1i8l
FIG. 3. Northern blot analysis of HSV-1 RNA transcripts. Total RNAwasextractedfrom 107WISH cells, either uninfected (cell ct)or
pretreated with IFNat6h postinfection (vir+ IFN). A10-,ug portion of each samplewas run on a1%agarosedenaturing gel. The samples
weretransferredtoNC and hybridizedtolabeledHSV-1 DEgene sequences(Aand B)orIEgene sequences(C and D).Twoexposuresof
eachblotarepresentedtorevealintactmessagesin the IFN-treated samples.(A)1-hexposure;(B) 20-hexposure;(C)3-hexposure;(D) 13-h
exposure.
tion of preexisting HSV IE messages. Degradation should result in a smear on the autoradiogram rather than intact bands. These results support the idea that IFN acts at(or prior to) the transcription ofIEgenes.
IFN effectontheHSV-1attachment and penetration. Even though the above data indicate thatIE transcription is the primary step inhibited, steps in viral infection that occur before transactivation could still cooperate in the general inhibitory effect that is observed in vivo. To test this, we measured (i) virus adsorption and penetration and (ii) viral uncoating and migration ofviral DNAto the nucleus. The firststagesof the HSV-1 infectious cycle involve the inter-action of the virus with the host cell surface receptor (attachment), fusion ofthe viral membrane with thehost cell membrane, and penetration ofthe viralparticle into the host cell. These events were studied by adsorbing 3H-labeled viral particlesto permissive cells and samples harvested at
various times after viral addition (the unadsorbed virus being washed off the cells with phosphate-buffered saline). The acid-precipitable radioactivity in the cells was then deter-mined. The effect of IFN was studied by comparing the adsorption-penetration curve of untreated cells with the
curvesobtained with cellspretreatedwith 1,000U ofIFN--y
per ml overnight. No difference in the adsorption-penetra-tion patterns of HSV-1 was found in the presence of IFN compared withanuntreatedcontrol.
Dot-blot ofDNAextractedfrom isolated nuclei. The trans-portof viral DNAtothe nucleus in thepresenceof IFNswas studied by harvesting total DNA from isolated nuclei 4 h postinfection and then quantitating the amount of HSV-1 DNA contained in the nuclei by molecular hybridization (19). At this time, HSV-1 DNA has not yet replicated in WISH cells. These experiments showed no difference be-tween the treated and untreated samples, indicating that IFNs donotinhibittransportof HSV-1 DNAtothe nucleus and that uncoating and transport of viral DNA occur nor-mally inbothcases(datanotshown).
IFN-induced inhibition of HSV-1 IE genetransactivation. Tocreateamodelsystemin which the transactivation event
was isolated from other viral functions, a chimeric plasmid containingareportergene(CAT)under the control of theIE
regulatorysequence wastransfected into WISH cells. Trans-activation was performed by adding intact HSV-1 at 60 h posttransfection for 6 h in the presence ofcycloheximide.
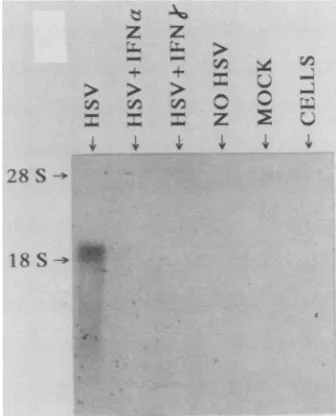
Figure4 shows aNorthernblot analysis ofthetotal RNAs extracted from transfected cells and probed with a 3p_
z z
r;L >
+ +
>
~~~
>2.8
18
FIG. 4. Northern blotanalysisof total RNA extracted from cells transfected withplEllOCAT andsuperinfected with HSV-1. Cells
were transfected with 2 ~Lg/100-mm plate with plElOOCAT DNA. Twelve hourslater,IFN-oLor--ywasaddedat1,000U/ml,and itwas
maintained onthe cells throughout the experiment. At60 h post-transfection,HSV-1 andcycloheximide(50 ~LgIml)wereadded for 6 h and cells were harvested for RNA extraction. No HSV, The nontransactivated control; mock, transfection with pBR322; cells,
notransfection.
D
!: f
*j;'': _ :I,
td
_A
1 ,,
28 5
-18 S
-VOL. 64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.97.520.80.271.2] [image:4.612.351.519.435.643.2]2592 DE STASIO AND TAYLOR
a. Cellsspotted x103
100 50 25 12 6
HSV- .SV
IFN HSV'- IFN HSVD. ;
IFN HSV'.
NOHSV- NOHSVV'
CONTROL- CONTROL0
FIG. 5. Dot-blot hybridization of WISH cells transfected with pIECAT and activated with HSV-1. Cellswerecotransfected with
pIEllOCATand pSAM-I (mouseaprt under the nativepromoter). After 24 h,twoof the samples weretreated with IFN. At 68 h, all samples wereinfected with HSV-1(MOI = 10). Fourhours later,
sampleswereharvestedandspottedontoNC filters. The filterswere
fixedasdescribed previously (19) andwereprobedwith P32-labeled
pIECAT (a) and P32-labeled pSAM-I (b).
labeledCAT probe. A marked inhibition of the IE transac-tivation is showninlanesHSV + IFN-aand HSV + IFN--y when compared with the positive control HSV. The con-trols, NO HSV (corresponding to a sample that was not superinfected with HSV-1), MOCK (corresponding to cells transfected with 2p,gofpBR322), and CELLS (correspond-ingtountreated WISH cells), didnotshowanyhybridization
to the probe. The positive signal observed for the HSV sample versus little or no signal inthe IFN lanes indicates inhibition of IE transactivation. Similar results were ob-tainedby usingaplasmid containing the HSV tkgenelinked to IE sequences. VP16 is not synthesized de novo under these conditions but is brought in by the virus,aswould be thecase duringnormal infection.
Inordertoconfirm that the IFN effectwasnotdueto an effect on transfection efficiency or a general transcription inhibition, similar transfection experiments weredone with themouseaprtgene. Cells werecotransfected with pIE110 CAT and pSAMI. After24h, 1,000U ofIFN (alandy) per mlwere addedto each plate, and 48 h later, HSVataMOI of 1wasadded.Cellswereharvested 4 h later andwerefixed in glutaraldehyde as described previously (19). Dot blots were probed separately with labeled pCAT and pSAM probes. After 20 h ofexposure,control cellstransfected with pCATandinfected withHSVgave astrongsignal; however, cellstreatedwith IFNgavenosignal whatsoever, indicating inhibition of CATtranscription (Fig. Sa). At later times of
exposure, some signal could be detected in IFN-treated cells. When a replica of the same filter was probed with pSAM, the mouse aprt gene, no signal could be detected until 48 h ofexposure. This probably reflects the weaker promoter strength ofaprt, a housekeeping gene. However IFN hadnoeffect on transcription of thisgenein the same transfectedcells (Fig. Sb).
DISCUSSION
Theevidencepresented in Resultssupportsthe hypothesis originally proposed by Oberman and Panet (15, 16) that HSV-1 is inhibited by IFN-ot at an early step in the virus replication cycle involving transactivation of IE genes by
VP16.Transactivationof IEgenes occursvia the formation
of a protein complex defined as IEC (20). This complex resultsfromprotein-protein interactionbetweenone or more
hostproteins (probablytheoctamericbinding factor) and the viraltransactivatorVP16.The latteris imported intothe host cell by the infecting virus and transported to the nucleus
along with the viral genome. It should beemphasizedthat in theabsenceoffunctionalVP16, low levels ofHSV-1 are still synthesized, inagreementwith the reported IFN effects.
Theexperiments reportedhere describe aspecificeffect of IFNs onthe IE transactivation event.Thespecificityof such aneffectis alsosupported bytheobservation that transcrip-tionof an unrelated gene (mouse aprt) isunaffectedby IFN treatmentin the presence of VP16. The inhibitory effectof IFNseems tobe directedattheonsetofIEtranscription, as reported also by other investigators (12, 15, 16). These investigators performed nuclear runoff assays to show that the IE inhibition observed was notdue to RNase L degra-dationofthe IE messages. The latter result is also confirmed by the Northern blotsreportedinthis work(Fig. 3),inwhich no degradation (resulting in smearing) is evident in the
IFN-treated sample.
Specific inhibitionof IE transactivationmight be achieved by anIFN-inducedgene(s) directly affecting IEC formation or modifying a correctly assembled IEC. The evidence
provided here does notdistinguish between these
possibili-ties.
Another possibility isan effecton VP16 transport to the
nucleiby IFNs. This possibility becomes important ifone considers that IEC transactivating ability lies in the VP16 acidic domain. If, in the presence ofIFN, VP16 is incor-rectly transported to the host nuclei, then IEC formation would be impaired. Indirect evidence from other laborato-ries does not support thisidea. HSV-1 inducedearly shutoff of hostprotein synthesiswas notaffectedby IFN pretreat-ment of the cells (15). Early shutoffin HSV-1 infection is inducedby the HSV protein imported into the cells by the infecting virions (9). This evidence suggests that IFNs do not affect the very early stages of HSV-1 infection, namely,
penetration and uncoating. VP16 is similar to the shutoff
protein in that it is both a structural and functional viral
protein. If the shutoffproteinis correctly imported intothe cells in the presence ofIFNs, sopresumably is VP16.
If transportof VP16tothenuclei is shown to beunaffected
by IFNs, modification(or lack ofmodification) ofIEC (one or more ofitscomponents)is themostlikely possibilityleft to explainthefailure of thecomplex to stimulate
transcrip-tion.
ACKNOWLEDGMENTS
This research was supported by Public Health Service grant AI21898 fromthe NationalInstitutes ofHealthand bythe United CancerCouncil(Indianapolis, Ind.).
LITERATURECITED
1. Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chlo-roform extraction. Anal. Biochem. 162:156-159.
2. DeMaeyer, E., and J. DeMaeyer-Guignard. 1988. Interferons and otherregulatorycytokines. John Wiley & Sons,Inc., New York.
3. Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabellingDNArestriction endonucleasefragments tohigh specific activity. Anal. Biochem. 132:6-13.
4. Friedman, A. D., S. J. Triezenberg, and S. L.McKnight. 1988. Expression ofatruncated viraltrans-activator selectively im-pedes lytic infection by its cognate virus. Nature (London) 335:452-454.
5. Greaves, R., and P. O'Hare. 1989. Separation of requirements for proteinDNAcomplex assembly from these forfunctional activityin theherpes simplexvirusregulatory proteinVmw65. J.Virol. 63:1641-1650.
6. Hayward, G. S. 1986. Herpesviruses: genome structure and J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.60.300.79.164.2]regulation.Cancer Cells (Cold Spring Harbor)4:50-77. 7. Kaufman, R. J., and P. Murtha. 1987. Translational control
mediated byeucaryotic initiation factor-2 is restricted to specific mRNAsintransfected cells. Mol. Cell. Biol. 7:1568-1571. 8. Kristie, T. M., and B. Roizman. 1988.Differentiation and DNA
contact points of host proteins binding at the cis site for virion-mediated induction of a genes of herpes simplex virus 1. J.Virol.62:1145-1157.
9. Kwong, A. D., J. A. Kruper, and N. Frenkel. 1988. Herpes simplex virusvirion hostshutoff function.J. Virol. 62:912-921. 10. Lopata, M. A., D. W. Cleveland, and B. Sollner-Webb. 1984. High level transient expression of a chloramphenicol acetyl transferasegeneby DEAE-dextran mediatedDNAtransfection coupledwithadimethylsulfoxyde or glycerolshock treatment. NucleicAcids Res. 12:5707-5717.
11. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: alaboratory manual. ColdSpringHarbor Laboratory, ColdSpringHarbor, N.Y.
12. Mittnacht, S., P.Straub, H. Kirchner, and H. Jacobsen. 1988. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology164:201-210.
13. Munoz, A., and L. Carrasco. 1987.Effects of interferononviral replication and on the cytopathic effects induced by animal viruses. In L. Carrasco (ed.), Mechanisms of viral toxicity. CRC Press, Inc., BocaRaton,Fla.
14. Nilsen, T. W., and C. Baglioni. 1979. Mechanism for discrimi-nation betweenviral andhost mRNAininterferon-treated cells. Proc.Natl. Acad. Sci. USA76:2600-2604.
15. Oberman,F., and A. Panet.1988. Inhibition of transcription of herpessimplex virus immediate earlygenesin interferon-treated human cells. J. Gen. Virol. 69:1167-1177.
16. Oberman,F., and A. Panet. 1989.Characterization oftheearly stepsof herpessimplex replication in interferon-treatedhuman cells. J.InterferonRes.9:563-572.
17. O'Hare, P., and C. R. Goding. 1988. Herpes simplex virus regulatory elements and the immunoglobulin octamerbinding
domain bind a common factor andare both targets for virion transactivation. Cell 52:435-445.
18. O'Hare, P., C. R. Goding, and A.Haigh. 1988. Direct combina-torial interaction between a herpes simplex virus regulatory protein and a cellularoctamer-bindingfactor mediatesspecific inductionof virus immediate-earlygene expression. EMBO J. 7:4231-4238.
19. Paeratakul, U., P. R. De Stasio, and M. W. Taylor. 1988. A fast andsensitivemethodfor detecting specific viral RNAin mam-maliancells. J.Virol. 62:1132-1135.
20. Preston, C. M., M. C. Frame, and M. E. M. Campbell.1988.A complexformed between cellcomponents andan HSV struc-turalpolypeptide bindsto aviral immediateearlygene regula-tory DNA sequence. Cell 52:425-434.
21. Ptashne, M. 1988. How eukaryotic transcriptional activators work.Nature(London)335:683-689.
22. Roizman, B., F. J. Kenkins, and T. M. Kristie. 1987. Herpes viruses:biology, generegulation,latencyandgenetic engineer-ing.In R. Perez-Bercoff (ed.), Molecular basis of viral replica-tion, NATO ASI series, vol. 136. Plenum Publishing Corp., NewYork.
23. Sadowski, I., J. Ma, S. Triezenberg, and M. Ptashne. 1988. GAL4-VP16 is an unusually potent transcriptional activator. Nature(London)335:563-564.
24. Schaffer, P. A., E. K. Wagner, G. B. Devi-Rao, and V. G. Preston.1987. Herpessimplexvirus,p.93-98.InS. J.O'Brein (ed.), Geneticmaps, vol. 4, 1987: acompilationoflinkageand restrictionmapsofgenetically studiedorganisms. ColdSpring HarborLaboratory, ColdSpring Harbor, N.Y.
25. Spear,P.G.,and B.Roizman.1972. Proteinsspecified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J.Virol. 9:143-159.
26. Triezenberg, S.J.,R.C.Kingsbury,andS. L.McKnight. 1988. Functional dissection of VP16, the transactivator of herpes simplex virus immediate early gene expression. Genes Dev.
2:718-729.