Type I Interferons and NK Cells Restrict Gammaherpesvirus Lymph
Node Infection
Clara Lawler,aCindy S. E. Tan,aJ. Pedro Simas,b Philip G. Stevensona
School of Chemistry and Molecular Biosciences, University of Queensland and Royal Children’s Hospital, Brisbane, Australiaa; Instituto de Medicina Molecular e Instituto de Microbiologia, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugalb
ABSTRACT
Gammaherpesviruses establish persistent, systemic infections and cause cancers. Murid herpesvirus 4 (MuHV-4) provides a
unique window into the early events of host colonization. It spreads via lymph nodes. While dendritic cells (DC) pass MuHV-4
to lymph node B cells, subcapsular sinus macrophages (SSM), which capture virions from the afferent lymph, restrict its spread.
Understanding how this restriction works offers potential clues to a more comprehensive defense. Type I interferon (IFN-I)
blocked SSM lytic infection and reduced lytic cycle-independent viral reporter gene expression. Plasmacytoid DC were not
re-quired, but neither were SSM the only source of IFN-I, as IFN-I blockade increased infection in both intact and SSM-depleted
mice. NK cells restricted lytic SSM infection independently of IFN-I, and SSM-derived virions spread to the spleen only when
both IFN-I responses and NK cells were lacking. Thus, multiple innate defenses allowed SSM to adsorb virions from the afferent
lymph with relative impunity. Enhancing IFN-I and NK cell recruitment could potentially also restrict DC infection and thus
improve infection control.
IMPORTANCE
Human gammaherpesviruses cause cancers by infecting B cells. However, vaccines designed to block virus binding to B cells
have not stopped infection. Using a related gammaherpesvirus of mice, we have shown that B cells are infected not via cell-free
virus but via infected myeloid cells. This suggests a different strategy to stop B cell infection: stop virus production by myeloid
cells. Not all myeloid infection is productive. We show that subcapsular sinus macrophages, which do not pass infection to B
cells, restrict gammaherpesvirus production by recruiting type I interferons and natural killer cells. Therefore, a vaccine that
speeds the recruitment of these defenses might stop B cell infection.
E
pstein-Barr virus (EBV) and Kaposi’s sarcoma-associated
her-pesvirus (KSHV) persist in B cells and cause cancers (
1
).
Re-ducing their B cell infections is therefore an important therapeutic
goal. Limited viral gene expression (
2
) makes established
infec-tions difficult to clear. The early events of host colonization may
provide better targets. However, control mechanisms must be
de-fined
in vivo
: inferring mechanisms from
in vitro
studies has
proven problematic because immune function and its evasion are
context dependent. Thus, EBV gp350-specific antibodies block B
cell infection, and CD8
⫹T cells kill infected B cells
in vitro
, but
vaccinations to induce these effectors have not reduced infection
rates (
3
).
The early events of human infections are difficult to analyze
because they predate clinical presentation (
4
). However,
gam-maherpesviruses long predate human speciation (
5
), and peak
viral diversity in genes that interact with host-diverse functions
suggests that viral coevolution has since acted to counter host
divergence. Therefore, human and other mammalian
gamma-herpesviruses should colonize their hosts in similar ways.
Mu-rid herpesvirus 4 (MuHV-4) realistically infects laboratory
mice (
6
) and so can experimentally reveal events of likely
rele-vance to EBV and KSHV. Eighty percent to 90% of its genes
have clear homologs in EBV and KSHV (
7
), and even where
there is genetic diversity, for example, in CD8
⫹T cell evasion,
function appears to be conserved.
EBV is hypothesized to enter new hosts by infecting B cells.
However, naive B cells rarely meet environmental antigens
di-rectly, with their default response to antigen alone being apoptosis
(
8
); rather, they meet antigens presented on myeloid cells in
lymph nodes (LN) (
9
). MuHV-4 host colonization conforms to
this paradigm, with infection first reaching B cells in LN via
den-dritic cells (DC) (
10
); submucosal lymphoid tissue is colonized
later (
11
). MuHV-4 exploits myeloid/lymphoid cell contact for
spread (
12
), making B cell infection difficult to block directly.
However, blocking myeloid infection could potentially restrict B
cell infection indirectly. Viral exploitation of endocytic scavenging
pathways (
13
,
14
) makes myeloid cell entry difficult to block, but
virus production by myeloid cells might be susceptible. Of note,
not all myeloid infection is productive: subcapsular sinus
macro-phages (SSM) communicate with B cells (
15
) and are infected by
MuHV-4 yet restrict its spread (
16
). To reveal mechanisms
capa-ble of
in vivo
infection control, we sought to understand how SSM
restrict MuHV-4 replication.
SSM are specialized sessile macrophages that filter the lymph;
splenic marginal zone (MZ) macrophages (MZM) analogously
filter the blood (
17
). Slow percolation of the lymph and blood past
Received6 June 2016Accepted22 July 2016 Accepted manuscript posted online27 July 2016
CitationLawler C, Tan CSE, Simas JP, Stevenson PG. 2016. Type I interferons and NK cells restrict gammaherpesvirus lymph node infection. J Virol 90:9046 –9057.
doi:10.1128/JVI.01108-16.
Editor:R. M. Longnecker, Northwestern University
Address correspondence to Philip G. Stevenson, [email protected]. Copyright © 2016, American Society for Microbiology. All Rights Reserved.
on November 7, 2019 by guest
http://jvi.asm.org/
their filtering macrophages promotes pathogen adsorption. A
po-tential hazard is that adsorbed pathogens then replicate in the
filtering macrophages. Host defense against this has been studied
by inoculating murine footpads (intrafootpad [i.f.] inoculation)
with vesicular stomatitis virus (VSV): SSM infection is productive,
but the resulting type I interferon (IFN-I) response protects
pe-ripheral nerves and prevents disease (
18
). SSM susceptibility yet
neuronal protection suggests that SSM respond weakly to IFN-I,
and weak MZM IFN-I responses are associated with enhanced
immune priming (
19
). IFN-I responses to vaccinia virus Ankara
also recruit NK cells, although the antiviral efficacy of this
re-sponse was not shown (
20
).
Extrapolating such results to natural infections is not
necessar-ily straightforward, as most viruses engage in host-specific IFN-I
evasion (
21
). VSV normally infects cows rather than mice,
vac-cinia virus is not mouse adapted, and the Ankara strain has lost
many immune evasion genes. In contrast, MuHV-4 evasion
ap-pears to be fully functional in laboratory mice (
6
). Natural
MuHV-4 entry is probably via the upper respiratory tract (
22
), but
i.f. infection is also productive (
16
) and allows comparison with
data from other SSM studies. Both intranasal (i.n.) and i.f.
inocu-lations lead to SSM infection that inhibits acute viral spread (
16
).
MuHV-4 evades IFN-I by targeting interferon regulatory
fac-tor 3 (IRF3) (
23
), TBK-1 (
24
), the IFN-I receptor (IFNAR) (
25
),
STAT-1/2 (
26
), as well as other pathways (
27
) and associated
de-fenses such as apoptosis/autophagy (
28
), NF-
B (
29
), and PML
(
30
,
31
). Nonetheless, disease in IFNAR-deficient mice (
32
,
33
)
indicates IFN-I-dependent restraint. IFN-I reduces MuHV-4
re-activation from latency in B cells (
34
), but heightened reactivation
normally attenuates infection (
35
), and the acute phenotypes of
IFNAR deficiency are more suggestive of increased lytic
replica-tion before B cell colonizareplica-tion. In the spleen, IFN-I restricts
mainly macrophage infection (
36
). Here we show that IFN-I and
NK cells are key components of the SSM barrier to MuHV-4
spread.
MATERIALS AND METHODS
Mice and immune depletions.C57BL/6J, LysM-cre (37), and CD11c-cre (38) mice were infected at 6 to 12 weeks of age. Experiments were ap-proved by the University of Queensland Animal Ethics Committee in accordance with Australian National Health and Medical Research Coun-cil guidelines. Virus was given i.f. in 50l (105PFU) under isoflurane
anesthesia. Phagocytic cells were depleted by i.f. administration of 50l clodronate-loaded liposomes (39) 3 and 5 days before infection, which was confirmed by CD169 loss around the subcapsular sinus (16). NK cells were depleted by intraperitoneal (i.p.) administration of 200g mono-clonal antibody (MAb) PK136 (anti-NK1.1; Bio-X-Cell) 1 and 3 days before infection and every 2 days thereafter. Plasmacytoid DC (pDC) were depleted by i.p. injection of 400g MAb BX444 (anti-CD317; Bio-X-Cell) 1 and 3 days before infection and every 2 days thereafter. IFN-I signaling was blocked by i.p. injection of 200g MAb MAR1-5A3 (anti-IFNAR; Bio-X-Cell) 1 day before infection and every 2 days thereafter. Experi-mental groups were compared statistically by Student’s two-tailed un-pairedttest.
Cells and viruses.BHK-21 cells and 3T3-50 cells, which express doxy-cycline-inducible MuHV-4 (MHV) open reading frame 50 (ORF50) (40), were grown in Dulbecco’s modified Eagle’s medium with 2 mM glu-tamine, 100 IU/ml penicillin, 100g/ml streptomycin, and 10% fetal calf serum (complete medium). MHV-green fluorescent protein (GFP) (41) expresses GFP from an EF1␣ promoter between ORFs 57 and 58. MHV-RG (11) has a viral M3 promoter driving LoxP-flanked mCherry upstream of GFP between ORFs 57 and 58. Cre switches its fluorochrome
expression irreversibly from mCherry to GFP (MHV-G). Neither MHV-RG nor MHV-G is attenuated in C57BL/6 mice, and neither virus outgrows the other in a mixed infection (36). MHV-M50 has 416 bp of the murine cytomegalovirus (MCMV) IE1 promoter in the 5=untranslated region of ORF50 exon 1. This deregulates the lytic switch, essentially abol-ishing viral latency (35). ORF50-negative (ORF50⫺) MuHV-4 has a 1,715-bp deletion in ORF50 exon 2, precluding lytic gene expression with-out complementation (40). The virus was grown and titers were deter-mined in 3T3-50 cells with doxycycline (1g/ml). Other viruses were grown and titers were determined in BHK-21 cells (42).
Infectivity assays.Infectious virus was quantified by a plaque assay (42). Virus dilutions were incubated with BHK-21 cells (2 h at 37°C), overlaid with complete medium– 0.3% carboxymethylcellulose, cultured for 4 days, fixed (1% formaldehyde), and stained (0.1% toluidine blue) for plaque counting. Total virus (latent plus infectious) was quantified by an infectious center (IC) assay (42). Freshly isolated LN or spleen cells were layered onto BHK-21 cell monolayers and cultured as described above for plaque assays. To measure Cre-dependent viral fluorochrome switching, plaque or IC assays were performed at limiting dilution in 96-well plates (16 wells per dilution). After 4 days, each well was scored under UV illu-mination for green (GFP positive [GFP⫹], switched) and red (mCherry⫹, unswitched) fluorescence. Percent switching was calculated as 100⫻ green titer/(red titer⫹green titer).
Viral genome quantitation.MuHV-4 genomic positions 24832 to 25071 were amplified by PCR (Rotor Gene 3000; Corbett Research) from 10 ng DNA (NucleoSpin Tissue kit; Macherey-Nagel). PCR prod-ucts quantified with Sybr green (Invitrogen) were compared to a stan-dard curve of a cloned template amplified in parallel and distinguished from paired primers by melting-curve analysis. Correct sizing was confirmed by electrophoresis and ethidium bromide staining. Cellular DNA in the same samples was quantified by amplifying a-actin gene fragment.
Immunostaining.Organs were fixed in 1% formaldehyde–10 mM sodium periodate–75 mML-lysine (18 h at 4°C), equilibrated in 30% sucrose (24 h at 4°C), and then frozen in OCT. Six-micrometer sections were blocked with 0.3% Triton X-100 –5% donkey serum (1 h at 23°C) and then incubated (18 h at 4°C) with primary antibodies to GFP (rabbit polyclonal antibody [PAb] or goat PAb; Abcam), B220 (rat MAb RA3-6B2; Santa Cruz Biotechnology), NKp46 (rat MAb 29A1.4; BioLegend), CD11c (hamster MAb N418), CD68 (rat MAb FA-11; Abcam), F4/80 (rat MAb CI:A3-1; Santa Cruz Biotechnology), mCherry (rabbit PAb; Ba-drilla), and CD169 (rat MAb 3D6.112; Serotec) and polyclonal rabbit sera to MuHV-4, raised by subcutaneous virus inoculation. This serum recog-nizes multiple lytic antigens, including the ORF65 capsid protein, the gp70 complement control protein, and gp150 (43). Sections were washed three times in phosphate-buffered saline (PBS); incubated (1 h at 23°C) with Alexa 568- or Alexa 647-donkey anti-rat IgG PAb, Alexa 488- or Alexa 568-donkey anti-rabbit IgG PAb (Life Technologies), Alexa 488-donkey anti-goat PAb, and Alexa 647-488-donkey anti-hamster IgG PAb (Ab-cam); washed three times in PBS; stained with 4= ,6-diamidino-2-phe-nylindole (DAPI); and mounted in Prolong Gold (Life Technologies). Fluorescence was visualized with a Zeiss LSM 510/710 confocal micro-scope.
Flow cytometry.To identify NK cells, dissociated spleen cells were blocked with anti-CD16/32 (BD Biosciences) and incubated with bio-tinylated anti-NKp46 MAb (BioLegend) and then with Alexa 488-conju-gated streptavidin (Invitrogen). To identify pDC, spleen cells were blocked with 5% donkey serum and then incubated with antibodies to CD11c and BST-2 (rabbit PAb; Pierce Biotechnology) or Siglec-H (rat MAb 440c; Abcam), followed by Alexa 647-donkey anti-hamster IgG PAb plus Alexa 488-donkey anti-rat PAb or Alexa 488-donkey anti-rabbit PAb (Life Technologies). Cells were then washed twice in PBS and analyzed on an Accuri flow cytometer (BD Biosciences).
IFN and NK Cells Restrict MuHV-4 in LN
on November 7, 2019 by guest
http://jvi.asm.org/
RESULTS
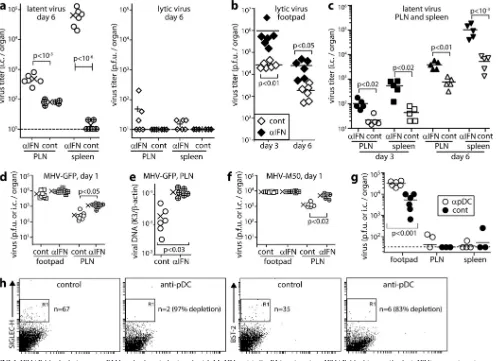
IFNAR blockade increases MuHV-4 dissemination via LN.
We
hypothesized that SSM are an important site of anti-MuHV-4
action for IFN-I. To test this hypothesis, we gave mice
IFNAR-blocking antibody or not i.p., inoculated them with MuHV-4 by
the i.f. route, and measured virus titers (
Fig. 1
). IFNAR blockade
significantly increased day 6 titers in popliteal LN (PLN) and
spleens (
Fig. 1a
). Assays of freeze-thawed samples established that
the increased infection was predominantly latent. IFNAR
block-ade also increased footpad virus titers (
Fig. 1b
), so as i.f. MCMV
spreads from footpads to PLN to spleen (
16
), the increased PLN
and spleen titers could potentially have been secondary effects.
However, PLN and spleen titers increased from day 3 to day 6,
whereas footpad titers decreased (
Fig. 1c
). Therefore, IFN-I
inde-pendently restricted lymphoid infection.
IFNAR blockade increases early LN infection.
Increasing
PLN virus titers from day 3 to day 6 implied more B cell
prolifer-ation in IFNAR-blocked mice, as this is how MuHV-4 amplifies its
latent load. Higher titers at day 3, when B cell infection is first
detected (
16
), suggested that this was due to more initial B cell
infection. PLN titers were also increased at day 1 (
Fig. 1d
).
In-creased viral genome copy numbers (
Fig. 1e
) indicated more PLN
FIG 1IFNAR blockade increases PLN and spleen infections by i.f. MuHV-4. (a) C57BL/6 mice given IFNAR-blocking antibody (␣IFN) or not (cont) were infected i.f. with MuHV-4 (105PFU). Six days later, PLN and spleen viruses were IC assayed for total virus and plaque assayed for infectious virus. Crosses show means, and other symbols show data for individual mice. IFNAR-blocking antibody increased total but not lytic titers. (b) Mice treated as described for panel a were plaque assayed 3 and 6 days later for infectious virus in footpads. Bars show means, and diamonds show data for individuals. IFNAR-blocking antibody increased titers at both time points. (c) Mice treated as described for panel a were IC assayed for reactivatable virus in PLN and spleen 3 and 6 days later. Bars show means; other symbols show data for individuals. IFNAR-blocking antibody increased infection at both time points in both sites. (d) Mice treated as described for panel a were tested 1 day later for infectious footpad virus by plaque assay and for reactivatable PLN virus by IC assay. Crosses show means; other symbols show data for individual mice. IFNAR-blocking antibody increased PLN but not footpad infection. (e) DNA from PLN in panel d was assayed for viral genome load by quantitative PCR. Viral load (K3) was normalized by cellular load (-actin) for each sample. Crosses show means; other symbols show data for individuals. IFNAR-blocking antibody increased viral genome loads. (f) Mice were given IFNAR-blocking antibody or not and then latency-deficient MHV-M50 (105PFU i.f.). One day later, footpad virus was plaque assayed and PLN virus was IC assayed. Crosses show means; other symbols show data for individuals. IFNAR-blocking antibody increased PLN but not footpad infection. (g) Mice were depleted of pDC (␣pDC) or not (cont) by 2 i.p. injections of anti-CD317/BST-2 MAb (400g/mouse) 48 h apart and then given MHV-GFP i.f. (105PFU). Three days later, footpad virus was plaque assayed, and PLN virus and spleen virus were IC assayed. Bars show means; other symbols show data for individual mice. pDC depletion increased footpad but not PLN or spleen infections. Two further experiments gave equivalent results. (h) pDC depletion efficacy was checked by flow cytometry of gated CD11c⫹spleen cells for the pDC markers Siglec-H and BST-2. n is the number of cells in the boxed region.on November 7, 2019 by guest
http://jvi.asm.org/
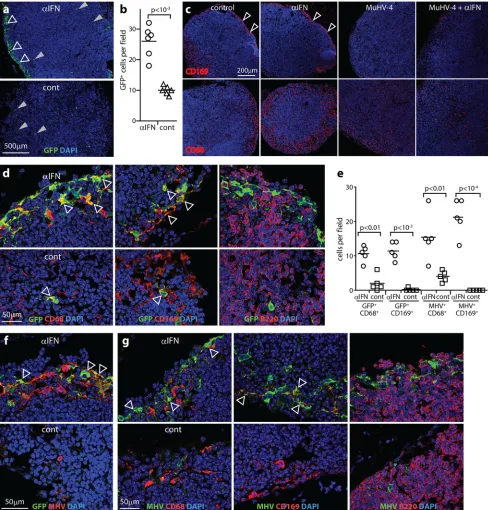
[image:3.585.42.543.67.428.2]FIG 2IFNAR blockade increases SSM infection. (a) Mice given IFNAR-blocking antibody (␣IFN) or not (cont) were infected i.f. with MHV-GFP (105PFU). One day later, PLN sections were stained for viral GFP. Nuclei were stained with DAPI. Gray arrows show example GFP⫹cells in the LN substance. Open arrows show increased GFP staining around the subcapsular sinus of mice treated with IFNAR-blocking antibody. Each image is representative of data for 6 samples per group. (b) GFP⫹cells were counted for samples as described for panel a. Bars show group means. Other symbols show mean counts for 3 randomly selected fields of view per section across 3 sections of individual mice. IFNAR-blocking antibody significantly increased GFP⫹cell numbers. (c) C57BL/6 mice were given IFNAR-blocking antibody or not and infected or not with MuHV-4 as described for panel a. Six days later, PLN sections were stained for CD169 (SSM) and CD68 (macrophages/DC). Infected mice lost CD169 expression around the subcapsular sinus (arrows), regardless of IFNAR blockade. CD68 staining around the subcapsular sinus was also reduced. (d) PLN from mice infected as described for panel a were stained 1 day later for GFP plus CD68, CD169, or B220 (B cells). Arrows show example dual-positive cells. (e) GFP⫹and viral lytic antigen (MHV)-positive cells colocalizing with myeloid cell markers were counted for 3 fields of view per section across 3 sections for each of 5 mice per group. Bars show group means; other symbols show individual mean counts. IFNAR-blocking antibody increased GFP⫹and MHV⫹myeloid (CD169⫹or CD68⫹) cell numbers around the subcapsular sinus. (f) PLN of mice infected as described for panel a were stained 1 day later for viral GFP and lytic antigens (MHV). MHV expression was minimal in control mice. Arrows show example GFP⫹MHV⫹cells in mice treated with IFNAR-blocking antibody. Each image is representative of data for 6 samples per group. (g) PLN of mice infected as described for panel a were stained for viral antigens and cell type markers. Arrows show examples of colocalization. Quantitation was as described for panel e.
IFN and NK Cells Restrict MuHV-4 in LN
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.46.534.67.577.2]infection and not just more
ex vivo
reactivation. IFNAR blockade
also increased day 1 PLN infection by MHV-M50. This virus has a
murine cytomegalovirus IE1 promoter inserted into the ORF50 5
=
untranslated region, essentially abolishing lymphoproliferation
through forced lytic reactivation (
35
) (
Fig. 1f
). Therefore, IFNAR
blockade increased PLN infection before the onset of virus-driven
lymphoproliferation. IFNAR blockade did not significantly
in-crease day 1 footpad infection by either wild-type (WT) or M50
MuHV-4. Therefore, it acted directly on PLN infection.
pDC are nonessential to restrict LN infection.
pDC produce
copious amounts of IFN-I (
44
). To test whether they were
re-quired for IFN-I to restrict MuHV-4 spread, we gave mice a
de-pleting antibody to CD317/tetherin/BST-2, which is expressed
constitutively by pDC and inducibly by other cell types (
45
) (
Fig.
1g
and
h
). This significantly increased day 3 virus titers in footpads
but not in PLN or spleens. Therefore, pDC were nonessential for
IFN-I to restrict acute lymphoid infection.
IFN-I restricts SSM infection.
We identified infected cells by
immunostaining of PLN sections for virus-expressed GFP and
lytic antigens (
Fig. 2
). Low-magnification images at day 1 (
Fig. 2a
)
showed many more GFP
⫹cells around the subcapsular sinus of
IFNAR-blocked mice (
Fig. 2b
). GFP
⫹cell numbers elsewhere in
the PLN remained low.
Inflammation is associated with CD169
⫹SSM displacement
(
46
). Virus infection ablated CD169 staining at day 6 more
dra-matically (
Fig. 2c
). The concomitant loss of subcapsular sinus
CD68 expression, which marks macrophages and DC (
47
), was
consistent with cell displacement or loss. Nonetheless, at day 1,
when CD169 loss was less marked, IFNAR blockade significantly
increased the number of CD68
⫹and CD169
⫹GFP
⫹cells around
the subcapsular sinus (
Fig. 2d
and
e
). B220
⫹B cells were closely
associated with GFP
⫹cells but remained GFP
⫺. While most
my-eloid cells express CD68, its restriction to endosomes and
lyso-somes limited detection sensitivity, as these are not always
cap-tured on sections. However, all GFP
⫹cells had a myeloid rather
than a lymphoid morphology, and the vast majority (
⬎
90%)
lo-calized to the subcapsular sinus. Thus, they appeared to be SSM
and possibly also other myeloid cells, such as dendritic cells, in the
same site.
IFNAR blockade also increased MuHV-4 lytic antigen staining
around the subcapsular sinus (
Fig. 2e
to
g
). Lytic antigen and GFP
staining only partly overlapped. GFP was expressed from an EF1
␣
promoter, which operates independently of the viral lytic cycle
(
45
), so GFP
⫹antigen-negative cells may have harbored latent
genomes. The presence of GFP
⫺antigen-positive cells indicated
that GFP expression could also be shut off during lytic infection.
No B220
⫹cells were viral antigen positive. Rather, IFNAR
block-ade increased lytic infection in myeloid cells.
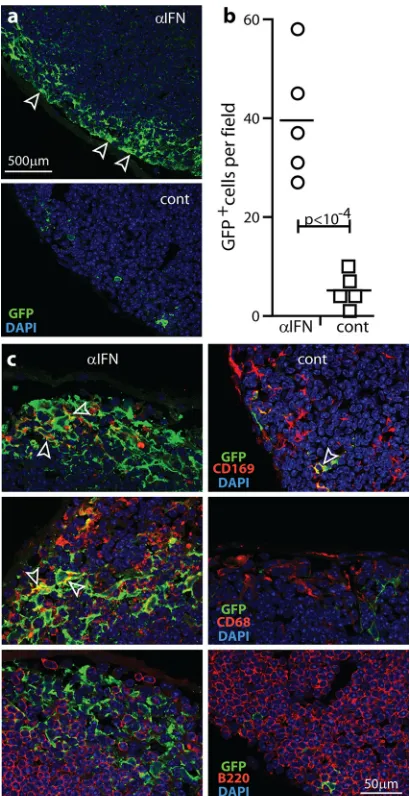
IFN-I protects SSM independently of lytic infection.
IFN-I
limits protein synthesis and thus should inhibit mainly viral lytic
replication. Increased viral lytic antigen expression in SSM after
IFNAR blockade was consistent with this idea. To test whether
IFN-I could also act before the initiation of lytic infection, we gave
mice anti-IFNAR antibody or not and then infected them by the
i.f. route with MuHV-4 lacking its essential ORF50 lytic
transac-tivator (
Fig. 3
). ORF50
⫺MuHV-4 does not express new lytic
genes without complementation. Thus, it is limited
in vivo
to lytic
cycle-independent GFP expression (from an EF1
␣
promoter).
IFNAR blockade again increased GFP expression around the
sub-capsular sinus (
Fig. 3a
and
b
) in CD68
⫹and CD169
⫹but not
B220
⫹cells (
Fig. 3c
). Therefore, IFN-I also restricted SSM
infec-tion before the initiainfec-tion of lytic gene expression.
Synergistic effects of IFN-I blockade and SSM depletion.
To
compare the loss of IFN-I-mediated SSM defense with complete
SSM loss, we gave mice either liposomal clodronate, anti-IFNAR
antibody, both, or neither and then administered MHV-GFP by
the i.f. route (
Fig. 4
). SSM depletion accelerates the spread of
MuHV-4 to the spleen (
16
), and both SSM depletion and IFNAR
blockade increased spleen infection after 6 days (
Fig. 4a
). SSM
depletion and IFNAR blockade together increased both PLN and
spleen infections significantly more than either one did alone.
FIG 3IFN-I restricts SSM infection independently of viral lytic gene expres-sion. (a) Mice were given IFNAR-blocking antibody (␣IFN) or not (cont) and then given i.f. ORF50⫺MuHV-4 (105PFU), which expresses lytic genes only with complementation. One day later, PLN sections were stained for viral GFP. Arrows show example GFP⫹cells. Each image is representative of results for 6 samples per group. (b) GFP⫹cells were counted across 3 randomly selected fields of view per section for 3 sections from each of 6 mice per group. Bars show group means, and other symbols show mean counts for individuals. IFNAR-blocking antibody increased GFP⫹cell numbers. (c) PLN of mice infected as described for panel a were stained for viral GFP plus CD68, CD169, and B220. Arrows show example dual-positive cells. No B cells were GFP⫹.on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.319.524.66.464.2]Therefore, SSM were not the only source or site of action of IFN-I,
and IFN-I was not the only SSM defense.
Immunostaining of PLN for viral GFP and lytic antigens (
Fig.
4b
) also showed that SSM depletion and IFNAR blockade
addi-tively increased infection. All mice showed more GFP
⫹than viral
antigen-positive cells, and most GFP
⫹cells were B220
⫹B cells
(
Fig. 4c
). Thus, by this time, increased myeloid infection had fed
through to increased B cell infection. The paucity of lytic
antigen-positive cells at day 6 compared to day 3 (
Fig. 2e
) implied that
other immune defenses had substituted for IFN-I to control lytic
infection.
In spleens, IFNAR blockade and SSM depletion individually
had little effect on GFP
⫹or virus-positive cell numbers but
to-gether caused a marked increase (
Fig. 4d
to
f
). This applied across
the red pulp, MZ, and white pulp (WP) (
Fig. 4e
), with WP B cells
being prominently infected (
Fig. 4f
). i.f. liposomal clodronate
does not deplete MZM (
39
), so it must have increased virus
seed-ing to the spleen. IFNAR blockade increased SSM infection (
Fig.
2
), but this evidently entailed exposure to additional immune
de-fenses, so SSM depletion more efficiently seeded PLN virus to the
spleen. However, IFNAR blockade also promotes splenic MZM
infection (
38
). Thus, together, SSM depletion and IFNAR
block-ade avoided virus holdup in the PLN and increased subsequent
replication in the spleen.
Functional tracking of MuHV-4 replication in SSM.
Viral
floxed reporter gene switching can track infection through specific
FIG 4IFNAR blockade and SSM depletion synergistically disseminate MuHV-4. (a) Mice were given liposomal clodronate to deplete SSM (clod), IFNAR-blocking antibody (␣IFN), both treatments (both), or neither treatment (cont) and then given MHV-GFP i.f. (105PFU). Six days later, footpad virus was plaque assayed, and PLN and spleen virus were IC assayed. Bars show means, and other symbols show individual titers. All treatments increased footpad and spleen titers; only dual treatment increased PLN titers. *,P⬍0.05; **,P⬍0.01; ***,P⬍10⫺3; ****,P⬍10⫺4. (b) GFP⫹and viral antigen (MHV)-positive cells were countedon PLN sections of mice treated as described for panel a. Bars show group means. Other symbols show mean counts for 3 fields of view per section across 3 sections of each mouse. All treatments increased GFP⫹and MHV⫹cell numbers, although MHV⫹cell numbers were low. (c) Example images from panel b show infected cells in PLN. Most GFP⫹cells were B220⫹(B cells, white arrows). Gray-filled arrows show lytically infected cells. (d) GFP⫹and MHV⫹cells were counted on spleen sections of mice treated as described for panel a. Bars show group means. Other symbols show mean counts for 3 fields of view per section across 3 sections of individual mice. Only dual treatment (both) increased spleen infection by this measure. (e) Splenic GFP⫹cells were further subdivided by site. Dual treatment increased infection in the red pulp (RP), MZ, and white pulp (WP). Single treatments had no significant effect. (f) Example images from panel e show infected B220⫹cells. Dashed lines correspond to the MZ. White arrows show infected WP B cells. The gray-filled arrow shows a lytically infected cell.
IFN and NK Cells Restrict MuHV-4 in LN
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.77.511.64.455.2]cell types of Cre-transgenic mice. Cre switches MHV-RG
irrevers-ibly from red (mCherry) to green (GFP) fluorescence. SSM
ex-press LysM (
16
), so to test whether IFN-I restricts MHV-RG
prop-agation in SSM, we gave IFNAR-blocking antibody or not and
then i.f. MHV-RG to LysM-cre mice (
Fig. 5
). IFN-I blockade
in-creased day 6 virus titers, most noticeably in spleens (
Fig. 5a
). It
also significantly increased the proportion of
fluorochrome-switched virus in footpads and PLN, which was otherwise
negligi-ble (
Fig. 5b
). Occasionally, mice had high levels of splenic virus
switching. This possibly reflected replication in splenic MZM, as
they also express LysM and switch
⬃
60% of the i.p. virus reaching
splenic B cells (
12
). The splenic virus of most IFNAR-blocked
mice was unswitched. Therefore, IFNAR blockade increased both
the productivity of LysM
⫹cell infection in footpads and PLN and
the rate of infection spread to the spleen but as separate effects:
most virus still reached the spleen via LysM
⫺cells.
MuHV-4 fluorochrome switching in infected cells.
Viral
fluo-rochrome switching can also be visualized in infected cells. The
M3 promoter driving fluorochrome expression is active mainly in
early/late lytic infection (
48–50
). IFNAR blockade increased
fluo-rochrome switching at day 6 in PLN and spleen cells of LysM-cre
mice (
Fig. 5c
and
d
). In both IFNAR-blocked and control mice,
cellular fluorochrome switching exceeded that of recovered
viri-ons. GFP
⫹PLN cells were difficult to type with certainty but
ap-peared to be myeloid, as none were B220
⫹(B cells). CD11c
⫹cells
were also unswitched, consistent with few DC expressing LysM
(
39
). Thus, IFNAR blockade increased viral lytic gene expression
in LysM
⫹cells, but LysM
⫺cells remained the main source of viral
propagation.
MuHV-4 fluorochrome switching in CD11c-cre mice.
We
next tracked MHV-RG replication in CD11c-cre mice (
Fig. 6
).
Again IFNAR blockade increased virus titers (
Fig. 6a
), but now it
also significantly increased the switching of virus recovered from
spleens (
Fig. 6b
). Although the proportion of PLN virus that was
switched was unchanged, PLN-infected cell switching increased
(
Fig. 6c
and
e
), and unlike LysM-cre mice, IFNAR-blocked
CD11c-cre mice had GFP
⫹PLN B cells (
Fig. 6f
). These results
were consistent with IFNAR blockade increasing the total amount
of B cell infection but not altering its predominant route, which
was via DC.
IFNAR blockade increased the fluorochrome switching of both
splenic virus (
Fig. 6b
) and splenic infected cells (
Fig. 6c
and
d
).
Most GFP
⫹spleen cells were located around WP follicles (
Fig. 6g
)
and were myeloid (CD11c
⫹CD169
⫹), although GFP
⫹MZ B cells
were also evident (
Fig. 6h
). Control mice also had GFP
⫹myeloid
cells and B cells but fewer (
Fig. 6d
). Thus, again, IFNAR blockade
increased virus spread but did not alter its predominant route.
NK cells are a second line of SSM defense.
The finding that
IFNAR blockade did not increase MuHV-4 passage through SSM
implied additional, IFN-I-independent restriction, before
adap-tive immunity comes into play (
51
). The important role of NK
cells in controlling murine cytomegalovirus (
52
) suggested that
they might also control MuHV-4. Although NK cells are activated
by IFN-I (
20
), IFNAR blockade increased NK cell recruitment to
MuHV-4-infected LN (
Fig. 7a
and
b
), implying IFN-I
indepen-dence in this context. To reveal NK cell function, we compared
their depletion with IFNAR blockade: C57BL/6 mice were
given anti-NK1.1 or anti-IFNAR antibody i.p. or left untreated
and then given MHV-GFP i.f. (
Fig. 7c
and
d
). After 1 day, both
treatments significantly increased virus titers, with IFNAR
blockade having a greater effect. PLN sections showed more
viral GFP
⫹cells after IFNAR blockade and a smaller but still
FIG 5IFNAR blockade increases virus production in but not transfer from LysM⫹cells. (a) LysM-cre mice were given IFNAR-blocking antibody (␣IFN) or not (cont) and then floxed color-switching MHV-RG (105PFU i.f.). Six days later, virus was plaque assayed (footpads) or IC assayed (PLN and spleens). Horizontal bars show means. Other symbols show data for individual mice. The dashed line indicates the assay sensitivity limit. IFNAR-blocking antibody increased infection in footpads and spleens but not PLN. (b) Viruses from panel a were assayed for fluorochrome switching. Bars show means; other symbols show data for individual mice. IFNAR-blocking antibody increased virus switching in footpads and PLN. ND, not determined, as there was insufficient virus. (c) Tissue sections of mice infected as described for panel a were analyzed for infected-cell fluorochrome expression. Bars show group means. Other points show means of data for 3 views per section for each of 3 sections per mouse. IFNAR-blocking antibody increased fluorochrome switching in both PLN and spleens. (d) Example PLN images show unswitched (mCherry⫹, white arrows) but not switched (GFP⫹) CD11c⫹cells in control mice and mice treated with IFNAR-blocking antibody. Gray arrows show example GFP⫹cells.on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.60.521.65.274.2]significant increase after NK cell depletion (
Fig. 7a
and
e
).
GFP
⫹cells of all groups clustered around the subcapsular
si-nus, and many were CD68
⫹and CD169
⫹. Thus, NK cell
deple-tion increased SSM infecdeple-tion.
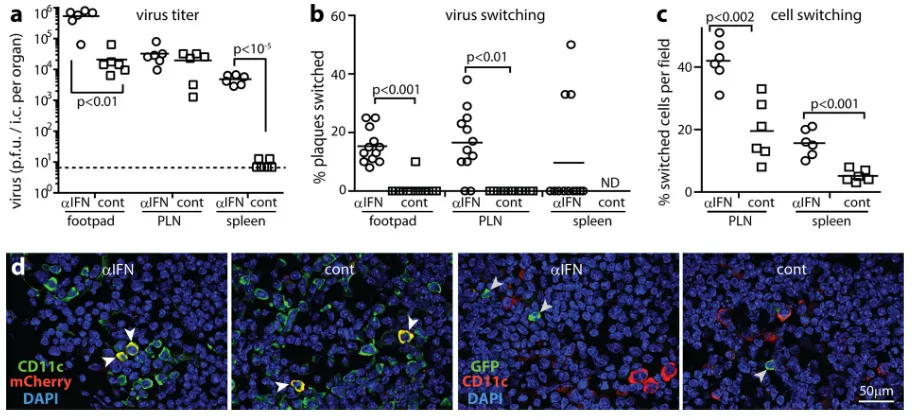
Without IFN-I and NK cells, SSM pass infection to B cells.
SSM attack by NK cells potentially explained the failure of
fluoro-chrome-switched virus to spread in IFNAR-blocked LysM-cre
mice (
Fig. 5
). To test this hypothesis, we gave LysM-cre mice both
FIG 6IFNAR blockade increases virus production in and transfer from CD11c⫹cells. (a) CD11c-cre mice were given IFNAR-blocking antibody or not and then MHV-RG i.f. (105PFU). Six days later, virus was plaque assayed (footpads) or IC assayed (PLN and spleens). Horizontal bars show means. Other symbols show data for individual mice. The dashed line indicates the assay sensitivity limit. IFNAR-blocking antibody significantly increased footpad and PLN but not spleen infections. (b) Viruses from panel a were assayed for fluorochrome switching. Bars show means; other symbols show data for individuals. IFNAR-blocking antibody significantly increased the switching of virus recovered from spleens. (c) Tissue sections of mice infected as described for panel a were analyzed for cellular fluorochrome expression. Bars show group means. Other points show mean counts for 3 views per section for 3 sections per mouse. IFNAR-blocking antibody increased infected-cell switching in both PLN and spleens. (d) Spleens were analyzed further for viral fluorochrome-positive cell types. IFNAR-blocking antibody increased B220⫹and CD11c⫹cell switching, although the increase was significant only for CD11c⫹. (e) PLN overview images show more GFP⫹ (switched) cells with IFNAR-blocking antibody, quantitated as described for panel c. Unswitched (mCherry⫹) cell numbers were similar to those of controls. Six mice per group gave equivalent results. (f) Higher-power images show both switched and unswitched B cells with IFNAR-blocking antibody and only unswitched B cells in controls. The images are representative of results for 6 mice per group. (g) Spleen overview images show IFNAR-blocking antibody increasing switched GFP⫹cell numbers in the MZ between WP follicles and the F4/80⫹red pulp (arrows). Six mice per group gave similar results, quantitated as described for panel c. (h) Higher-power spleen images show example GFP⫹and mCherry⫹cells. GFP⫹cells were evident in all mice, but IFNAR-blocking antibody gave significantly more CD11c⫹GFP⫹cells than controls, quantitated as described for panel d.IFN and NK Cells Restrict MuHV-4 in LN
on November 7, 2019 by guest
http://jvi.asm.org/
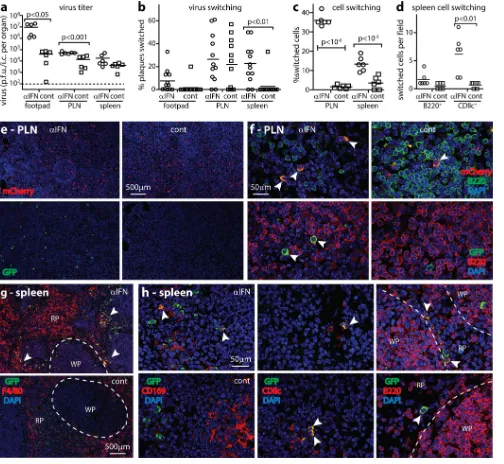
[image:8.585.42.546.66.530.2]NK-depleting and IFNAR-blocking antibodies before infecting
them with MHV-RG. After 4 days, PLN virus titers of
antibody-treated mice exceeded those of controls (
Fig. 8a
), and 25% of the
recovered virus was fluorochrome switched (
Fig. 8b
). Spleen virus
titers also increased, with
⬎
50% switching. Virus from footpads
showed negligible switching. Thus, when both IFN-I and NK cells
were disabled, more virus was produced and a greater proportion
passed through LysM
⫹cells, presumably SSM. Day 4 would
nor-mally be too early for virus to have passed through MZM (
Fig. 5
).
Thus, the switching of splenic virus probably reflected seeding
from the PLN, while PLN virus also included seeding from
foot-pads. However, accelerated MZM infection may have
contrib-uted, as SSM and MZM infections are likely to have similar
im-mune restraints.
The greater virus switching of antibody-treated mice argued
that IFN-I and NK cells regulate MuHV-4 production in LysM
⫹cells. However, while PLN sections of antibody-treated mice
showed more GFP
⫹cells than those of controls, their GFP
⫹/
mCherry
⫹cell ratios were similar (
Fig. 8c
). Therefore, IFN-I and
NK cells also regulated LysM
⫺infection. Most
fluorochrome-pos-itive PLN cells were myeloid rather than lymphoid (
Fig. 8d
and
e
),
consistent with fluorochrome expression being lytic while B cell
infection was mainly latent (
Fig. 2
). Nonetheless GFP
⫹B cells
(B220
⫹) were evident in antibody-treated mice (
Fig. 8e
and
f
).
Thus, when IFN-I and NK cells were lacking, SSM passed
MuHV-4 to B cells.
DISCUSSION
Extracellular fluid returning to the blood provides viruses with a
ready-made vehicle of systemic spread. LN are a key checkpoint,
and myeloid cells are the gatekeepers: migratory DC survey
cell-associated antigens, and sessile SSM survey the afferent lymph.
MuHV-4 infects both cell types, but only DC pass infection to B
cells. IFN-I and NK cells protected SSM against productive
infec-tion by virions adsorbed from the lymph. Other innate immune
effectors (
53
) may also contribute—the immune response is
in-herently multilayered, and with shared induction pathways,
indi-vidual effectors rarely act alone— but IFN-I and NK cells had key
roles.
A previous study of lung infection (
54
) found no significant
NK cell contribution to MuHV-4 control. However, LN infection
was not measured. The defensive role of NK cells identified here
was consistent with human genetic deficiency phenotypes (
55
),
prominent NK cell responses to EBV (
56
), and NK cell-mediated
defense against EBV in chimeric mice (
57
). Protection by IFN-I
argued against filtering macrophages being deliberately virus
per-missive (
19
); rather, the immune response consistently inhibits
infection as it escalates from IFN-I to NK cells to adaptive
re-sponses, with each gaining functional prominence if upstream
containment fails.
Host defense against viremia has anatomical as well as
func-tional layers. Most extracellular fluid traverses more than one
FIG 7IFN-I and NK cells control PLN infection. (a) C57BL/6 mice were given IFNAR-blocking antibody (␣IFN) or NK-depleting antibody (␣NK) or left untreated (cont) and then infected i.f. with MHV-GFP (105PFU). One day later, PLN sections were stained for viral GFP plus NKp46⫹NK cells (NK), myeloidcells (CD68), or SSM (CD169). Nuclei were stained with DAPI. Open arrows show NKp46⫹NK cells around the subcapsular sinus. Gray-filled arrows show CD68⫹and CD169⫹infected cells. (b) NKp46⫹NK cells were counted on PLN sections of mice treated as described for panel a. Bars show group means. Circles show mean counts for 3 fields of view per section from 3 sections per mouse. IFNAR-blocking antibody significantly increased NK cell recruitment above controls, whereas NK depletion significantly reduced it. (c) Spleen cells of mice given anti-NK1.1 depleting antibody (PK136) (200g/mouse in 2 injections 48 h apart) or not (control) were analyzed 24 h later by flow cytometry for expression of the NK cell marker NKp46. n is the number of cells in the boxed region. (d) PLN of mice treated as described for panel a were IC assayed for recoverable virus 1 day after i.f. MHV-GFP. Crosses show means; other symbols show data for individuals. Both NK cell-depleting antibody and IFNAR-blocking antibody increased titers. IFNAR-blocking antibody had a significantly greater effect. (e) GFP⫹cells on PLN sections described for panel a were counted for 3 views per section across 3 sections per mouse. Circles show mean counts for individuals. Bars show group means. Both IFNAR-blocking antibody and NK cell-depleting antibody increased GFP⫹cell numbers. IFNAR-blocking antibody had a significantly greater effect.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.46.542.66.308.2]lymph node; for example, footpad-inoculated MuHV-4 passes
from the PLN to the para-aortic LN (
16
), and splenic MZM
filter the blood. Invasive virus inoculations are often more
pathogenic than mucosal inoculations because they bypass the
outer defenses: i.p. MuHV-4 reaches the spleen directly (
12
),
and in this context, IFNAR blockade greatly increases
macro-phage infection (
36
), consistent with IFNAR
⫺/⫺mice
suc-cumbing more rapidly to i.p. than to i.n. infection (
32
,
33
).
Although invasive inoculations cause more disease, host
colo-nization is not necessarily enhanced, as viral genes now operate
outside their normal evolutionary context. Thus, protecting
against disease after an invasive inoculation is not the same as
protecting against natural infection. For example, recombinant
gp350 protected tamarins against EBV-induced disease but did
not prevent natural human infection (
3
). Such outcomes
em-phasize the need to develop vaccine strategies that allow for
viral immune evasion.
Natural MuHV-4 infection is probably nasal (
22
); we studied
i.f. infection because the complexity of i.n. infection makes
pri-mary and secondary LN effects difficult to separate, but the SSM
barrier is relevant to both (
16
), and increased spleen infection by
i.n. MuHV-4 in IFNAR
⫺/⫺mice (
33
) is consistent with IFN-I also
restricting MuHV-4 passage through mucosa-associated LN.
When the host response meets viral evasion, the outcome can
depend on cell type, and IFN-I and NK cells evidently restricted
MuHV-4 less in DC than in SSM. Most DC infection is initially
latent (
58
). It may become lytic
in vivo
only after DC have
mi-grated away from inflammatory infection sites and IFN-I
signal-ing has subsided. Migratory DC may also be less IFN-I responsive
than SSM (
59
). Nonetheless, antiviral states are inducible, and
vaccine-primed T cells could potentially recruit IFN-I and NK cell
responses upon virus challenge. CD4
⫹T cells control long-term
MuHV-4 infection (
60
), interact with LN DC (
61
), and protect via
IFN-II (
62
), which potentiates IFN-I (
63
). The efficacy of innate
immunity in restricting SSM infection suggested that recruitment
of these defenses might also be able to limit DC infection and thus
reduce host colonization.
ACKNOWLEDGMENTS
We thank Orry Wyer for technical support and Helen Farrell for helpful discussion.
This work was supported by National Health and Medical Research Council grants 1060138, 1064015, and 1079180; Australian Research Council grant FT130100138; Queensland Health; the Sakzewski
Founda-FIG 8SSM pass on infection when both IFN-I and NK cells are disabled. (a) LysM-cre mice were given IFNAR-blocking (␣IFN) and NK cell-depleting (␣NK) antibodies or not and then infected i.f. with MHV-RG (105PFU). Four days later, infectious virus from footpads was plaque assayed, and reactivatable virus from PLN and spleen virus were IC assayed. Crosses show means; other symbols show data for individuals. IFNAR-blocking and NK cell-depleting antibodies significantly increased PLN and spleen titers. The baseline is the assay sensitivity limit. (b) Virus described for panel a was typed for fluorochrome expression. IFNAR-blocking and NK cell-depleting antibodies increased fluorochrome switching in PLN, and in mice treated with IFNAR-blocking and NK cell-depleting antibodies, spleen virus was significantly more switched than PLN virus. Control mice yielded insufficient spleen virus for assays (ND, not determined). (c) PLN sections of mice treated as described for panel a were stained for virus-expressed GFP (switched) and mCherry (unswitched). Total GFP⫹cells were counted for 3 fields of view per PLN section across 3 sections for each of 6 mice (left-hand graph). Bars show mean counts⫾standard errors of the means for individual mice. IFNAR-blocking and NK cell-depleting antibodies significantly increased total GFP⫹cell numbers. However, the ratios of GFP⫹cells to mCherry⫹cells (percentages of switched cells) (right-hand graph) were not significantly different. Circles and squares show mean counts for individuals, and bars show group means. (d) PLN sections of mice treated as described for panel a were stained for GFP and CD169 to identify fluorochrome-switched, infected SSM. Arrows show examples. Total GFP expression was quantitated as described for panel c. (e) PLN sections of mice treated as described for panel a were stained for GFP and B220 to identify fluorochrome-switched, infected B cells. Symbols show mean counts for 2 fields of view per mouse. Bars show group means. Control mice lacked GFP⫹ B cells. (f) The arrow shows an example GFP⫹B220⫹cell in PLN of an IFNAR-blocked, NK cell-depleted mouse, quantitated as described for panel d.
IFN and NK Cells Restrict MuHV-4 in LN
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.42.542.67.324.2]tion; and BELSPO (collaborative grant BelVir) (to P.G.S.). J.P.S. was sup-ported by FCT SFRH/BSAB/113927/2015.
FUNDING INFORMATION
This work, including the efforts of Philip G. Stevenson, was funded by Department of Health | National Health and Medical Research Council (NHMRC) (1060138, 1064015, and 1079180). This work, including the efforts of Philip G. Stevenson, was funded by Federaal Wetenschapsbeleid (BELSPO) (BELVIR). This work, including the efforts of J. Pedro Simas, was funded by Ministry of Education and Science | Fundação para a Ciên-cia e a Tecnologia (FCT) (SFRH/BSAB/113927/2015). This work, includ-ing the efforts of Philip G. Stevenson, was funded by Australian Research Council (ARC) (FT130100138).
The funders had no role in study design, data collection and interpreta-tion, or the decision to submit the work for publication.
REFERENCES
1.Cesarman E, Mesri EA.2007. Kaposi sarcoma-associated herpesvirus and other viruses in human lymphomagenesis. Curr Top Microbiol Immunol
312:263–287.
2.Joseph AM, Babcock GJ, Thorley-Lawson DA.2000. Cells expressing the Epstein-Barr virus growth program are present in and restricted to the naive B-cell subset of healthy tonsils. J Virol74:9964 –9971.http://dx.doi .org/10.1128/JVI.74.21.9964-9971.2000.
3.Balfour HH.2014. Progress, prospects, and problems in Epstein-Barr virus vaccine development. Curr Opin Virol6:1–5.http://dx.doi.org/10 .1016/j.coviro.2014.02.005.
4.Dunmire SK, Grimm JM, Schmeling DO, Balfour HH, Jr, Hogquist KA.
2015. The incubation period of primary Epstein-Barr virus infection: viral dynamics and immunologic events. PLoS Pathog11:e1005286.http://dx .doi.org/10.1371/journal.ppat.1005286.
5.McGeoch DJ.2001. Molecular evolution of the gamma-Herpesvirinae. Philos Trans R Soc Lond B Biol Sci356:421– 435.http://dx.doi.org/10 .1098/rstb.2000.0775.
6.Stevenson PG, Simas JP, Efstathiou S.2009. Immune control of mam-malian gamma-herpesviruses: lessons from murid herpesvirus-4. J Gen Virol90:2317–2330.http://dx.doi.org/10.1099/vir.0.013300-0. 7.Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto
AJ, Speck SH.1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol71:5894 –5904.
8.Goodnow CC.1992. Transgenic mice and analysis of B-cell tolerance. Annu Rev Immunol 10:489 –518.http://dx.doi.org/10.1146/annurev.iy .10.040192.002421.
9.Garside P, Brewer JM.2008. Real-time imaging of the cellular interac-tions underlying tolerance, priming, and responses to infection. Immunol Rev221:130 –146.http://dx.doi.org/10.1111/j.1600-065X.2008.00587.x. 10. Gaspar M, May JS, Sukla S, Frederico B, Gill MB, Smith CM, Belz GT,
Stevenson PG.2011. Murid herpesvirus-4 exploits dendritic cells to infect B cells. PLoS Pathog7:e1002346.http://dx.doi.org/10.1371/journal.ppat .1002346.
11. Frederico B, Milho R, May JS, Gillet L, Stevenson PG.2012. Myeloid infection links epithelial and B cell tropisms of murid herpesvirus-4. PLoS Pathog8:e1002935.http://dx.doi.org/10.1371/journal.ppat.1002935. 12. Frederico B, Chao B, May JS, Belz GT, Stevenson PG.2014. A murid
gamma-herpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe15:457– 470.http://dx.doi .org/10.1016/j.chom.2014.03.010.
13. Lawler C, Milho R, May JS, Stevenson PG.2015. Rhadinovirus host entry by co-operative infection. PLoS Pathog11:e1004761.http://dx.doi .org/10.1371/journal.ppat.1004761.
14. Rosa GT, Gillet L, Smith CM, de Lima BD, Stevenson PG.2007. IgG fc receptors provide an alternative infection route for murine gamma-herpesvirus-68. PLoS One2:e560.http://dx.doi.org/10.1371/journal.pone .0000560.
15. Carrasco YR, Batista FD.2007. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the sub-capsular sinus of the lymph node. Immunity27:160 –171.http://dx.doi .org/10.1016/j.immuni.2007.06.007.
16. Frederico B, Chao B, Lawler C, May JS, Stevenson PG.2015. Subcap-sular sinus macrophages limit acute gammaherpesvirus dissemination. J Gen Virol96:2314 –2327.http://dx.doi.org/10.1099/vir.0.000140.
17. Kuka M, Iannacone M.2014. The role of lymph node sinus macrophages in host defense. Ann N Y Acad Sci1319:38 – 46.http://dx.doi.org/10.1111 /nyas.12387.
18. Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH.2010. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature465:1079 –1083.http://dx.doi.org/10.1038 /nature09118.
19. Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, Klingel K, Sauter M, Kandolf R, Gailus N, van Rooijen N, Burkart C, Baldus SE, Grusdat M, Löhning M, Hengel H, Pfeffer K, Tanaka M, Häussinger D, Recher M, Lang PA, Lang KS. 2011. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol13:51–57.http://dx.doi.org/10.1038/ni .2169.
20. Garcia Z, Lemaître F, van Rooijen N, Albert ML, Levy Y, Schwartz O, Bousso P.2012. Subcapsular sinus macrophages promote NK cell accu-mulation and activation in response to lymph-borne viral particles. Blood
120:4744 – 4750.http://dx.doi.org/10.1182/blood-2012-02-408179. 21. Randall RE, Goodbourn S.2008. Interferons and viruses: an interplay
between induction, signalling, antiviral responses and virus countermea-sures. J Gen Virol89:1– 47.http://dx.doi.org/10.1099/vir.0.83391-0. 22. Milho R, Frederico B, Efstathiou S, Stevenson PG.2012. A
heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog 8:e1002986. http://dx.doi.org/10.1371/journal .ppat.1002986.
23. Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, Song MJ, Sanchez DJ, O’Connell RM, Cheng G, Sun R.2009. Conserved herpes-viral kinase promotes herpes-viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe5:166 –178.http://dx.doi .org/10.1016/j.chom.2008.12.013.
24. Kang HR, Cheong WC, Park JE, Ryu S, Cho HJ, Youn H, Ahn JH, Song MJ.2014. Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. J Virol88:6832– 6846.http://dx.doi.org/10 .1128/JVI.03460-13.
25. Leang RS, Wu TT, Hwang S, Liang LT, Tong L, Truong JT, Sun R.2011. The anti interferon activity of conserved viral dUTPase ORF54 is essential for an effective MHV-68 infection. PLoS Pathog7:e1002292.http://dx.doi .org/10.1371/journal.ppat.1002292.
26. Liang X, Shin YC, Means RE, Jung JU.2004. Inhibition of interferon-mediated antiviral activity by murine gammaherpesvirus 68 latency-associated M2 protein. J Virol78:12416 –12427.http://dx.doi.org/10.1128 /JVI.78.22.12416-12427.2004.
27. Sheridan V, Polychronopoulos L, Dutia BM, Ebrahimi B.2014. A shutoff and exonuclease mutant of murine gammaherpesvirus-68 yields infectious virus and causes RNA loss in type I interferon receptor knockout cells. J Gen Virol95:1135–1143.http://dx.doi.org/10.1099/vir.0.059329-0.
28. Ku B, Woo JS, Liang C, Lee KH, Hong HS, E X, Kim KS, Jung JU, Oh BH.2008. Structural and biochemical bases for the inhibition of au-tophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog4:e25.http://dx.doi.org/10.1371/journal.ppat.0040025. 29. Zhao J, He S, Minassian A, Li J, Feng P.2015. Recent advances on viral
manipulation of NF-B signaling pathway. Curr Opin Virol15:103–111.
http://dx.doi.org/10.1016/j.coviro.2015.08.013.
30. Gaspar M, Gill MB, Lösing JB, May JS, Stevenson PG.2008. Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS One
3:e2781.http://dx.doi.org/10.1371/journal.pone.0002781.
31. Ling PD, Tan J, Sewatanon J, Peng R.2008. Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J Virol82:8000 – 8012.http://dx.doi.org/10.1128/JVI.02752-07.
32. Weck KE, Dal Canto AJ, Gould JD, O’Guin AK, Roth KA, Saffitz JE, Speck SH, Virgin HW.1997. Murine gamma-herpesvirus 68 causes se-vere large-vessel arteritis in mice lacking interferon-gamma responsive-ness: a new model for virus-induced vascular disease. Nat Med3:1346 – 1353.http://dx.doi.org/10.1038/nm1297-1346.
33. Dutia BM, Allen DJ, Dyson H, Nash AA.1999. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology261:173–179.http://dx.doi.org/10.1006/viro.1999.9834. 34. Mandal P, Krueger BE, Oldenburg D, Andry KA, Beard RS, White DW,
Barton ES.2011. A gammaherpesvirus cooperates with interferon-alpha/ beta-induced IRF2 to halt viral replication, control reactivation, and
on November 7, 2019 by guest
http://jvi.asm.org/
imize host lethality. PLoS Pathog7:e1002371.http://dx.doi.org/10.1371 /journal.ppat.1002371.
35. May JS, Coleman HM, Smillie B, Efstathiou S, Stevenson PG.2004. Forced lytic replication impairs host colonization by a latency-deficient mutant of murine gammaherpesvirus-68. J Gen Virol85:137–146.http: //dx.doi.org/10.1099/vir.0.19599-0.
36. Tan CS, Lawler C, May JS, Belz GT, Stevenson PG. 2016. Type I interferons direct gammaherpesvirus host colonization. PLoS Pathog12:
e1005654.http://dx.doi.org/10.1371/journal.ppat.1005654.
37. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I.1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res8:265–277. http://dx.doi.org/10.1023 /A:1008942828960.
38. Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J signaling controls the homeostasis of CD8⫺dendritic cells in the spleen. J Exp Med
204:1653–1664.http://dx.doi.org/10.1084/jem.20062648.
39. Van Rooijen N, Sanders A.1994. Liposome mediated depletion of mac-rophages: mechanism of action, preparation of liposomes and applica-tions. J Immunol Methods174:83–93.http://dx.doi.org/10.1016/0022 -1759(94)90012-4.
40. Milho R, Smith CM, Marques S, Alenquer M, May JS, Gillet L, Gaspar M, Efstathiou S, Simas JP, Stevenson PG.2009. In vivo imaging of murid herpesvirus-4 infection. J Gen Virol90:21–32.http://dx.doi.org/10.1099 /vir.0.006569-0.
41. May JS, Stevenson PG. 2010. Vaccination with murid herpesvirus-4 glycoprotein B reduces viral lytic replication but does not induce detect-able virion neutralization. J Gen Virol91:2542–2552.http://dx.doi.org/10 .1099/vir.0.023085-0.
42. de Lima BD, May JS, Stevenson PG.2004. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J Virol78:5103–5112.http://dx.doi.org/10.1128/JVI.78.10.5103 -5112.2004.
43. Gillet L, Adler H, Stevenson PG.2007. Glycosaminoglycan interactions in murine gammaherpesvirus-68 infection. PLoS One2:e347.http://dx .doi.org/10.1371/journal.pone.0000347.
44. Swiecki M, Colonna M.2015. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol15:471– 485.http://dx.doi.org/10.1038 /nri3865.
45. Asselin-Paturel C, Brizard G, Pin J-J, Briere F, Trinchieri G. 2003. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol171:6466 – 6477.http://dx.doi.org/10.4049/jimmunol.171.12.6466.
46. Gaya M, Castello A, Montaner B, Rogers N, Reis e Sousa C, Bruckbauer A, Batista FD.2015. Host response. Inflammation-induced disruption of SCS macrophages impairs B cell responses to secondary infection. Science
347:667– 672.http://dx.doi.org/10.1126/science.aaa1300.
47. Martinez-Pomares L, Platt N, McKnight AJ, da Silva RP, Gordon S.
1996. Macrophage membrane molecules: markers of tissue differentiation and heterogeneity. Immunobiology 195:407– 416.http://dx.doi.org/10 .1016/S0171-2985(96)80012-X.
48. Simas JP, Swann D, Bowden R, Efstathiou S.1999. Analysis of murine gammaherpesvirus-68 transcription during lytic and latent infection. J Gen Virol80:75– 82.http://dx.doi.org/10.1099/0022-1317-80-1-75.
49. van Berkel V, Preiter K, Virgin HW, Speck SH.1999. Identification and initial characterization of the murine gammaherpesvirus 68 gene M3, en-coding an abundantly secreted protein. J Virol73:4524 – 4529.
50. Marques S, Efstathiou S, Smith KG, Haury M, Simas JP.2003. Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J Virol77:7308 –7318.http://dx.doi.org/10.1128/JVI.77.13.7308-7318.2003. 51. Stevenson PG, Doherty PC.1998. Kinetic analysis of the specific host
response to a murine gammaherpesvirus. J Virol72:943–949.
52. Lisnic´ B, Lisnic´ VJ, Jonjic´ S.2015. NK cell interplay with cytomegalovi-ruses. Curr Opin Virol15:9 –18.http://dx.doi.org/10.1016/j.coviro.2015 .07.001.
53. Kastenmüller W, Torabi-Parizi P, Subramanian N, Lämmermann T, Germain RN.2012. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell150:1235– 1248.http://dx.doi.org/10.1016/j.cell.2012.07.021.
54. Usherwood EJ, Meadows SK, Crist SG, Bellfy SC, Sentman CL. 2005. Control of murine gammaherpesvirus infection is independent of NK cells. Eur J Immunol35:2956 –2961.http://dx.doi.org/10.1002/eji.200526245. 55. Carneiro-Sampaio M, Coutinho A.2007. Immunity to microbes: lessons
from primary immunodeficiencies. Infect Immun75:1545–1555.http: //dx.doi.org/10.1128/IAI.00787-06.
56. Hislop AD.2015. Early virological and immunological events in Epstein-Barr virus infection. Curr Opin Virol15:75–79.http://dx.doi.org/10.1016 /j.coviro.2015.08.002.
57. Chijioke O, Müller A, Feederle R, Barros MH, Krieg C, Emmel V, Marcenaro E, Leung CS, Antsiferova O, Landtwing V, Bossart W, Moretta A, Hassan R, Boyman O, Niedobitek G, Delecluse HJ, Capaul R, Münz C.2013. Human natural killer cells prevent infectious mononu-cleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep
5:1489 –1498.http://dx.doi.org/10.1016/j.celrep.2013.11.041.
58. Smith CM, Gill MB, May JS, Stevenson PG.2007. Murine gammaher-pesvirus-68 inhibits antigen presentation by dendritic cells. PLoS One
2:e1048.http://dx.doi.org/10.1371/journal.pone.0001048.
59. Moltedo B, Li W, Yount JS, Moran TM.2011. Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog7:e1002345.http://dx.doi .org/10.1371/journal.ppat.1002345.
60. Cardin RD, Brooks JW, Sarawar SR, Doherty PC.1996. Progressive loss of CD8⫹T cell-mediated control of a gamma-herpesvirus in the absence of CD4⫹T cells. J Exp Med184:863– 871.http://dx.doi.org/10.1084/jem .184.3.863.
61. Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT.2004. Cognate CD4⫹T cell licensing of dendritic cells in CD8⫹T cell immunity. Nat Immunol5:1143–1148.http://dx.doi .org/10.1038/ni1129.
62. Christensen JP, Cardin RD, Branum KC, Doherty PC.1999. CD4⫹T cell-mediated control of a gamma-herpesvirus in B cell-deficient mice is mediated by IFN-gamma. Proc Natl Acad Sci U S A96:5135–5140.http: //dx.doi.org/10.1073/pnas.96.9.5135.
63. Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol75:163–189.
IFN and NK Cells Restrict MuHV-4 in LN
on November 7, 2019 by guest
http://jvi.asm.org/