Effects of
Insertional and Point
Mutations
on
the Functions of the
Duck
Hepatitis
B
Virus Polymerase
LUNG-JI CHANG,1t RUSSELL C. HIRSCH,2 DON GANEM,"3 ANDHAROLD E. VARMUSl2* Departments of Microbiology andImmunology,1 Medicine,3 and Biochemistry and Biophysics,2
University of California, SanFrancisco, California 94143-0502 Received 4 June1990/Accepted 16 August1990
The polymerase (P)geneof hepadnaviruses encodesalargepolypeptide thatappearstoparticipate in several
stepsinthevirallifecycle: packaging of viral RNA, providing the primer for synthesis of minus-strand DNA,
synthesizing minus-strand DNA from an RNA template and plus-strand DNA from a DNA template, and
degrading viral RNA in RNA-DNA hybrids. To assist in the assignment of these functionstodomains of the duckhepatitisBvirus polymerase protein,wehave constructed aseriesof substitution mutations andalarge insertion mutation, based in parton amino acidsequence comparisonswith other proteins known toexhibit
reversetranscriptase (RT) and RNase H activities. We foundthatchanges in highly conserved sequences in
putativeRTand RNase Hdomainsin the carboxy-terminal half of the protein dramatically reduced synthesis of both strands of viral DNA without major effects on RNA packaging into subviral cores. Thus we can uncoupleRNApackagingand DNAsynthesisbutcannot separateRT and RNase H activitiesashas beendone
with human hepatitis B virus. The viability ofamutantwithalargeinsertion (123 amino acids)upstreamof
theRT and RNase H domain indicates that a hinge region may separate parts of the polymerase protein
implicatedinprimingandpolymerization.
Replication ofhepadnaviruses involves synthesis of the viral DNA genome from an RNA pregenome packaged in cytoplasmic core particles (16). Several of the functions required forreverse transcription of viral RNA-packaging
of viral RNA (8), priming of DNA synthesis (1), DNA
polymerization on RNA and DNAtemplates (reverse tran-scriptase [RT]), anddigestion of RNAin RNA-DNAhybrids (RNase H)-are believed to be encoded in a long open reading frame calledPthatoverlaps the 3' end of thecore(C) geneandall of thesurfaceantigengene(7). Unlike retroviral
polgenes, which areexpressed by synthesis ofcore
(gag)-pol fusion proteins, the hepadnavirus pol genes are ex-pressed by translational initiationatastartcodonnearthe5'
end of the P reading frame that is preceded by several upstreamAUGs in PmRNA(5, 14).
Comparison ofproteins with RT or RNase H activities
encoded by hepadnaviruses, retroviruses, and othergenetic
elements (see Fig. 1) indicates that a domainof the
hepad-navirus P protein likely to be essential for RT function is
positioned in thecarboxy-terminal halfof the duckhepatitis
B virus (DHBV) Pgene product (i.e., residues 477 to 515) upstream ofa region resembling known RNase H domains
(i.e., residues 665 to 755). Experiments with antipeptide
antibodies have shown that protein linked to the 5' end of
minus-strand DNA, believed to prime synthesis of that
strand,is derived from the Pgeneproduct (1).Theregionof
the P protein that facilitates packaging of viral RNA into
cytoplasmic core particleshas notbeenidentified, although
several P frameshift mutants are deficient in this recently
described function(8).
Totest some of these predictions about Pgene function, we have constructed a set of substitution mutations in conserved regions of the P gene of DHBV and one large
* Correspondingauthor.
tPresent address: Laboratory of Molecular Microbiology,
Na-tional Institute ofAllergy and InfectiousDiseases, Bethesda, MD 20205.
insertion mutation inaputatively functionlessspacerregion. The mutant DNAs have been tested for their abilities to
directassembly ofparticles containingviral RNA(the pack-agingfunction) andtosynthesizetheminus andplusstrands ofviralDNA(theRT andRNase Hfunctions).Incontrast to
arecentreportthat the RT and RNase H domains of human
hepatitisBvirus(HBV)canbedissociated by mutation (12), we have found that DHBV mutations in two conserved
RNase H motifs aswell as anRTmotif impairsynthesis of
both DNA strands. The packaging function, however, is unaffected by these changes. Finally, the insertion mutant
appears abletoperformallknownfunctions of the
polymer-ase, implyingthat the insertion is situated in a dispensable
and thusmanipulable regionof thegene. MATERIALSANDMETHODS
Plasmidconstructionandmutagenesis. A369-bp proteinA
fragment(HindIII-SalI) was isolatedfromplasmid pRIT-2T (Pharmacia) and end-filled with Klenow fragment of DNA
polymerase I; thefragmentthenwas ligatedtotheDHBV3
genomic vector (15) blunted with Asp718 (nucleotide [nt] 1290)and Klenow. Site-specificmutagenesis wasperformed as described previously except that a subclone of the
pTZ18U vector (Bio-Rad) containing the DHBV3 genomic
sequence was used (5). For thesynthesis of the YXDD (nt
1703)mutant, asynthetic30-mer(TATATGCATGCCTTCC TCCTCTGCCACCCA) was used. For thesynthesis of
mu-tants ataminoacid 666(nt2165)and755(nt 2432),a20-mer [GTAGCTACG(C/G)A(T/A)GCTACCCCI and a 24-mer
[TATAATCCTGCT(I/A/C)A(AII/_i)GGCCCATCC]
wereused(themismatchednucleotidesareunderlined).The clon-ing junctions and mutation sites of the final plasmids were
verified byDNA sequencing.
Transfection ofHepG2cells.HepG2cellsweremaintained
in Dulbecco modifiedEaglemediumsupplementedwith10%
heat-inactivated fetal calf serum (GIBCO Laboratories). Calcium-phosphatetransfectionwasperformedasdescribed previously (5). The protein A-polymerase mutant was
con-5553
0022-538X/90/115553-06$02.00/0
Copyright C) 1990,American Society for Microbiology
on November 10, 2019 by guest
http://jvi.asm.org/
structedas adimerand used fortransfection
directly.
Other mutantplasmids
contained DHBV monomer and therefore weredigested
andreligated
so as to present circularized monomers oroligomers
for transfection.Preparation
of viral nucleic acids. Viral cores were har-vested from transfectedHepG2
cellsby
immunoprecipita-tion with anticore antisera as
previously
described (5). Toexclude
input
plasmid
DNA as a contaminant in these assays, the cores were treated with DNase I and RNase Abeforethenucleic acidswereextracted. To harvest
particles
fromculture
media,
mediawerecollectedonday
3or6aftertransfection,
spuninaclinicalcentrifuge (800
x g, 5min)toremovedetached
cells,
furtherclarifiedby centrifugation
inaSorvallSS-34rotor
(10,000
rpm, 10min),
andlayered
onto2 ml of15% sucrose. The viral
particles
werepelleted
in aSW41rotor at
39,000
rpm for4h. Aftercentrifugation,
the supernatantwascarefully
removed. Beforefurtheranalysis,
the
pellet
wassuspended
in 30,ul ofHBV-RTbuffer(50
mMTris
[pH
7.6],
10 mMMgCl2,
0.1%2-mercaptoethanol,
1% TritonX-100)
at4°C overnight.
Southern blot
analysis, primer extension,
and RNasemap-ping.
Southern transfer wasperformed
aspreviously
de-scribed(5).
Preparation
of radiolabeledRNAwasperformed
as
previously
described(4).
In some cases, the blot filterscontaining
viral DNA were firsthybridized
to the DHBVprobe
or aplus-stranded
RNAprobe (nt
1658 to14)
in thebuffer described
previously by
Church and Gilbert (6). Tostrip
off theprobe,
the filter was incubated in a solutioncontaining
80%formamide,
1%sodiumdodecyl sulfate,
12.5mM
EDTA,
and 10mMTris(pH 7.5)
at65°C
for30 minto1 h. Afterprehybridization,
thefilterwasrehybridized
with aminus-stranded RNA
probe (DHBV
nt 14 to1658).
Forprimer
extension,
viralcoreRNAwashybridized
to a5'-end32P-labeled
oligonucleotide primer
(DHBV
nt2785 to2765)
ina solution
containing
80%formamide,
0.4MNaCl,
1mMEDTA,
and 40 mM PIPES[piperazine-N,N'-bis(2-ethane-sulfonic
acid)]
(pH 6.5),
at95°C
for3 min and thenplaced
onice for10 min. After
precipitation
with ethanol andsuspen-sionin 10 ,ul of avian
myeloblastosis
virus-RT buffer(50
mMTris
[pH 8.3],
6 mMMgCl2,
40 mMKCl,
1 ,ug ofbovineserum
albumin,
0.5 mMdeoxynucleoside triphosphate),
theRNA-primer
hybrid
was incubatedwith avianmyeloblasto-sis virus reverse
transcriptase
at37°C
for 45 min. Theproducts
wereprecipitated
once withethanol andanalyzed
on an 8%
sequencing
gel
next to asequencing
laddergenerated
with thesamephosphorylated primer.
For RNasemapping,
the viralcoreswereharvested andthenucleic acidcontents were
digested
with DNase I beforehybridization
with a
specific
antisenseRNAprobe,
which could annealto the 5' end of the viral pregenomes. The RNAprobe
wasmade
by
T7 RNApolymerase
(Promega)
and incubatedwith thecore pregenomesat85°C
for5 min, whichwasfollowedby
incubation at30°C overnight
in the 80% formamidehybridization
buffer described above. The RNA-RNAhy-bridswere
digested
with RNaseA(40
,ug/ml)
and RNaseT1(2
,ug/ml)
at30°C
for1h.This reactionwasterminatedbytheadditionof sodium
dodecyl
sulfate(to
1%)
andproteinase
K(50
pug)
followedby
an additionalincubation at 37°C for 15 min. Afterphenol
extraction and ethanol precipitation,theduplex
RNAstogether
with unlabeled DNA markers wereanalyzed
on a5%polyacrylamide gel.
Endogenous polymerase
assay. For the assayof viralpoly-merase
activity,
viral materialspelletedfrom 10 ml of culturemediawere
suspended
in 30 ,ulof HBV-RT buffer. To the mixturedATP, TTP,
dGTP(0.5 mMeach), dCTP(12puM),
and 1 ,ul of[a-32PIdCTP
(3,000
Ci/mmol;
Amersham) wereadded. The reaction was performedat 37°C for 3 h. At the end, cold dCTPwasaddedto0.2 mM and further incubated for 30 min. The reaction was stopped by the addition of sodiumdodecyl sulfate(to1%),10,ug of yeasttRNA,and 20 ,ug of proteinase K inafinal volume of100
pd
and incubated at 37°C for 30 min. The 32P-labeled viral DNA was then isolated by phenol-chloroform extraction and ethanol pre-cipitation. The reaction product wasanalyzedbyelectropho-resis on a 0.7% agarose gel, dried onto 3MM paper, and
subjected toautoradiography. RESULTS
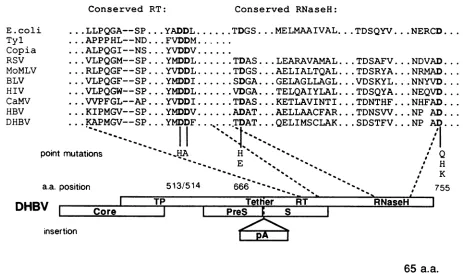
Experimental strategy. Site-directed mutagenesis tech-niques were used togenerate several substitution mutations at three highly conserved positions in putative RT- and RNase H-coding domains of cloned DHBV DNA (Fig. 1).
The mutant DNAs were introduced into the human hepa-tomacell lineHepG2 by calcium-phosphate-mediated
trans-fection to observe effects on P gene functions. Tojudge RNApackaging efficiency, core particles were isolated from cytoplasmic fractions by immunoprecipitation, and prege-nomic RNA was measured either by primer extension assay
or by RNase mapping (8). Synthesis of viral DNA was
assessed by gel electrophoresis and Southern blotting of DNA extracted from thecytoplasmiccores;when appropri-ate, probes that distinguish minus and plus strands of viral DNA were also used (data not shown). In some cases, we
also tested the culture medium for released virus particles that could incorporate radioactive nucleotides into DNA, a measureofendogenous DNA polymerase activity. Since the RTsofhepadnaviruseshave notbeendissociatedfrom their native templates without loss of enzymatic activity (13) and since we have been unsuccessful in assaying hepadnaviral RTactivityasdescribedbyBavand and Laub (2), itwasnot possible to test our mutants for polymerizing activity on
heterologous templates or to test directly for RNase H activity. Instead we sought indirect evidence for impaired RNase H in selectedcasesby looking for synthesis of minus strands butnotplus strands, the phenotype which would be
expected if RNase H were unable to remove pregenomic RNAfrom RNA-DNAhybrids. Nospecific tests for primer functionwereperformed.
A mutation in the conserved RTmotif, YXDD, inactivates
DHBV DNA polymerase. As an initial test of our ability to
perturb P gene functions with substitution mutations, we
changed the most highly conserved motif among RTs
(YXDD)by alteringthe YMDD sequence encodedby wild-type (wt) DHBV at amino acid positions 511 to 514 to
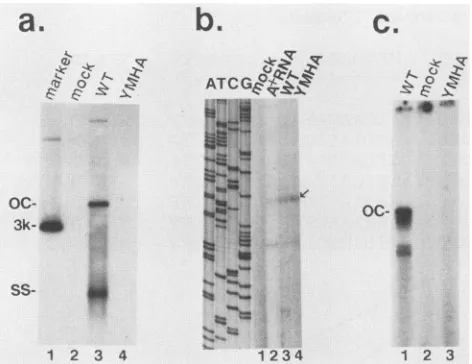
YMHA(Fig. 1). Aswehavepreviouslyshown(8), synthesis and packaging ofpregenomic RNA occurred normally in
cells transfected with the mutant genome (Fig. 2b, lane 4,
analyzed by primer extension). However, core particles from cells that received the mutant genome contained no
viral DNA (Fig. 2a, lane 4), which is consistent with the
prediction that the RT function was inactivated, thereby
preventingthesynthesisof either minus or plus strands. This conclusion was confirmed by the absence of endogenous DNApolymerase activityinextracellular particles (Fig. 2c, lane 3). This YMDD to YMHAmutation also changes two amino acidsatthecarboxy terminus ofthesurface antigen;
however, interruption of translation of the surface antigen frame in other mutants did not affect polymerase function
(see below) and cannot account for the results with this
mutant.
Mutations in twoconserved RNase H motifs inactivate RT
on November 10, 2019 by guest
http://jvi.asm.org/
Conserved RT: Conserved RNaseH: ...LLPQGA--SP...YADDL...TDGS...MELMAAIVAL. ...APPPHL--ND ...FVDDM...
...ALPQGI--NS... YVDDV.
...VLPQGM--SP
...YMDDL...TDAS... LEARAVAMAL....RLPQGF--SP... YVDDL...TDGS... AELIALTQAL.
...VLPQGF--SP
...YMDDI...SDGA ...GELAGLLAGL....VLPQGW--SP... YMDDL...VDGA ...TELQAIYLAL.
...VVPFGL--AP ...YVDDI...TDAS... KETLAVINTI.
...KIPMGV--SP ...YMDDV...ADAT ...AELLAACFAR.
...KAPMGV--SP .
YMDDF...T.DAT..
QELIMSCLAK.nutations 'HA H "
".
E "
)Sition 513/514 666 ' "
..TDSQYV.. .NERCD...
..TDSAFV ...NDVAD...
..TDSRYA.. .NRMAD... ..VDSKYL...NNYVD....
..TDSQYA...NEQVD....
..TDNTHF...NHFAD...
..TDNSVV...NP AD...
..SDSTFV...NP AD...
Q H K
K
I 755
DHBV
I
Iinsertion
Tetner
LI
PreS ICore S
RT
I65
a.a.FIG. 1. (Top) Sequence alignments of the conserved RT and RNase H domains of known polymerases. Alignmentsweregeneratedwith
the program Genalign from the package of sequence analysis programs ofHugo Martinez, Department of Biochemistry, University of
California, San Francisco. Alignmentswere made withcomplete sequencesandpiecewise by aligning 50-amino-acid stretchesatatime.
Alignmentswereoptimized visually by givinggreaterweighttoregions of high conservation.The most-conserved amino acid stretches in both
the RT and RNase Hgenes are listed; hyphens indicate spaceofasingle, nonconserved amino acid. Conserved amino acids of DHBV P
protein chosen formutagenesisareshowninboldface; the substituted aminoacids and their residue numbersareshown below. RSV,Rous sarcoma virus; MoMLV, Moloney murine leukemia virus; BLV, bovine leukemia virus; HIV, humanimmunodeficiency virus; CaMV, cauliflowermosaic virus. (Bottom) Coding organization ofthe DHBVgenome.Open boxes denoteopenreading frames,with the Pgeneand
itsputative domains indicated atthetop.pA, ProteinAcoding region inserted into the indicated siteinopenreadingframe P in theprotein
Amutant shown inFig. 4;a.a.,amino acid.
function. We next asked whether changes in highly
con-servedresidueswithinmotifs foundinseveral known RNase H domains would affect the synthesis of viral DNA. In
particular, we wanted to verify the importance ofthe
con-served sequences and to ask whether we could generate mutants specifically deficient inRNase H function without
loss of the abilitytosynthesize minus-strand DNA.
Two amino acidsuniversally conservedamongknownand
putativeRNase Hswereselectedastargetsformutagenesis (Fig. 1). TheAspresidueatposition666waschangedtoHis
(D666H)orGlu(D666E),and theAspresidueatposition755
waschangedtoGln (D755Q), His (D755H),orLys (D755K). Surprisingly, all five of these mutants were severely or completely deficient in DNA-synthetic capacity, as
mea-sured both by Southern blotting of nucleic acids from
cytoplasmic cores(Fig. 3a andb) andby endogenous
poly-merase assay with particles pelleted from culture medium
(Fig. 3c). The only viral DNA observed was a markedly
reducedamountof minus-strand DNA incoreparticlesfrom
cellstransfected withmutant D666E, indicating slight resid-ual RT activity (Fig. 3a, lane 3; data not shown). These
resultscannotbe ascribedtoafailuretopackage pregenomic RNA, since primer extension assays showed no difference between mutant and wt packaging efficiencies (data not shown). We conclude that the conserved residues in the putativeRNase H domain areessential forviralreplication
and that mutations at positions 666 and 755 affect RT
function, thereby preventingadissociation of RT and RNase
Hactivities (see Discussion).
Alarge insertionupstreamof the RT domain iscompatible
with Pprotein functions.Thesequencesbetween the RT and protein primer regionsofhepadnavirusPgeneproductsare poorly conserved, have no known functions, and might be dispensable for virus replication. To begin to assess these possibilities,we constructed aninsertionmutantby placing a 369-bp sequence encoding bacterial protein A into the
DHBV Pgene4bpdownstream of the translationinitiation
codon for the major surface antigen gene (Fig. 1). (This mutant had the additional potentialvirtue ofdirecting
syn-thesis ofaprotein A-P fusionproductthatcould bereadily purified by binding to immunoglobulin G-Sepharose; al-though this strategy has been successfully appliedto prod-uctsof in vitrosynthesis,wehave been unabletodetectthe predicted fusion protein in lysates ofHepG2 or Cos7 cells
transfected with the mutant gene under the control of a cytomegalovirus promoter, presumably because of low
lev-elsofexpression [L.-J. Chang, unpublished results].)
When expressed in HepG2 cells in the context of the
complete DHBV genome, the protein A insertion mutant
appeared to retain most or all viral functions. Pregenomic
and subgenomic RNAs (Fig. 4a) and core protein (not shown)werepresentatlevels similartothosefound in cells
transfected with wild-type DHBV DNA. The pregenomic
RNAwasalso detectedin thecytoplasmic-core particlesof
E.coli Tyl
Copia
RSV MoMLV BLV HIV
CaMV
HBV DHBV
point n
a.a. pc
RNaSeH I
T..
TPI
-..O...
I
pA
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.68.531.77.350.2]a.
oc-3k-
ss-b.
C.
-k- 7
1, (.)
.,\ 0
k.-.46mm
oc-I
1 2 3 4 1234
FIG. 2. Analysis of the YMDDtoYMHAmutant.(a)Southern blot analysis ofviral DNA in cytoplasmic-core particles. HepG2 cellsweretransfected withnoDNA(lane 2),wtDHBVDNA(lane
3), or mutant DHBV DNA (lane 4). Cytoplasmic cores were
prepared 4 days posttransfection, and total nucleic acids were
extracted as described in Materials and Methods; samples were
electrophoresed through 1.2% agarose gels, transferred to nylon filters, and hybridized to 32P-labeled DHBV DNA. Lane 1, 3-kbp DHBV linear DNA marker. (b) Analysis of encapsidated RNA. Cellsweretransfected withnoDNA(lane 1),wtDHBV DNA(lanes 2 and3),orYMHAmutantDNA(lane 4) and either totalpoly(A)+ RNA(lanes 1 and2)orRNA fromcytoplasmiccores(lanes 3 and 4) was prepared. These RNAs were annealed to a 5' end-labeled
oligonucleotide homologous tont 2658to 2641 nearthe 5' end of
pregenomic RNA; following extension with avian myeloblastosis virus polymerase, the labeled products were analyzed on an 8%
sequencing gelalongsideaDNA-sequencing ladder generated with
thesameprimer, shown in the four lanesonthe left. Arrowdenotes theposition of the 5' end of pregenomic RNA (3). (c) Endogenous polymerase assay. Particles were pelleted by ultracentrifugation
from themedium of HepG2 cells transfected with wtDHBV DNA (lane 1), noDNA(lane 2),orYMHAmutantDNA(lane 3). Pellets
were incubated with [32P]dCTP and unlabeled dATP, dGTP, and dTTP; labeled DNA productswerethen extracted and analyzedby
agarose gel electrophoresis and autoradiography. OC, Open
(re-laxed) circular DNA; SS, single-stranded DNA.
the protein A Pmutant (Fig. 4b), suggesting that the
inser-tion didnotpreventpackaging. Totestits effecton polymer-ase function, we analyzed the DNA in the mutant cores. Interestingly, the proteinAinsertionmutantretained
DNA-synthetic capacity, since cytoplasmic cores contained both open circular DNA and partially single-stranded
intermedi-ates (data not shown); in the culture media, the released mutantparticlescontainedopencircular DNA(Fig. 4c, lane 2). (The reduced mobility of the mutant DNA, compared
withDNAfound inwtcores,is attributabletothe size ofthe insertion mutation.) The retention ofDNA polymerase ac-tivity by the insertion mutant wasconfirmed byan
endoge-nous polymerase assay on particles harvestedfrom culture medium (Fig. 4d). Thus the region between the protein
primer and RT domains appears dispensable for P gene
functionandsubjecttoextensive manipulation;moreover, a
10% enlargement of the viral genome is compatible with packagingand complete DNAsynthesis.
Thepresence of viral DNA in themedium of cells
trans-fected with amutantthatdisrupts surface antigen synthesis
was a surprise, particularly in view ofour recent findings that Sproteinsarerequired for HBVDaneparticle assembly
and export (V. Bruss and D. Ganem, unpublished data). However, wehave also found thatHepG2 cells transfected with DHBV DNArelease significant quantities of viralcores
(V. Bruss, unpublished data); presumably the active parti-clesdetected in thisexperimentweresuchreleasedcores.
DISCUSSION
The generation of duplex DNA from a single-stranded
RNA template requiresthattheresponsible machinerycarry outmultiple reactions (7). These include minus-strand prim-ing and elongation onanRNAtemplate, the removal of the RNA from the RNA-DNA hybrid during DNA synthesis, and the priming and elongation of plus-strand DNA on a DNAtemplate. Inaddition, in viruses and retrotransposons
all ofthese reactionsareprecededby the selective encapsi-dation of the RNA template in a subviral nucleoprotein
complexorcapsid.For hepatitis B viruses, all of thesesteps
require the participation of product(s) of the Pgene. In this study, with the homologies that exist between known and presumed reverse transcriptasesas aguide, we
have attempted agenetic dissection of the functional
orga-nization of the DHBV P gene product. This approach is conceptually straightforward for those activities (RT and RNase H) that have been mapped to specific conserved regions of well-characterizedenzymes.Forexample,
exper-iments with murineleukemia virus polymerase expressed in
bacteria (17) have indicated that the RT and RNase H activities of this enzyme can be cleanly dissociated by mutationand that the two activitieslikelyreside in separate
protein domains separated byafunctionless spacerregion. Our study indicates thatsomebut notall of the activities of the DHBVenzyme can be dissociatedbymutation. First, the fact that multiple mutations affecting viral DNA synthe-sis remain competent for RNA packaging indicates that thesefunctionsareseparable.However,wedo notyetknow
whether the encapsidation function of the protein mapsto a discreteregionor(ifso)where thatregion maybe. Wehave recently examinedaseries offrameshift mutations
through-out the gene and found all of them to be defective for
encapsidation (8); this might implythatmultiple regions of
the chain arerequired or thattheactivity might mapto the
extreme C terminus. However, since mutant proteins are often unstable and since we have no way at present to measurethelevels ofPproteins, this result may simplybe a trivial consequence ofdegradation of the mutant
polypep-tides.
Ourattemptstoseparatethe RTand RNaseHfunctionsof
theDHBVenzyme, onthe otherhand, were to noavail. In
these studies we used as an indirect indicator of these activities the structure of theproductDNAs in mutant core
particles. Mutants with lesions affectingRTalone shouldbe
unable toproduce either strand of DNA, while pureRNase H mutants should accumulate RNA-DNA hybrids
contain-ing only minus-strand DNA. As expected, mutation of the YXDDmotif conserved in all RTsinactivated the DHBVRT
altogether. However, lesions in motifs implicated in RNase Hactivity also resultedin severedefects in minus-aswellas
plus-strand DNA synthesis. Again, this result could mean
either that these activities might not reside in independent domains or that the mutations we have chosen may have drastic effectsonprotein structure orstability.
Interestingly,recentstudiesindicate thatRTandRNase H
functionscanbe separated by mutation in human HBV (12). By using similar assays, Radziwill et al. have found that almost two-thirds of deletion mutations in the RNase H
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.73.309.76.258.2]a.
1 2 3
b.
AT,4 o
/
a-ow
12
CG
Q a
oc-
ss-1 2 3 4
do
o-p
CO CD to ( Q. Al (c otn I)
C} a a Q a
oc
p
3.:
.~~~~~~~~~~~~~~~~~~~.~-7
1 2 3 4 5 6
FIG. 3. Analysis of the RNaseH homologyregion mutants. (a) Southern blotanalysis of viralDNA incytoplasmic cores.Cells were
transfected with wt DHBVDNA(lane 1), D666Hmutant DNA(lane2),orD666Emutant DNA(lane 3);3days laterDNAextractedfrom cytoplasmic cores was analyzed by agarose gel electrophoresis and Southern hybridization with a 32P-DHBV DNA probe. (b) Primer extensionanalysis of viralRNApackaged intocytoplasmiccoresaftertransfection with D666H andD666E mutantDNAs. Two lanes from
asequencing ladderareshowntotheleft,asdescribed in the legendtoFig. 2b. Thearrowindicates thepositionofproductsexpectedfrom full-length RNA. (c)Southernblotanalysis of additionalmutants.Cellsweretransfected withwtDHBV DNA(lane1)ortheindicatedmutant
DNAs(lanes 2 through 4) and analyzedasdescribed forpanelA.Hybridizingmaterialnearthebottom of thegelrepresentsDpnI-digested
plasmidDNAusedfor transfection. (d)Endogenous polymerase assay. Cellsweretransfected withwtDHBV DNA(lane 1)ortheindicated mutant DNA(lanes2through 6);4days later virusparticleswerepelleted from
tfie
medium of each culture andanendogenouspolymerasereaction wascarried out. Labeled productDNAwasanalyzedas described in thelegendtoFig. 2c.OC,Open circular DHBVDNA;SS,
single-stranded unit-lengthDHBV DNA.
a.
-p4- ..
i24k- "
1.4
-1 2
b.
SZ~
C
622fb-
S2Tbr>)-b-
4bp-1 2 3
d.
A%tr
'S
I
[image:5.565.91.472.71.271.2]1 2 1 2
FIG. 4. Analysis of theproteinA(pA) insertionmutant.(A)Northern immunoblot of viralRNA. Poly(A)+RNAfrom cells transfected withwtDHBVDNA(lane 1)orproteinAmutant DNA(lane 2)wasseparatedon a1.6% agarose-2.2Mformaldehyde gel,transferredto a
nylon filter, andhybridized to
32P-DHBV
DNA. Positions of RNA size markersare shown at left. k, Kilobase. (b) RNase mappingof encapsidated viralRNA. Nucleic acidswere purifiedfromcytoplasmiccoresisolated from cells transfected withwtDHBV DNA(lane1), proteinAmutantDNA(lane 2),or noDNA(lane 3). Thesesampleswereannealedtoauniformlylabeled antisenseRNAprobespanningnt3021to2352,generatedbyin vitrotranscription withT7 RNApolymerase. Hybridswere digestedwithRNase, and theprotected duplex
fragmentwasanalyzed by electrophoresison a5%polyacrylamidegel.Positions of molecularweightmarkersareindicatedatleft.(candd)
Viral DNA synthesis. Pelleted material from culture medium of cells transfected withwtDHBV DNA(lanes 1)orproteinAmutantDNA (lanes2)wasanalyzedbySouthernhybridizationto32P-DHBVDNAinpanelcorbyendogenouspolymeraseassaywith
[32P]dCTP
inpaneld,asdescribed in thelegendtoFig.3d.
., zo
(O
A.. .o co -,. C.l
1"N rN
Co
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.565.109.453.404.638.2]domain of the HBV P gene selectively affect plus- rather than minus-strand synthesis (12). As in our experiment, mutations that changed the highly conserved YXDD motif in the RT domain impaired synthesis of both strands. A parallel difference exists among retroviral polymerases; unlike their murine leukemia virus counterparts (17), the RT and RNase Hfunctions of humanimmunodeficiencyvirushave not been separatedby mutation (9, 11).
Inspection of the sequence of hepadnavirus genomes reveals extensive divergence within the region of the P gene overlying the pre-S region of the overlapping S open reading frame. Thus this region ofthe P protein can tolerate many amino acid changes, which is compatible with the possibility that it encodes afunctionless spacer domain. Such a domain would separate the region of the protein linked to the 5' end ofminus-strand DNA (1), where it presumably functions as a primer (7), from the region of homology to other RT domains. The fact that our insertion of 369 nt in this region remained competent for all polymerase functions is consis-tent withthisnotion, as are several observations by others. Li et al. (10) recently noted that when DHBV genomes bearing frameshift mutations in the distal pre-S region were
inoculated intoducks, pseudorevertant viruses bearing com-pensatoryframeshift mutations several codons downstream wereregularly isolated. These mutants substitute a stretch of irrelevant amino acids for the authentic P sequences in this region yet grow and spread normally in vivo. Similarly, Schaller and co-workers (12) have shown that HBV genomes with deletions in this region are replication competent in
culturedhepatomacells,implyingthat thisregion is dispens-able.
These studies indicate both the power and the limitations of purely genetic analyses of protein function. Further progress in understanding the many activities of this com-plex enzyme will likely depend on either the purification of the native enzyme or the expression of large quantities of active protein by recombinant vectors and on the develop-ment ofbiochemical assays for the individual steps in the reverse transcription pathway.
ACKNOWLEDGMENTS
We thankRobin Colgrove forhelp with the sequence inspection andTitia DeLange, PeterPryciak, and Kuen-Teh Jeangfor helpful
discussions.
This workwas supported byPublic Health Service grants from
the National Institutes ofHealth. H.E.V. isan American Cancer
Society Professor of Molecular Virology.
LITERATURECITED
1. Bartenschlager, R., and H. Schaller. 1988. The aminoterminal
domainof the hepadnavirusP-geneencodestheterminalprotein
(genomic-linked protein) believed to prime reverse
transcrip-tion. EMBOJ.7:4185-4192.
2. Bavand, M. R.,and0. Laub. 1988. Twoproteins withreverse
transcriptase activities associated with hepatitis B virus-like
particles. J.Virol. 62:626-628.
3. Buscher, M., W. Reiser, H. Will,and H. Schaller. 1985.
Tran-scripts and the putative RNA pregenome of duck hepatitis B virus:implications for reversetranscription. Cell 40:717-724. 4. Chang, L.-J., D. Ganem, and H. E. Varmus. 1990.Mechanism
of translation of thehepadnaviral polymerase (P) gene. Proc. Natl. Acad. Sci. USA 87:5158-5162.
5. Chang, L.-J., P. Pryciak, D. Ganem, and H. E. Varmus. 1989. Biosynthesis of the reverse transcriptase of hepatitis B virus involves de novo translational initiation notribosomal
frame-shifting. Nature(London)337:364-367.
6. Church, G. M., and W. Gilbert. 1984. Genomic sequencing. Proc. Natl. Acad. Sci. USA81:1991-1995.
7. Ganem, D., and H. E. Varmus. 1987.The molecularbiology of
thehepatitis-Bviruses. Annu. Rev. Biochem. 56:651-693.
8. Hirsch, R.C., J. E. Lavine,L.-J. Chang, H. E. Varmus, and D. Ganem.1990.Polymerase gene products of hepatitis B virusare required for genomic RNA packaging as well as for reverse
transcription. Nature(London) 344:552-555.
9. Hizi, A., S. H. Hughes, and M. Shaharabany. 1990. Mutational
analysis of theribonuclease H activity of human
immunodefi-ciencyvirus 1 reversetranscriptase. Virology 175:575-580. 10. Li,J.-S., L. Cova, R. Buckland, V.Lambert, G. Deleage, and C.
Trepo. 1989. Duck hepatitis B virus can tolerate insertion, deletion, and partial frameshift mutation in the distal pre-S region.J. Virol. 63:4965-4968.
11. Prasad, V., and S.Goff. 1989. Linkerinsertionmutagenesisof the human immunodeficiency virus reverse transcriptase ex-pressed in bacteria: definition of the minimal polymerase do-main. Proc.Natl. Acad. Sci. USA86:3104-3108.
12. Radziwill, G., W. Tucker, and H. Schaller. 1990. Mutational
analysis of the hepatitis B virus P gene product: domain structure andRNase Hactivity. J. Virol. 64:613-620.
13. Radziwill, G., H. Zentgral, H. Schaller, and V. Bosch. 1988. The duck hepatitis-B virus DNA polymerase is tightly associated with the viral corestructure and unable to switch to an exoge-noustemplate. Virology 163:123-132.
14. Schlicht,H. J., G. Radziwill, and H. Schaller. 1989. Synthesis and encapsidation of the duck hepatitis B virus reverse
tran-scriptase does not require the formation of core/polymerase fusionproteins. Cell 56:85-92.
15. Sprengel,R., C.Kuhn, H. Will, and H. Schaller.1985.
Compar-ative sequence analysis of duck and human hepatitis-B virus genomes. J.Med. Virol. 15:323-333.
16. Summers, J., and W. Mason. 1982.Replication of thegenome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell29:403-415.
17. Tanese, N., and S. Goff. 1988. Domain structure ofMoloney murine leukemia virusreversetranscriptase: mutational
analy-sis and separate expression of the DNA polymerase and RNaseHactivities. Proc. Natl.Acad. Sci USA85:1777-1781.