Large-Scale Production and Structural and Biophysical
Characterizations of the Human Hepatitis B Virus Polymerase
Judit Vörös,aAnnika Urbanek,bGilles Jean Philippe Rautureau,a* Maggie O’Connor,bHenry C. Fisher,cAlison E. Ashcroft,c Neil Fergusonb
School of Biomolecular and Biomedical Science, University College Dublin, Dublin, Irelanda
; School of Medicine and Medical Science, University College Dublin, Dublin, Irelandb
; Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, United Kingdomc
ABSTRACT
Hepatitis B virus (HBV) is a major human pathogen that causes serious liver disease and 600,000 deaths annually. Approved therapies for treating chronic HBV infections usually target the multifunctional viral polymerase (hPOL). Unfortunately, these therapies— broad-spectrum antivirals—are not general cures, have side effects, and cause viral resistance. While hPOL remains an attractive therapeutic target, it is notoriously difficult to express and purify in a soluble form at yields appropriate for struc-tural studies. Thus, no empirical strucstruc-tural data exist for hPOL, and this impedes medicinal chemistry and rational lead discov-ery efforts targeting HBV. Here, we present an efficient strategy to overexpress recombinant hPOL domains inEscherichia coli, purifying them at high yield and solving their known aggregation tendencies. This allowed us to perform the first structural and biophysical characterizations of hPOL domains. Apo-hPOL domains adopt mainly␣-helical structures with small amounts of
-sheet structures. Our recombinant material exhibited metal-dependent, reverse transcriptase activityin vitro, with metal binding modulating the hPOL structure. Calcomine orange 2RS, a small molecule that inhibits duck HBV POL activity, also in-hibited thein vitropriming activity of recombinant hPOL. Our work paves the way for structural and biophysical characteriza-tions of hPOL and should facilitate high-throughput lead discovery for HBV.
IMPORTANCE
The viral polymerase from human hepatitis B virus (hPOL) is a well-validated therapeutic target. However, recombinant hPOL has a well-deserved reputation for being extremely difficult to express in a soluble, active form in yields appropriate to the struc-tural studies that usually play an important role in drug discovery programs. This has hindered the development of much-needed new antivirals for HBV. However, we have solved this problem and report here procedures for expressing recombinant hPOL domains inEscherichia coliand also methods for purifying them in soluble forms that have activityin vitro. We also pres-ent the first structural and biophysical characterizations of hPOL. Our work paves the way for new insights into hPOL structure and function, which should assist the discovery of novel antivirals for HBV.
H
epatitis B virus (HBV) is a highly infectious, species-specific pathogen that causes serious liver diseases, including cancer, and causes 600,000 deaths annually (1). While excellent prophy-lactic HBV vaccines exist, these are ineffective against extant chronic HBV infections which affect 350 million people world-wide. Approved therapies for chronic HBV infections include im-muno-modulators (e.g., interferon therapies) and nucleoside an-alogues that inhibit the reverse transcriptase domain of hPOL, the multifunctional viral polymerase of human HBV. These reverse transcriptase inhibitors, currently our best weapons against HBV, are not complete cures and have unwanted side effects (2, 3). While reverse transcriptase inhibitors are initially very effective at treating chronic HBV infections, they select for resistant viral strains (albeit with distinct emergence rates for each drug). This resistance leads to problems of multidrug-resistant HBV strains and complications in treating people coinfected with HIV (who are also treated with the same broad-spectrum reverse transcrip-tase inhibitors) (3–5). Thus, novel antivirals are needed to ensure future combination therapies can more effectively treat chronic HBV infections.HBV is the smallest DNA virus known to infect animals, and the partially double-stranded DNA 3.2-kb genome encodes just seven proteins. hPOL, the only HBV enzyme, mediates diverse functions, including protein-primed initiation of reverse
tran-scription, RNase H activity, and RNA- and DNA-dependent DNA polymerization (6–8). While the reverse transcriptase (RT) and RNase H (RH) domains of hPOL exhibit significant homology to counterparts from other viruses and bacteria, hPOL differs by having its enzymatic modules arranged in tandem within a single polypeptide and having a terminal protein domain (TP) at its N terminus (8–12). The TP domain is a key player in the protein-primed initiation of reverse transcription that is essential for HBV replication (13–15). Protein priming involves hPOL interacting with host chaperones (including Hsp40, Hop, Hsc70, and Hsp90), the HBV capsid-forming protein, and epsilon (a stem-loop
struc-Received5 September 2013Accepted9 December 2013 Published ahead of print18 December 2013
Editor:D. S. Lyles
Address correspondence to Neil Ferguson, [email protected].
J.V., A.U., and G.J.P.R. contributed equally to this work.
* Present address: Gilles Jean Philippe Rautureau, Ecole Normale Supérieure de Lyon, Centre de RMN à Très Hauts Champs, UMR 5280 CNRS/ENS, Villeurbanne, France.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02575-13
on November 7, 2019 by guest
http://jvi.asm.org/
ture near the 5=end of the viral pregenomic RNA) (Fig. 1) (6–8,
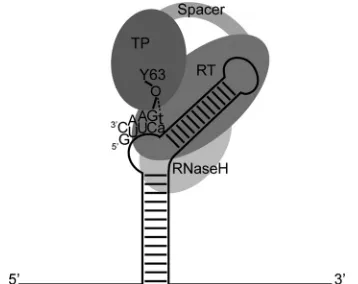
16–19). The RT domain of hPOL catalyzes an epsilon-templated reaction, wherein a single nucleotide is covalently attached to Y63 of hPOL TP (Y96 in duck HBV POL), to which a further 2 to 3 dNMPs are thereafter incorporated (Fig. 1) (13–15). This oligo-nucleotide is then translocated to a 3=direct repeat sequence on the pregenomic RNA, where reverse transcription continues and the RH domain simultaneously degrades the RNA template. How-ever, a short RNA oligonucleotide evades degradation and primes plus-strand DNA synthesis via a complex process reported to in-volve two further template switches (6–8,20).
Despite being such an important target, our understanding of hPOL structure-function and the ability to generate novel antivi-rals targeting its diverse functions currently lags behind that of important enzymes from other pathogenic viruses (21–25). This situation arises from the extreme difficulties in obtaining suffi-cient yields of soluble, active recombinant hPOL that are needed to drive forward structural biology and high-throughput drug dis-covery programs (26–31). Consequently, most insights on hPOL functions and interactions have been obtained through complex virology studies, often performed in high-containment laborato-ries, including cellular HBV replication assays,trans -complemen-tation studies, and immunoprecipi-complemen-tation experiments (13,19,32– 35). While these approaches do not directly yield high-resolution mechanistic details, they have delivered impressive insights into the various hPOL activities needed for HBV replication.
Historical difficulties in obtaining sufficient quantities of re-combinant hPOL forin vitrostudies led to the widespread adop-tion of duck HBV POL (dPOL) as a model system. dPOL shares ⬃26% homology to hPOL, and recombinant dPOL is easier to express (inE. colior insect cells) at yields appropriate forin vitro functional assays. Recombinant dPOL requires cell extract supple-ments in order to exhibitin vitroactivity in functional reconstitu-tion assays, which led to host chaperones being identified as es-sential cofactors (36–38). dPOL expressed inE. coli, supplemented with appropriate divalent metal ions, recombinant host
chaper-ones, and an ATP-regenerating system, was later shown to medi-atein vitropriming and elongation reactions (36,37,39–41). A mini-dPOL variant, which lacked the dispensable spacer domain and the RH domain, was shown to have chaperone-independent activity and mediate cryptic primingin vitro(where deoxyribo-nucleotides are covalently attached to tyrosine residues of the RT domain instead of TP) (42–44).
Unfortunately, it has proved challenging to mirror this success with recombinant hPOL. hPOL is reportedly expressed poorly by E. coli(if at all), with only a few reports citingin vitroactivity and little (or no) follow-up of these studies (16,27,28,45). Hu and coworkers, however, recently showed small quantities of recom-binant hPOL could be expressed in mammalian cells (46). This material faithfully recapitulated hPOL activityin vitro, including the dependence of protein-priming and DNA elongation reac-tions on manganese and magnesium salts, respectively, as per re-ports for dPOL (17,46). While this is a landmark achievement, the protein yields obtained were not sufficient for the high-through-put screening and structural biology programs that typically sup-port medicinal chemistry efforts. This situation contrasts mark-edly with the drug discovery programs that have successfully targeted other pathogenic viruses (e.g., human immunodeficiency virus and herpes simplex virus [47–49]).
Here, we report effective strategies for high-level expression of recombinant hPOL constructs inE. coliand their subsequent pu-rification in soluble forms amenable to most biophysical and structural methodologies. Rather than deleting the dispensable spacer domain and religating remaining POL sequences, as per-formed by a number of laboratories (37,39–41,44), we expressed recombinant TP and an RT-RH concatemer as independent poly-peptides. Microscale thermophoresis and isothermal titration cal-orimetry showed a direct, specific interaction between recombi-nant TP and RT-RH constructs in vitro, consistent with these recombinant domains being correctly folded, as they have to in-teract during protein priming (Fig. 1). Apo-TP and apo-RT-RH were largely␣-helical with smaller amounts of-sheet structures. The structures of TP and RT-RH were modulated by binding di-valent metal cations that modulate hPOL activity (31,46,50). Our recombinant hPOL materials were active inin vitroreconstitution assays that included human chaperone molecules, an ATP-regen-erating system, and appropriate divalent cations (and were also inhibited by a known dPOL inhibitor). Thus, our work makes it possible to rapidly produce soluble hPOL constructs (on the scale of hundreds of milligrams to grams) that faithfully recapitulate key functional activities of dPOL and hPOL. These factors, to-gether with our hPOL constructs being soluble and well-behaved under a wide range of experimental conditions (even at high pro-tein concentrations), opens the door for detailed structure-func-tion analyses of hPOL and also high-throughput lead discovery efforts. These findings should aid the quest for novel antivirals that more effectively treat chronic HBV infections.
MATERIALS AND METHODS
Reagents.All reagents were of AnalaR grade and purchased from Sigma Chemical Co. The amphipathic polymer (NV-10) was purchased from Expedeon (United Kingdom). Epsilon RNA from human HBV (bases 1822 to 1989; GenBank accession numberU87746.3) and a similar-sized control (mock) RNA (UAUAGGGAGA CCACAACGGU UUCCCU CUAG AAAUAAUUUU GUUUAACUUU AAGAAGGAGA UAUACA UAUG AUGGAACUAA GCCUGGCUCU GGUAAAUAGC UCCAAU
FIG 1Schematic representation of protein-primed initiation of reverse tran-scription mediated by hPOL. The epsilon stem-loop found on HBV pre-genomic RNA is represented by a solid black line (internal base pairing is shown as short cross-hatches). The hPOL TP, spacer, RT, and RH domains are shaded. During protein-primed initiation of reverse transcription, the enzy-matic activity of RT covalently attaches a deoxyribonucleotide to the side chain hydroxyl group of Y63 in TP (8,13). This priming reaction is guided by the epsilon template. The first nucleotide incorporated into TP has been reported to be G (as shown here), whereas others have shown that T can also be used (shown in lowercase) (17,31,46,87).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.75.254.67.209.2]GUGC GAUGAGAAUU) were transcribedin vitroby using a MegaScript kit (Ambion). Hsp40 and HOP DNAs were obtained from the Arizona State University Biodesign Institute Plasmid Repository.
Protein expression and purification. (i) Terminal protein domain.A gene encoding residues 1 to 192 of human HBV polymerase (subtypeayw) was subcloned into pET21a (Novagen) to generate pET21a_TP192 encod-ing TP1–192with an N-terminal T7 tag (MASMTGGQQMG) and a
C-ter-minal six-histidine tag. E. coli C41(DE3) cells transformed with pET21a_TP192 were grown at 37°C to an optical density at 600 nm
(OD600) of 0.6 to 0.8 and induced overnight with 1 mM isopropyl--D
-thiogalactopyranoside (IPTG). Cell pellets were resuspended in 20 mM sodium phosphate buffer (pH 7.2) containing 2 mM dithiothreitol, 1 mg/ml lysozyme, DNase I, and protease inhibitor tablets. Following son-ication, cell lysates were centrifuged at 8,000⫻gfor 30 min to pellet TP1–192-containing inclusion bodies. Inclusion bodies were washed three
times with 50 mM Tris-HCl buffer (pH 8.0), 1 mM EDTA, 1% (vol/vol) Triton X-100, followed by two washes with distilled water. Inclusion bod-ies were solubilized in 50 mM Tris-HCl buffer (pH 8.0), 200 mM NaCl, 10 mM imidazole, and 6 M guanidine hydrochloride. This solution was passed through a 0.22-m filter, and TP1–192was then purified using denaturing Ni-affinity chromatography. Denatured TP1–192was
solubi-lized and renatured by sequential dialysis steps into TMK buffer (20 mM Tris-HCl [pH 7.5], 2.5 mM MgCl2, 50 mM KCl, 5 mM
-mercaptoetha-nol, and NV-10 at a 10-fold weight excess to recombinant protein). The remaining impurities were removed by gel filtration chromatography.
(ii) Reverse transcriptase-RNase H concatemer. Bioinformatics analyses identified 4 putative N termini for the hPOL RT-RH concatemer (residues 283, 303, 320, and 346) and 3 C termini (residues 778, 783, and 832). DNA sequences containing all possible combinations of these boundaries were generated using PCR. Each construct was engineered to have a 3=epsilon stem-loop sequence outside the coding region, as this had been reported to facilitate hPOL expression (51,52). These cassettes were subcloned to generate RT-RH expression vectors containing an N-terminal tag (either maltose-binding protein or a T7 tag) and a C-N-terminal six-histidine tag. These constructs were used in expression tests, where the effects of cell line, growth temperature, medium type, and induction time on the expression levels and ratio of soluble-to-insoluble RT-RH were assessed. The construct with the best-behaving properties that was used to produce recombinant protein for biophysical studies and functional as-says was pET21a_RT-RH303–778(pET21a containing an RT-RH303–778
cassette with T7 and six-histidine tags at its N and C termini, respectively).
E. coliBL21(DE3) cells were transformed with pRT-RH303–778and
grown at 42°C until the OD600reached⬃0.9. Cells were induced at 30°C
with 1 mM IPTG and grown for a further 3 h. Cell pellets were resus-pended and lysed in 50 mM sodium phosphate buffer (pH 8.0; containing 300 mM NaCl and 1 mg/ml lysozyme). Following lysis, inclusion bodies containing RT-RH303–778were harvested by centrifugation at 8,000⫻g
for 15 min. Inclusion bodies were washed three times with 50 mM sodium phosphate buffer (pH 8.0; containing 300 mM NaCl and 1% [vol/vol] Triton X-100) and twice with distilled water. The pellet was solubilized in 20 mM sodium phosphate buffer (pH 7.5), 8 M guanidine hydrochloride and centrifuged to remove insoluble material. Imidazole was added to a final concentration of 5 mM, and RT-RH303–778was purified using
dena-turing Ni-affinity chromatography. RT-RH303–778was refolded as de-scribed for TP1–192.
(iii) Human chaperones.Human Hsc70 cloned into pET21a was transformed intoE. coliBL21(DE3). These cells were grown at 22°C and induced with 1 mM IPTG when the OD600reached⬃0.7. Purification was
carried out as reported elsewhere (53). Human Hop cloned into pET21a was transformed intoE. coliBL21(DE3) cells. These cells were grown at 37°C and induced with 0.5 mM IPTG when the OD600reached⬃0.6. Hop
was purified according to reported protocols (54).E. coliC41(DE3) cells transformed with pHsp90were grown at 37°C until the OD600reached
⬃0.6, and then the cells were induced for 4 h with 0.5 mM IPTG. Hsp90 was purified as previously reported (55).
Human Hsp40 was cloned into pET21a and overexpressed at 37°C in C41(DE3) cells (via an overnight induction with 0.5 mM IPTG). Cells were lysed in 20 mM HEPES buffer (pH 7.2) containing 100 mM NaCl, 5 mM-mercaptoethanol, and protease inhibitor cocktail. Ammonium sulfate was added to the cleared cell lysate to a final concentration of 40% (wt/vol). Hsp40-containing pellets harvested by centrifugation were re-suspended and dialyzed in the lysis buffer prior to purification on a MonoS cation exchange column (GE Healthcare, PA).
Circular dichroism.Protein solutions contained 50 mM EDTA prior to refolding to ensure chelation of residual metals in buffers. EDTA was removed during refolding by dialysis into circular dichroism (CD) sample buffer (20 mM Tris-HCl [pH 7.5], 50 mM KCl, 5 mM -mercaptoetha-nol, and a 10-fold [wt/wt] excess of NV-10 [relative to protein]). CD spectra were recorded on a spectropolarimeter (model 810; Jasco, MD) equipped with a Peltier temperature controller. Far-UV CD spectra were recorded at 20°C by using a 1-mm-path-length cuvette and a protein concentration of⬃0.15 mg/ml. Four spectra were acquired, from 190 to 270 nm, by using a 50-nm/min scan speed. Buffer contributions were subtracted from all reported spectra.
Size exclusion chromatography–multiangle light-scattering mea-surements.Size exclusion chromatography combined with multiangle light scattering (SEC-MALS) was performed at 20°C with a Shimadzu high-performance liquid chromatography (HPLC) apparatus connected to Wyatt Dawn HeleosII light scattering and Optilab rEX instruments (Wyatt Technology, CA). SEC was performed by using 24 ml analytical Superose 6 (TP1–192) or Superdex 200 (RT-RH303–778) columns (GE
Healthcare, PA) in a column oven. Ten-microliter protein samples were injected at protein concentrations of up to 300M (TP1–192) and 165M
(RT-RH303–778). The manufacturer’s software was used to correct the UV,
differential refractive index, and light-scattering signals for baseline drifts and band broadening, thus allowing determination of the molar masses of species eluting from columns.
SEC-MALS analyses of protein-NV-10 conjugates.The specific re-fractive index increment (dn/dc) value for NV-10 was determined in batch mode using the manufacturer’s Astra software (Wyatt Technology, CA). A dilution series of NV-10 was made containing 0.03125, 0.0625, 0.125, 0.25, 0.5, 1.0, 2.0, and 4.0 mg/ml NV-10 dissolved in water. Five-milliliter samples were injected into the Optilab rEX flow cell, and the refractive index at 658 nm was recorded. The Astra template was used to fit the linear data obtained, and the data yielded adn/dcvalue of⬃0.1368 ml · g⫺1. To
obtain the extinction coefficient for NV-10, UV absorbance spectra were acquired for samples from the same dilution series and, where relevant, corrected for contributions from light scattering. This yielded an extinc-tion coefficient at 280 nm of⬃163.2 M⫺1· cm⫺1for NV-10. To obtain the
respective molar masses of recombinant proteins, NV-10, and protein– NV-10 conjugates, SEC-MALS data were acquired as described in the previous section. The manufacturer’s protein-conjugate template was used to analyze the data, together with thedn/dcvalues and extinction coefficients of NV-10 and either TP1–192or RT-RH303–778.
Mass spectrometry.Electrospray ionization-ion mobility spectrome-try-mass spectrometry (ESI-IMS-MS) analyses were performed using a Synapt G2-S mass spectrometer (Waters Corp., Manchester, United Kingdom). Protein/amphipol samples were analyzed in positive ion mode with a capillary voltage of 1.75 kV. A cone voltage of 100 V and trap wave guide energy of 25 V (2.45⫻106Pa argon) were used to extract TP
1–192
from the amphipol. A helium pressure of 2.33⫻106Pa at the entrance to
the IMS cell was used for collisional cooling prior to IMS separation. The N2flow rate in the IMS cell was set to 90 ml/min (3.01⫻105Pa), and
separations were performed with a wave height ramped from 10 to 40 V and a wave speed of 750 m/s. RNA samples were analyzed in 50 mM ammonium acetate and 50 mM piperidine-imidazole buffers (Sigma-Al-drich Company Ltd., United Kingdom). RNA samples were analyzed us-ing negative-ion ESI-MS with a capillary voltage of 0.8 to 1.2 kV and a cone voltage of 30 to 90 V.
Vörös et al.
on November 7, 2019 by guest
http://jvi.asm.org/
Microscale thermophoresis.Samples for microscale thermophoresis (MST) were labeled using the Red–N-hydroxysuccinimide protein-label-ing kit (NanoTemper Technologies, Germany). A 0.05% (vol/vol) con-centration of Tween 20 was added to the sample buffer (20 mM HEPES buffer, 50 mM KCl, 2.5 mM MgCl2, 5 mM-mercaptoethanol, 10-fold
[wt/wt] excess of NV-10) to prevent protein adsorption to capillary side-walls. Labeled TP1–192at 40 nM was titrated with unlabeled RT-RH303–778
by a series of 16 1:2 serial dilutions (where the highest concentration of RT-RH303–778was⬃5.2M). A 4-l aliquot of each sample was aspirated
into a hydrophilic capillary, and measurements were performed at 20°C with a Monolith NT.115 (NanoTemper Technologies, Germany). The excitation light-emitting diode was set to 50%, and laser powers of 40% and 80% were used (for heating and cooling phases of 30 s and 5 s, respec-tively).
Isothermal titration calorimetry.Isothermal titration calorimetry (ITC) measurements were performed at 20°C using a MicroCal ITC200
calorimeter (GE Healthcare, PA). TP1–192and RT-RH303–778samples
were dialyzed against 20 mM HMK buffer (pH 7.5; 20 mM HEPES buffer, 50 mM KCl, 2.5 mM MgCl2, 5 mM-mercaptoethanol) prior to
measure-ments. The concentration of RT-RH303–778in the calorimeter cell was
⬃20M, into which a stock of⬃100M TP1–192was titrated.
Experi-mental parameters were as follows: a first injection of 3l followed by nine injections of 4l, 150 s between injections, 1,000-rpm syringe stir-ring speed, and high feedback mode. Sequential titrations were acquired and concatenated (as previously described [56]) to ensure binding satu-ration. Buffer titrations were performed to correct for heat-of-dilution effects. Concatenated, referenced titrations were analyzed using Origin7.0 (OriginLab Corp., MA).
In vitrohPOL protein-priming assay.hPOL priming activity assays were performed with recombinant proteins expressed inE. coli(as de-scribed above) at the following concentrations (unless stated otherwise): 0.6M TP1–192, 2M RT-RH303–778, 1.4M Hsc70, 1.9M Hsp40, 0.6
M Hsp90, 0.6M Hop, and 2M epsilon RNA. These assay mixtures contained an ATP-regenerating system to facilitate chaperone activity (5 mM ATP, 25 mM creatine phosphate, and 10 U/ml creatine phosphoki-nase). Ten-microliter reaction mixtures were incubated for 2 h at 30°C in TMK buffer for reconstitution of priming complexes. Priming was initi-ated by the addition of 10l priming buffer (20 mM Tris-HCl [pH 8], 20 mM NH4Cl, 10 mM MgCl2, 0.4% [vol/vol] Triton X-100, 20 mM
-mer-captoethanol, 0.5l of [␣-32P]dGTP or [␣-32P]dTTP [3,000 Ci/mmol; 10
mCi/ml]) and various concentrations of MnCl2. Priming reaction
mix-tures were incubated at 37°C for 2 h and stopped by the addition of 20l SDS-PAGE loading buffer. Twenty-microliter samples were loaded onto 15% SDS-PAGE or 8% urea-PAGE gels, and32P-labeled proteins were
identified by autoradiography.
RESULTS
Reappraising the structural boundaries of hPOL domains.The domain boundaries of hPOL were originally mapped by system-atically truncating its N or C termini to determine which con-structs retained functional activity in cellulartrans -complemen-tation assays (34). hPOL was found to have four domains (Fig. 2A): the TP domain conserved across the hepadnaviridae (resi-dues 1 to 177), the spacer region (resi(resi-dues 178 to 335), linking TP to the RT domain (residues 336 to 678) and the C-terminal RH domain (residues 679 to 832). Importantly, these studies also re-vealed the following: (i) the spacer domain is dispensable and not needed for hPOL activity (10); (ii) the TP domain did not need to be part of the same polypeptide chain as RT and RH to mediate protein-priming and elongation activities (and vice versa); (iii) independently expressed hPOL domains could function intrans (13,34).
While the hPOL domains described above interacted intrans and restored hPOL activity, this does not mean that these domains
were independently stable, fully folded structural entities. Overtruncation of proteins can destabilize them or cause them to degrade or aggregate, whereas “excess” sequence can promote ag-gregation and hamper protein crystallization. We hypothesized that the known difficulties in expressing hPOL inE. colimight arise from classical domain boundaries not corresponding to bona fide structural boundaries.
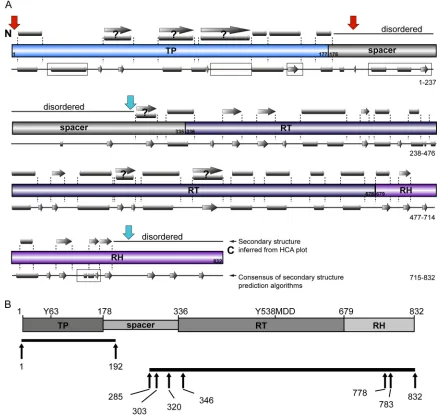
To test this hypothesis, we used secondary structure prediction tools and hydrophobic cluster analysis (HCA) (57,58) to help identify plausible structural boundaries for hPOL domains (Fig. 2A). Secondary structure prediction algorithms are relatively ro-bust for identifying tracts of potential␣-helical,-sheet, turn, and coil-like regions in protein sequences (57). HCA applies a differ-ent approach and examines the patterns of hydrophobic clusters in polypeptide sequences, with a view to finding clusters indicative of different types of structural motifs. While HCA makes no ref-erence to other sequence features of the protein, such as sequence homology or conserved functional motifs, its pattern recognition capabilities can often help to identify the structural boundaries of protein domains (58).
Our combined analyses highlighted the following (Fig. 2A): (i) the reported C terminus of the TP domain (residue 177) falls in the middle of a predicted␣-helical element; (ii) the spacer domain contains many proline and glycine residues and is therefore likely to be disordered (44,59–61); (iii) the classical N-terminal bound-ary of the RT domain (residue 336) falls within the middle of a predicted␣-helix; (iv) we found no convincing evidence of the RT and RH domains being discrete entities in hPOL.
Taking into account our bioinformatics analyses findings, re-ported functional studies, and the ability of independently ex-pressed POL domains to interact intrans, we redesigned hPOL domain boundaries with a view to expressing TP and an RT-RH concatemer as independent polypeptides (Fig. 2B). Specifically, we extended the C terminus of TP by 15 residues to ensure capture of all putative structural elements within TP (Fig. 2A). The situa-tion was more complex for RT-RH, where the putative N- and C-terminal boundaries were ambiguous. Thus, we identified four potential N-terminal boundaries located within predicted un-structured regions (residues 285 and 303 [Fig. 2B]) or at the be-ginning (residue 320) and end (residue 346) of a putative␣-helix preceding the RT domain. As the C terminus of RH was rich in proline and hydrophobic residues, we considered it unlikely to be folded and more likely a cause of poor expression and protein aggregation. Thus, we designed three putative C termini for our RT-RH constructs: the authentic C terminus (residue 832) and truncations to either residue 778 or 783 (Fig. 2B). The latter trun-cations preserve reported RH catalytic residues (62) but eliminate the C terminus, which we predicted to be unstructured.
High-level expression of recombinant TP and RT-RH inE. coli.We used PCR amplification to generate an extended TP vari-ant (TP1–192) and RT-RH constructs containing various
combina-tions of alternative N and C termini. These species were subcloned into a range of bacterial expression vectors to generate a range of RT-RH and TP constructs with six-histidine tags or fusion protein partners (e.g., maltose-binding protein) at the N or C terminus. The overall aims were to increase the yield and solubility of recom-binant TP1–192 and RT-RH expression. Small-scale expression
tests were used to triage which incubation temperature, induction time,E. colistrains, and expression constructs had the most
on November 7, 2019 by guest
http://jvi.asm.org/
tive protein expression properties (described in Materials and Methods).
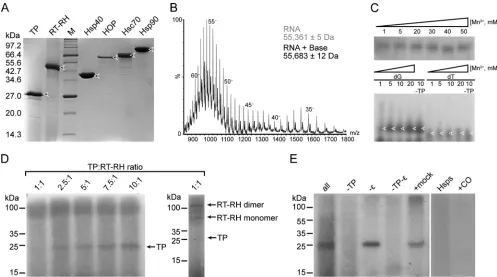
TP1–192with a six-histidine tag at its C terminus was expressed
best inE. coliC41(DE3) with an overnight induction of protein expression at 37°C. Under these conditions, TP1–192was expressed
at very high levels (Fig. 3A) but accumulated intracellularly as insoluble inclusion bodies. While we also checked the effects of adding various fusion protein partners to TP1–192, we found that
these fusion partners did not significantly improve the yields of soluble recombinant TP1–192(data not shown). Initial expression
tests of RT-RH285– 832, RT-RH303–778, and RT-RH303–783showed
that these constructs also were expressed, albeit at much lower levels than TP1–192(Fig. 3B). Addition of an MBP fusion partner
marginally improved the expression levels of RT-RH285– 832,
RT-RH303–778, and RT-RH303–783(Fig. 3B, lanes 1 to 6) compared to
constructs lacking fusion partners (Fig. 3B, lanes 7 to 9). Notably, the expression of RT-RH285– 832constructs was extremely variable
and unreliable, in marked contrast to RT-RH303–778 and
RT-RH303–783(which gave reproducible expression levels). Thus, we
focused downstream efforts on improving the expression of RT-RH303–778, as it expressed robustly and at slightly higher levels than
RT-RH303–783.
FIG 2Reappraisal of the boundaries of hPOL structural domains. (A) Annotation of secondary structure predictions based on HCA (gray symbols above the schematic presentation of classical hPOL domain boundaries) and using sequence-based algorithms (gray symbols below the schematic presentation of classical hPOL domains) (57,58). The boundaries of classical domains are numbered. Predicted␣-helical (gray cylinders) and-strand (gray arrows) secondary structures are shown. Ambiguous, but predicted to be structured, regions are indicated by question marks. Disagreements between the HCA and the consensus of conventional algorithms are indicated by boxed areas on the conventional sequence-based algorithms. Red arrows indicate the newly identified putative domain boundaries of TP, whereas the cyan arrows indicate the new domain boundaries for RT-RH. (B) Optimizing hPOL domain boundaries for expression inE. coli. (Upper graphic) Classical hPOL domain boundaries as reported in the literature (10,34). Functionally important residues in the TP and RT domains are indicated. (Lower graphic) Black bars indicate the TP and concatemeric RT-RH constructs we designed, with optimized domain boundaries based upon hydrophobic cluster analyses (58), secondary structure prediction algorithms (57), and removal of motifs that could cause aggregation or proteolysis.
Vörös et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.71.511.68.486.2]All RT-RH constructs were expressed as insoluble inclusion bodies, despite our employing a wide range of different vectors, fusion protein partners, culture conditions, and cell lines to try and achieve soluble expression (Fig. 3Cand unpublished data). Insoluble expression of RT-RH was not ideal, since this made necessary additional downstream steps to solubilize inclusion bodies and refold RT-RHin vitro. Such strategies typically cause significant protein losses, which would undermine the goal of pu-rifying hPOL domains at high yield. To offset these potential losses, we heat shocked E. coli cultures prior to induction of RT-RH expression (63), thereby inducing a chaperone response that increased RT-RH303–778and RT-RH303–783expression to very
high levels (⬃40% of total cellular protein), albeit still in an insol-uble form (Fig. 3C).
Purification of recombinant TP1–192and RT-RH303–778.Since
recombinant TP1–192and RT-RH303–778formed inclusion bodies
inE. coli, we thereafter concentrated on purifying constructs that lacked fusion proteins, as these would have complicatedin vitro refolding steps, but which contained C-terminal six-histidine tags (to simplify protein purification [see Materials and Methods]). Inclusion bodies containing TP1–192or RT-RH303–778were
solu-bilized using chemical denaturants and purified under denaturing conditions via nickel affinity chromatography, thus yielding de-natured, histidine-tagged TP1–192and RT-RH303–778. A huge range
of experimental conditions, additives, and solubilization strate-gies were screened in order to remove chemical denaturants and refold our hPOL constructs (unpublished data). Despite extensive efforts, we found very few conditions that allowed hPOL domains to be isolated in soluble forms and at high yields.
However, TP1–192and RT-RH303–778could be efficiently
solu-bilized and refolded in buffers containing a molar excess of NV-10, an amphipathic carbohydrate polymer (amphipol). These polymers are thought to improve protein solubility by binding to exposed hydrophobic patches on proteins, displaying in their place hydrophilic carbohydrate moieties that mask sticky, poten-tially aggregation-prone patches (64). By using this refolding strategy, and also gel filtration chromatography (see Materials and Methods), we purified TP1–192and RT-RH303–778to homogeneity
(as judged by SDS-PAGE analysis [Fig. 4AandB]). The yield of soluble, pure TP1–192and RT-RH303–778was⬃50 mg and⬃23 mg,
respectively, from each liter ofE. coliculture. This is the highest yield ever reported for purification of recombinant hPOL con-structs (26,29,30,65). Critically, recombinant TP1–192and
RT-RH303–778were soluble over a wide pH range and at high protein
concentrations (up to 10 mg/ml).
Coarse-grain structural characterizations of TP1–192and RT-RH303–778. The availability of pure, soluble hPOL domains
al-lowed us to perform the first structural characterizations of TP1–192and RT-RH303–778. Far-UV CD spectroscopy is a sensitive
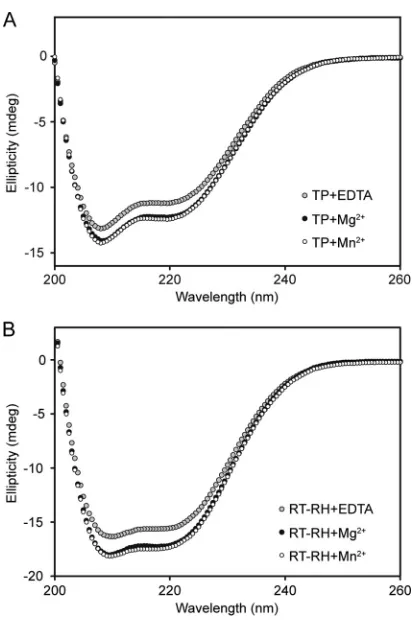
way of measuring chirality adopted by polypeptide backbones (and, by corollary, their secondary structure composition). Re-combinant TP1–192had a far-UV CD spectrum with pronounced
negative minima at 208 and 222 nm (Fig. 5A), consistent with
TP1–192 having an extensive␣-helical structure and additional
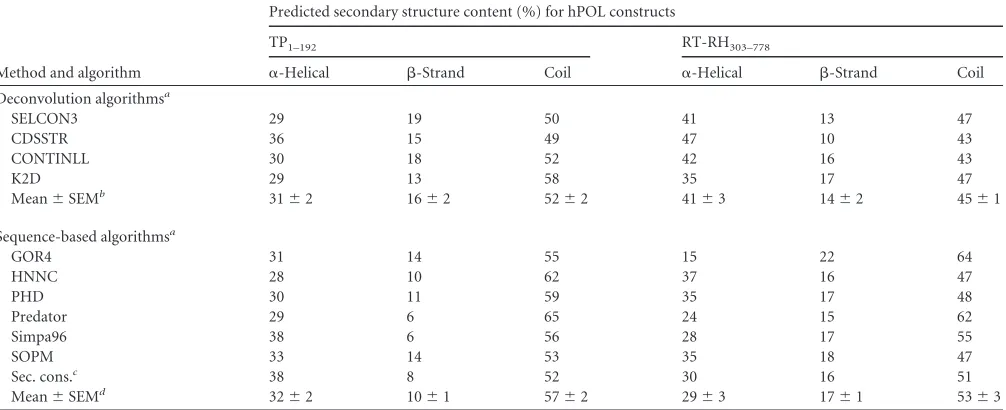
contributions from-strand and coil-like structures. Deconvolu-tion of the far-UV CD spectrum suggested apo-TP1–192contained
⬃31%␣-helix and⬃16%-sheet, values that agreed well with the sequence-based structural predictions (32% ␣-helix and 10% -sheet, respectively) (Table 1) (57,66).
The far-UV CD spectrum of RT-RH303-778also had the double
FIG 3Coomassie blue-stained SDS-PAGE gels of recombinant TP1–192and RT-RH constructs expressed inE. coli. (A) Lane 1, noninduced C41(DE3) cells containing pET21a_TP192, which contains a TP1–192insert with a C-terminal six-histidine tag (see Materials and Methods); lane 2, overnight expression of pET21a_TP1–192in C41(DE3) cells. The arrow indicates TP1–192. Lane M, protein size markers. (B) Expression of RT-RH constructs in BL21(DE3) cells. RT-RH285– 832(lanes 1, 4, and 7), RT-RH303–778(lanes 2, 5, and 8), and RT-RH303–783(lanes 3, 6, and 9) results are shown, where these constructs had an N-terminal six-His-tagged MBP fusion partner (6His-MBP-X), an N-terminal MBP fusion partner, and a C-terminal six-His tag (MBP-X-6His) or a C-terminal 6-His tag (X-6His), as indicated above the gel. Arrows indicate expression of different RT-RH constructs. Notably, RT-RH constructs had a higher mobility than expected from their theoretical mass, even though they were confirmed to be full length based on proteomic analyses (unpublished data). (C) BL21(DE3) cells transformed with 6His-MBP-RT-RH303–778were grown at 30°C (left panel) and at 42°C (right panel, to stimulate chaperone expression) prior to expression at 30°C. Postinduction, samples were taken at the times (in min) indicated at the bottom of these gels. Total material (T) and soluble fractions (S) of cell lysates were analyzed using SDS-PAGE. Arrows indicate expected products; lane M contains protein size markers. These data are representative of the heat shock-induced enhancement of protein expression observed for all RT-RH303–778and RT-RH303–783constructs.
on November 7, 2019 by guest
http://jvi.asm.org/
minima at 208 and 222 nm characteristic of␣-helical proteins (Fig. 5B). Spectral deconvolution algorithms predicted the spec-trum was dominated mainly by␣-helical contributions (⬃41%) and smaller amounts of-sheet (⬃14%). These estimates, while broadly consistent with the values obtained from sequence-based secondary structure predictions for RT-RH303-778(⬃29%␣-helix
and⬃17%-sheet) (Table 1), had larger discrepancies than for TP1-192(Table 1). Discrepancies of this magnitude are often
en-countered, and the magnitude of the discrepancies for
RT-RH303-778were considerably less than we have determined for
other proteins we are studying (barley chymotrypsin inhibitor 1, hepatitis B virus core protein, human superoxide dismutase 1, and the EAR domain of human␥2-adaptin [data not shown]) (56,
67–69). The far-UV CD spectra of apo-TP1-192 and
apo-RT-RH303-778were essentially invariant between pH 5.5 and 9.5,
con-sistent with their structures not varying significantly in this pH range (data not shown).
Manganese and magnesium salts have been reported to mod-ulate hPOL-mediated protein-priming and elongation activities, respectively (31,46). To test whether the structures of hPOL do-mains were modulated by these divalent cations, we measured the far-UV CD spectra of TP1-192and RT-RH303-778in the presence of
excess manganese chloride or magnesium chloride. These ions each induced significant increases in the secondary structure con-tents of TP1-192and RT-RH303-778(Fig. 5AandB). The
magni-tudes of these changes were basically identical when manganese
chloride or magnesium chloride was added. These data show re-combinant hPOL domains have secondary structure composi-tions in line with sequence-based prediccomposi-tions and adopt 3D con-formations where metal-binding residues can coordinate the metal ions needed for hPOL activity.
Characterization of the oligomeric states of recombinant TP1–192and RT-RH303–778.Current mechanistic models of hPOL/
dPOL function represent POL as a single polypeptide that medi-ates different functions by adopting different conformations (8). However, to the best of our knowledge, there are no reports dem-onstrating that hPOL and dPOL are monomeric. Thus, we used SEC-MALS to characterize the solution properties of TP1–192and
RT-RH303–778.
In order to determine the oligomeric state of TP1–192, we
per-formed a conjugate analysis, an approach more commonly em-ployed in SEC-MALS studies of membrane proteins embedded within detergent micelles (70). Conjugate SEC-MALS analyses are very powerful, since they allow the absolute determination of the molar mass of a complex, and also its component species, provid-ing one knows their respective molar extinction coefficients and
FIG 4Purification of recombinant TP1–192and RT-RH303–778. (A) Silver-stained SDS-PAGE gels from the purification procedure for TP1–192. Purified inclusion bodies (lane 1) were solubilized in 6 M guanidine-HCl and loaded onto a metal affinity column. The resultant flowthrough (lane 2), wash (lane 3), nonspecific wash (lane 4), and elution fractions (lanes 5 and 6) are shown. The arrow indicates pure protein used forin vitrorefolding. The masses (in kDa) of molecular size markers (lane M) are indicated on the left. (B) Coo-massie blue-stained SDS-PAGE gel showing purification of recombinant RT-RH303–778. Lane 1, purified inclusion bodies containing RT-RH303–778; lane 2, flowthrough from Ni-affinity column; lane 3, wash of Ni-affinity column; lane M, protein marker; lanes 4 to 8, elution fractions from Ni-affinity column; lane 9, pool of purified and refolded RT-RH303–778.
FIG 5Far-UV CD spectroscopy of recombinant TP1–192and RT-RH303–778. (A) The far-UV CD spectrum of apo-TP1–192was consistent with it being largely␣-helical and having smaller amounts of-sheet and coil-like struc-tures (Table 1). Magnesium chloride and manganese chloride induced very similar increases in the secondary structure of TP1–192. (B) The far-UV CD spectrum of apo-RT-RH303–778was consistent with it being mainly␣-helical but having a smaller amount of-sheet structure (as per sequence-based structure predictions [Table 1]). Magnesium chloride and manganese chloride induced identical increases in the secondary structure of RT-RH303–778. All spectra were acquired at 20°C using a 0.15-mg/ml protein solution (equivalent to⬃6M TP1–192and⬃2.7M RT-RH303–778) with divalent metal ions at a final concentration of 2.5 mM (described in Materials and Methods).
Vörös et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.318.524.62.372.2] [image:7.585.59.267.68.303.2]specific refractive index increment (dn/dc, the incremental change in refractive index,n, as a function ofc, the macromolecular con-centration). We determined the dn/dc value for NV-10 to be 0.1368 ml · g⫺1(described in Materials and Methods), which is in
close accord with values of⬃0.14 typically reported for other car-bohydrate polymers (71). Similarly, we determined the molar ex-tinction coefficient of NV-10 to be 163 M⫺1· cm⫺1(when cor-rected for contributions from light scattering).
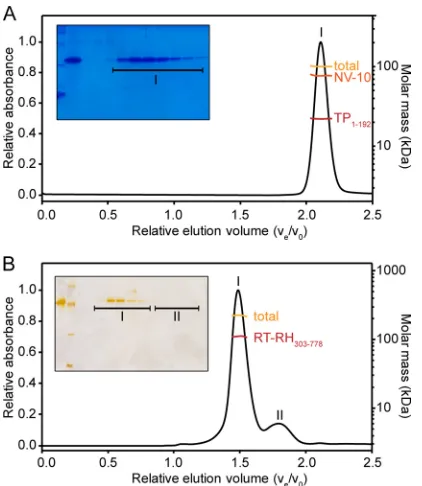
TP1–192 solubilized in NV-10-containing buffers eluted as a
single, monodisperse peak in SEC-MALS experiments that SDS-PAGE analysis revealed was pure TP1–192(Fig. 6). The apparent
molar mass of the complex formed between NV-10 and TP1–192
was⬃100 kDa, of which the protein component had a molar mass of⬃22.4 kDa. These data provide strong support for sole species populated in solution being a TP1–192monomer (theoretical mass
of⬃24.6 kDa) in complex with 17 molecules of NV-10 (⬃4.5 kDa/molecule).
We also employed IMS-MS to characterize the oligomeric state of TP1–192. IMS-MS is a gas-phase technique that allows different
polypeptide conformers (and complexes thereof) to be resolved (72, 73), providing they have significantly different mobility through the ion mobility drift cell (arising from their distinct ori-entationally averaged collision cross-sections [74]). We found that signals arising from NV-10 dominated the mass spectra in IMS-MS experiments for TP1–192in NV-10-containing volatile
buffers (Fig. 7A), consistent with the amphipol being present in significant molar excess to protein. High energies were needed to dissociate NV-10 from TP1–192, whereupon we observed a
charge-state distribution consistent with a TP1–192monomer (Fig. 7Aand B). These data support TP1–192being a monomer when interacting
with the NV-10 molecules needed for its solubility.
Recombinant RT-RH303–778 reproducibly yielded two large
elution peaks in SEC-MALS experiments (Fig. 6B, peaks I and II).
SDS-PAGE analysis showed that RT-RH303–778was present only in
elution peak I (Fig. 6B, inset), and no protein was detected in peak II. Control experiments, where only NV-10 was injected onto the column, showed that peak II corresponded to NV-10 oligomers that form in solution at the high concentrations employed, as reported for other amphipols (75).
Conjugate analysis of SEC-MALS experiments on RT-RH303–778showed that peak I had a total molar mass of⬃221 kDa,
of which 111 kDa came from the protein component and the remaining mass arose from bound NV-10 (Fig. 6B). These data show clearly that RT-RH303–778, with a theoretical mass of⬃55.7
kDa, formed dimers in solution, wherein each monomer was dec-orated with 12 NV-10 molecules. Independent experiments showed that RT-RH303–778was strictly monodisperse (Mw/Mn⫽
1.000), even at⬃10 mg/ml, and we did not observe significantly populated monomers or higher-order oligomers (data not shown). As RT-RH303–778was not detectable in IMS-MS
experi-ments, this approach could not be used for RT-RH303–778.
Epsilon-independent interactions between recombinant hPOL domains.We used microscale thermophoresis (MST) and isothermal titration calorimetry (ITC) to establish whether TP1–192and RT-RH303–778interactin vitroin the absence of other
[image:8.585.43.546.79.284.2]viral or host proteins. MST measures the intrinsic property of biomolecules to align along a laser-induced temperature gradient of only a few degrees Celsius. This property (thermophoresis) de-pends upon the size, shape, and charge of molecules, making MST exquisitely sensitive to structural changes and complex formation (76). Thus, binding affinities can be obtained by measuring changes in the thermophoresis of a target molecule as a function of increased ligand concentration. ITC measures the heat evolved or taken up when one molecule binds another and can yield accurate measurements of binding affinity and sometimes binding en-thalpy and stoichiometry (77).
TABLE 1Secondary structure compositions of TP1–192and RT-RH303–778, predicted using spectral deconvolution and sequence-based algorithms
Method and algorithm
Predicted secondary structure content (%) for hPOL constructs
TP1–192 RT-RH303–778
␣-Helical -Strand Coil ␣-Helical -Strand Coil Deconvolution algorithmsa
SELCON3 29 19 50 41 13 47
CDSSTR 36 15 49 47 10 43
CONTINLL 30 18 52 42 16 43
K2D 29 13 58 35 17 47
Mean⫾SEMb 31⫾2 16⫾2 52⫾2 41⫾3 14⫾2 45⫾1
Sequence-based algorithmsa
GOR4 31 14 55 15 22 64
HNNC 28 10 62 37 16 47
PHD 30 11 59 35 17 48
Predator 29 6 65 24 15 62
Simpa96 38 6 56 28 17 55
SOPM 33 14 53 35 18 47
Sec. cons.c 38 8 52 30 16 51
Mean⫾SEMd 32⫾2 10⫾1 57⫾2 29⫾3 17⫾1 53⫾3 a
We used a range of different spectral deconvolution and secondary structure prediction algorithms to avoid bias and ensure that the comparison of predictions was less subjective.
bMean values (⫾standard errors of the means) for the secondary structures of TP1–192and RT-RH303–778, predicted using deconvolution of far-UV CD spectra (and a reference
data set from 43 soluble proteins [66,88]).
cSec. cons. refers to the secondary structure content of the consensus sequence generated using the prediction algorithms used. d
Average values (⫾standard errors of the means) for the secondary structures of TP1–192and RT-RH303–778, predicted from their sequences by use of various secondary structure prediction algorithms (57).
on November 7, 2019 by guest
http://jvi.asm.org/
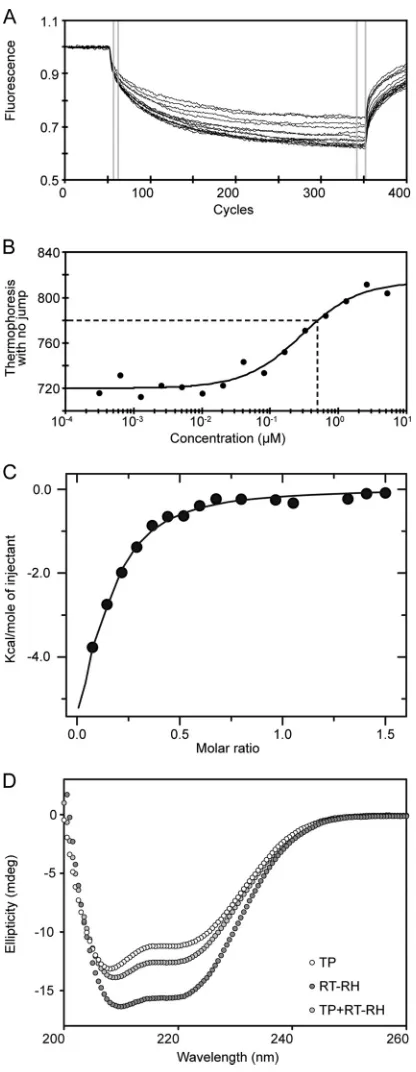
We performed MST experiments in which TP1–192was titrated
against RT-RH303–778, followed by the reverse experiment, in
which RT-RH303–778was titrated against TP1–192. In each case, we
saw clear evidence for interactions between these domains (with apparentKdvalues in the range of 0.5 to 3M (Fig. 8AandB). In
subsequent ITC experiments, we titrated the more soluble hPOL construct, TP1–192, into the calorimeter cell that contained
RT-RH303–778at low protein concentrations. TP1–192bound to
RT-RH303–778(Fig. 8C) with an apparentKdof⬃5M (in the same
affinity range as we determined via MST [Fig. 8B]). These data show recombinant TP1–192and RT-RH303–778can form a complex
in vitroin the absence of other viral or host-derived proteins, consistent with results from independent far-UV CD spectros-copy experiments (Fig. 8D).
In vitroreconstitution of an active ribonucleoprotein com-plex. To further validate our recombinant TP1–192 and
RT-RH303–778 constructs, we used them in in vitro reconstitution
assays reported to support dPOL- or hPOL-mediated protein-priming activity (17, 36, 37, 40,41, 46). Recombinant human Hsp90, Hsc70, Hop, and Hsp40 were overexpressed inE. coliand
isolated in-house to high purity (Fig. 9A).In vitrotranscription was used to generate recombinant epsilon RNA (see Materials and Methods).
We used negative ion mass spectrometry to verify the integrity of our epsilon RNA preparations (Fig. 9B). This analysis showed that the predominant RNA ions had an apparent molecular mass of 55,361 Da, lower than the mass we expected for the designed 174-mer epsilon transcript (theoretical mass of 56,231 Da). The measured mass of epsilon, however, was consistent with epsilon being a 172-mer. A second group of lower-intensity RNA ions (consistent with an additional mass of 322 Da) corresponded to epsilon with an additional nucleotide (i.e., a 173-mer [Fig. 9B]). Electrospray ionization-mass spectrometry under native-like con-ditions showed fewer charge states (12⫺, 13⫺, and 14⫺ions) and indicated a folded RNA, corresponding to an epsilon monomer (data not shown).
Recombinant TP1–192,RT-RH303–778, human chaperones, and
epsilon RNA were mixed in the presence of an ATP-regenerating system (to support chaperone nucleotide requirements) (see Ma-terials and Methods). This assay was supplemented with either [␣-32P]dGTP or [␣-32P]dTTP to probe incorporation of
radiola-beled dGMP or dTMP, respectively, into hPOL domains, as
ex-FIG 6SEC-MALS analysis of recombinant TP1–192and RT-RH303–778. (A) A 10-l aliquot of a 300M sample of TP1–192was injected onto a 24-ml ana-lytical Superose 6 column. The UV absorbance at 280 nm was plotted against relative elution volume (Ve/V0). The TP1–192preparation contained only a single elution peak, corresponding to TP1–192extensively decorated with NV-10 molecules. Conjugate analysis (see Materials and Methods) showed that the total mass of this complex was⬃100 kDa (gold line), but the complex contained only TP1–192monomers (⬃25 kDa; red line) bound to many NV-10 molecules (orange line). (Inset) SDS-PAGE showed that the elution peak con-tained pure TP1–192. (B) A 10-l aliquot of a 165M sample of RT-RH303–778 was injected onto a 24-ml analytical Superdex 200 column. UV absorbance at 280 nm was plotted against relative elution volume. Only two major elution peaks were observed for recombinant RT-RH303–778(peaks I and II). Peak I contained a complex with a molar mass of⬃221 kDa (gold line), which con-tained only dimeric RT-RH303–778(red line). For clarity, the molar mass of the NV-10 component was omitted here, as it would obscure the results for RT-RH303–778. (Inset) Only peak I contained protein. Peak II contained NV-10 oligomers.
FIG 7Ion mobility spectrometry-mass spectrometry analysis of TP1–192. (A) Three-dimensional IMS-MS DriftScope plot of 20M TP1–192in 100 mM ammonium acetate buffer (pH 5.5; containing NV-10 at an⬃20-fold molar excess over TP1–192). The mass-to-charge ratio (m/z) is shown on theyaxis, and the drift time (corresponding to the transit time through the IMS drift cell) is shown on thexaxis. The relative ion intensity is shown by the colors: yellow and black represent the most and least intense ions, respectively. The mass spectrum comprises mainly intense signals arising from the amphipol (circled in red). Careful examination of the DriftScope plot, however, shows a band of signals arising from TP1–192that have the bow shape characteristic of a protein charge-state distribution (circled in white). Very similar IMS-MS data were obtained using 100 mM ammonium bicarbonate buffer (pH 7.8). (B) The signals giving rise to the TP1–192charge-state distribution (shown in panel A) were extracted by using DriftScope (the manufacturer’s software; Waters UK Ltd.) and plotted as a separate mass spectrum. The TP1–192charge-state distri-bution encompassed the 14⫹through 19⫹ions and was consistent with a polypeptide with a mass of 24,655 Da (which agreed well with the theoretical monomer mass of 24,645 Da). These data show that TP1–192is monomeric and forms a complex with NV-10 molecules.
Vörös et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.317.525.63.281.2] [image:9.585.60.271.65.308.2]pected forin vitroprotein priming (17,46). Autoradiography of SDS-PAGE gels was used to analyze protein-priming reactions and showed protein incorporation of [␣-32P]dTTP (Fig. 9C). The
extent of labeling was significantly higher with [␣-32
P]dGTP than [␣-32P]dTTP (Fig. 9C), consistent with the nucleotide preferences
reported for hPOL priming (31). We next varied the relative ratios of manganese chloride or magnesium chloride concentrations in assay mixtures (from 1:6 up to 8:1) (Fig. 9C), as this has been shown to favor protein priming when manganese chloride is in molar excess (31). Increasing the relative concentration of man-ganese chloride to magnesium chloride significantly increased protein labeling in priming assays (Fig. 9C).
Protein size markers revealed that the protein labeled in our initial priming assays was consistent with dimeric RT-RH303–778,
not TP1–192, as expected for authentic protein priming. This
sug-gests that RT-RH303–778can label itself, rather than TP1–192, under
the conditions of our initial assays. dPOL-mediated “cryptic priming” has been reported, wherein [␣-32
P]dGMP/dTMP is co-valently attached to tyrosine residues of RT (42,43). Consistent with this hypothesis, removing TP1–192from ourin vitropriming
reactions still yielded radiolabeling of RT-RH303–778 (Fig. 9C).
Thus, while RT-RH303–778clearly hadin vitroprotein-priming
ac-tivity, this appeared to be cryptic priming.
Since SEC-MALS analysis showed that RT-RH303–778 forms
stable dimers, we hypothesized that cryptic priming of
RT-RH303–778arises from the close proximity of one RT-RH303–778
monomer to the active site of the other monomer, thus effectively outcompeting TP1–192in priming reactions. To test this
hypothe-sis, we performed furtherin vitropriming assays wherein the mo-lar ratio of TP1–192to RT-RH303–778was varied (from 1:1 to 10:1).
Using this approach, we found that cryptic priming of
RT-RH303–778 prevailed when RT-RH303–778 was equimolar with
TP1–192, but authentic priming on TP1–192became prevalent when
TP1–192was in excess (Fig. 9D). We also observed labeling of
di-meric and monodi-meric RT-RH303–778, even though denaturing
SDS-PAGE was used. It is plausible that cryptic priming labels a dimeric RT-RH303–778 conformer that is refractory to complete
denaturation, consistent with the high-molecular-mass species observed in our earlier experiments (Fig. 9C).
We next performed further experiments to ensure that the ob-served activities were substrate specific and not due to low-abun-dance, contaminating enzymes present in our protein prepara-tions. Removing TP1–192 from our assay mixtures caused the
reappearance of the high-molecular-mass RT-RH303–778species
formed by cryptic priming (Fig. 9E). Removing epsilon from the priming assay mixture still yielded labeled TP1–192. It is not clear
whether this is due to free nucleotides bound to TP1–192being able
to prime protein labeling or copurification of the epsilon RNA that was part of the noncoding region of our RT-RH constructs (see Materials and Methods). However, mock RNA, of a similar size to epsilon, reduced labeling of TP1–192in vitro(Fig. 9E). This
observation is consistent with reports of POL being able to bind noncognate RNA substrates (which then reduce its activity in FIG 8Binding studies of TP1–192and RT-RH303–778. (A) Normalized
fluores-cence data for the raw MST signal acquired for each titration point. Initial fluorescence was recorded for 5 s before the establishment of a temperature gradient (of⬃6°C) for 30 s. The differences in normalized fluorescence inten-sities in the selected data range (gray bars) were used to determine the binding curve shown in panel B (76). (B) MST binding curve acquired by titrating nonlabeled RT-RH303–778into labeled TP1–192. A 1:2 dilution series was ap-plied, with the highest RT-RH303–778concentration being 5.2M, whereas the concentration of labeled TP1–192was kept at 40 nM. The fitted MST data yielded apparentKdvalues in the range of 0.5 to 3M (dashed lines indicate
theKdfrom the titration). (C) ITC binding curve for the interaction between
TP1–192and RT-RH303–778at 20°C. A stock of⬃100M TP1–192was titrated against a stock of⬃20M RT-RH303–778in the calorimeter cell. The apparent
Kdof this interaction (⬃5M) agreed well with that determined using MST
(panel B). (D) Far-UV CD spectra of recombinant TP1–192(white circles, ⬃0.075 mg/ml), RT-RH303–778(dark grey circles,⬃0.075 mg/ml), and a 2:1 molar ratio of TP1–192and RT-RH303–778(light grey circles,⬃0.15 mg/ml total protein concentration).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.60.268.70.606.2]vitro) (78). Control priming reaction mixtures, in which only the recombinant chaperones and the ATP regeneration system were present, yielded no labeled proteins, demonstrating that the activ-ity we saw was not due to a contaminatingE. colienzyme.
Having successfully demonstrated both authentic and cryptic in vitropriming activities for our recombinant hPOL constructs, we checked whether Calcomine orange 2RS, a molecule reported to inhibit dPOL activityin vitroand in cellular viral replication assays (79), had any effect onin vitropriming activity. Reassur-ingly, this reagent completely inhibitedin vitropriming activity (Fig. 9E). In summary, our recombinant hPOL constructs specif-ically incorporated dGMP/dTMP via cryptic and authentic prim-ing. Thesein vitroenzymatic activities faithfully recapitulated the metal-dependent priming activity reported for dPOL and hPOL (expressed in mammalian cells) and were ablated by a reported
POL inhibitor (79). To the best of our knowledge, this is the first time this has been achieved using recombinant hPOL and human chaperone proteins expressed solely inE. coli.
DISCUSSION
Independent hPOL domains facilitate high-level protein ex-pression inE. coli.A few laboratories have successfully produced recombinant full-length dPOL, hPOL, or POL “miniproteins” wherein the functionally dispensable spacer region has been re-moved (13,16,17,26,27,36,37). To date, these strategies have not yielded advances in structural biology or biophysical studies of POL. To address this, we used structural prediction algorithms and hydrophobic cluster analysis to design independently folding hPOL domains (Fig. 2AandB). Our protein design and expres-sion strategies were successful and allowed us to express these
FIG 9In vitroprotein-priming activity of recombinant TP1–192and RT-RH303–778. (A) SDS-PAGE analysis of hPOL constructs and chaperones used in functional assays. Protein identities and the molecular mass (in kDa) of size markers (lane M) are indicated. (B) ESI-MS result for a 40M solution of epsilon RNA in a 50 mM piperidine-imidazole solution. This (denatured) mass spectrum showed that the predominant RNA ions had a molecular mass of 55,361 Da, consistent with being an epsilon 172-mer rather than the expected 174-mer (theoretical mass of 56,235 Da). Less-prominent ions were also observed, with an additional mass of 322 Da, consistent with an epsilon 173-mer also being present. (C, upper gel) Results ofin vitropriming using [␣-32P]dTTP in the presence of increasing manganese chloride concentrations and a fixed magnesium chloride concentration of 6 mM (thus, the Mn/Mg ratio varied from 1:6 to 8.3:1). SDS-PAGE and autoradiography showed incorporation of radiolabeled dTMP, consistent with protein priming, into a protein with a mass of⬃100 kDa. (Lower gel) Results ofin vitropriming followed by urea-PAGE and autoradiography. The effects of adding [␣-32P]dGTP versus [␣-32P]dTTP and increasing manganese concentrations were compared, with the magnesium concentration held at 5 mM (thus, the Mn/Mg ratio varied from 1:5 through 4:1). Arrows indicate labeled proteins. Removing TP did not reduce protein labeling, consistent with cryptic priming by RT (42,
43). (D)In vitropriming in the presence of [␣-32P]dGTP and with molar ratios of TP
1–192:RT-RH303–778that ranged from 1:1 to 10:1. SDS-PAGE and autoradiography showed incorporation of radiolabeled dGMP into three species: TP1–192 (mass of⬃25 kDa), as well as monomeric and dimeric RT-RH303–778(molecular masses of⬃55 and⬃100 kDa, respectively). The signal intensity for the authentic priming reaction (i.e., labeling of TP1–192) was significantly increased at higher molar ratios of TP1–192:RT-RH303–778. This is consistent with authentic priming outcompeting the cryptic priming when TP1–192is present in molar excess. Protein identities and molecular mass markers are indicated. (E) Specificity ofin vitropriming reactions. [␣
-32P]dGTP was used as the radioactive substrate, with products resolved using SDS-PAGE and labeled species detected using autoradiography. Shown is a comparison of a full reaction mixture (all) versus control reaction mixtures that lacked TP, lacked epsilon, lacked TP and epsilon, or included a similar-sized, unrelated RNA instead of epsilon (mock). These data show that the product of the authentic priming reaction was strictly TP dependent but was not strictly epsilon dependent (46). No priming product was observed for the reaction mixture containing only recombinant human chaperones and the ATP regeneration systems (Hsps), demonstrating that the labeling reaction was dependent on the presence of hPOL activities. Similarly, priming activity was ablated when we used Calcomine orange RS (⫹CO), a known inhibitor of hPOL activity (79). The molecular masses are indicated.
Vörös et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.42.539.65.342.2]constructs inE. colito high levels (e.g.,⬃40% of the cellular pro-tein for RT-RH303–778[Fig. 3C]).
By using the protocols we developed, academic and commer-cial researchers can now rapidly obtain high-level expression of recombinant hPOL domains inE. coli. However, despite exami-nation of a comprehensive range of expression conditions, vec-tors, and fusion proteins, we had great difficulty in directly ex-pressing TP1–192or RT-RH constructs in a soluble form. All hPOL
constructs we examined accumulated as insoluble inclusion bod-ies inE. coli, suggesting this is an inherent feature of our hPOL constructs.
We noted some interesting properties of RT-RH constructs that appeared to correlate with protein expression levels: (i) inclu-sion of the “full-length” RH domain caused RT-RH expresinclu-sion to be poor and irreproducible (in marked contrast to constructs with truncated C termini). This suggests the C terminus compromises hPOL expression, perhaps due to it being unstructured and aggre-gation prone, as per our predictions. Alternatively, exogenous RNase H activity may be toxic toE. coli. (ii) Extending the N terminus of RT to include an extra predicted helical element (i.e., beginning at residue 303) significantly improved the yield and reproducibility of protein expression. However, further extension at the N terminus (i.e., beginning at residue 283) significantly reduced expression. (iii) Expressing RT-RH as a concatemer was key to high-level expression inE. coli, consistent with our hypoth-esis that RT-RH forms a contiguous structural unit. Despite ex-tensive efforts, we have been unable to express RH as an indepen-dent domain, or to reproduce others’ reports of this (62,65,80), despite using the sameE. colistrains, expression vectors, and do-main boundaries (unpublished data). The reasons for this are cur-rently unclear but may be due to procedural differences between laboratories.
Amphipols as a tool for studying challenging proteinsin vitro.We used chemical denaturants to solubilize inclusion bodies and keep aggregation-prone hPOL domains denatured and solu-ble during initial protein purification steps (Fig. 4). However, the only way we were able to keep TP1–192and RT-RH303–778soluble
during subsequentin vitrorefolding steps was to have NV-10 (an amphipathic carbohydrate polymer) present at a minimum of a 10-fold (wt/wt) excess relative to hPOL domains. While am-phipols are typically used to solubilize membrane proteins (64), we demonstrated that they efficiently solubilized recombinant hPOL constructs (Fig. 6). Employing the procedures we have de-veloped, a single researcher can rapidly purify soluble TP1–192and
RT-RH303–778on a scale of hundreds of milligrams. These
con-structs are amenable toin vitrostudy over a wide pH range and at concentrations appropriate to most biophysical and structural studies (Fig. 5to8).
Our SEC-MALS analyses (Fig. 6) showed that TP1–192and
RT-RH303–778, even when refolded, were extensively decorated with
amphipols. These data are consistent with hPOL domains having multiple solvent-exposed hydrophobic patches to which the am-phipols bind (and therefore help to solubilize these proteins). These sticky patches most likely contribute to the challenges in expressing hPOL constructs inE. coliand their accumulation as inclusion bodies. We speculate that these hydrophobic patches may overlap with the binding sites of host chaperones (Fig. 10). If true, this might explain why low levels of soluble hPOL can be expressed in mammalian cells (where host chaperones are pres-ent). Consistent with this hypothesis, hPOL expressed in
mamma-lian cells appears to copurify with host chaperones (17). High-level expression of hPOL constructs may stress the E. coli chaperone machinery unless, as shown here, chaperone expres-sion is upregulated by a heat shock prior to expresexpres-sion (Fig. 3C).
Structural characterization of recombinant hPOL domains.
Far-UV CD spectroscopy showed that TP1–192and RT-RH303–778
were largely␣-helical but had small amounts of-sheet structure (Fig. 5). Spectral deconvolution algorithms showed that the mea-sured secondary structure compositions of TP1–192 and
RT-RH303–778were broadly consistent with the structure predicted
from their polypeptide sequences (Table 1). While these data sug-gested our experimental procedures yielded correctly folded hPOL domains, it was possible that TP1–192 and RT-RH303–778
contained a similar secondary structure but different tertiary structures compared tobona fide, correctly folded hPOL domains. However, the observations that recombinant TP1–192 and
RT-RH303–778both bound manganese chloride and magnesium
chlo-ride, divalent ions needed forin vitroactivity (Fig. 5AandB), that they interactedin vitro(Fig. 8), and that they had priming activi-ties (Fig. 9) suggested our hPOL constructs were correctly folded. Manganese chloride and magnesium chloride induced signifi-cant changes in the far-UV CD spectra of TP1–192 and
RT-RH303–778compared to the respective apo-proteins (Fig. 5Aand B), consistent with metal binding inducing structural rearrange-ments. Since each metal induced very similar changes in the far-UV CD spectra, these compounds may compete for binding to the same sites on TP1–192 or RT-RH303–778. Alternatively, these
metals may bind to distinct sites that, by coincidence, invoke sim-ilar spectral changes. Higher-resolution experiments are needed to distinguish between these possibilities.
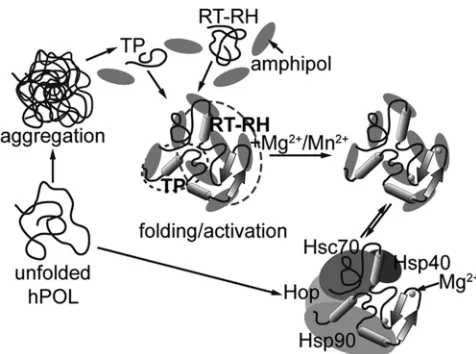
SEC-MALS and IMS-MS showed that TP1–192 exists as a
FIG 10Schematic ofin vitroreconstitution of functional hPOL complexes. Unfolded hPOL has an extreme tendency to aggregate and form inclusion bodies. This aggregation can be attenuated in the presence of host chaperones that bind to and presumably stabilize hPOL in a conformation that can then be activated (lower pathway). The strategy we employed here entailed expression of independent TP1–192and RT-RH303–778constructs and NV-10 (shaded el-lipses) to ameliorate aggregation (upper pathway). hPOL domains thus treated were structured, could interact with each other, bound metals, and hadin vitro
activities in the presence of recombinant host chaperones (middle pathway). It is possible that amphipols and host chaperones have the same or overlapping binding motifs. The secondary structure and their partitioning into TP and RT-RH domains shown here are purely schematic.