Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Attachment and Growth of Human Rotaviruses RV-3
and S12/85 in Caco-2 Cells Depend on VP4
CARL D. KIRKWOOD,1† RUTH F. BISHOP,1ANDBARBARA S. COULSON2*
Department of Gastroenterology and Clinical Nutrition, Royal Children’s Hospital,1and Department of Microbiology and Immunology, The University of Melbourne,2Parkville 3052, Victoria, Australia
Received 4 May 1998/Accepted 24 July 1998
Studies with human neonatal rotaviruses RV-3 and S12/85 and their reassortants showed that VP4 is a determinant of rotavirus attachment to and growth in Caco-2 cells. The binding of these viruses to MA104 and Caco-2 cells correlated with their growth ability. Virus sensitivity to trypsin and the VP4 fusion region may be implicated in these processes.
Rotaviruses are triple-layered, icosahedral particles contain-ing a segmented double-stranded RNA genome. Both the outer capsid proteins VP4 and VP7 elicit neutralizing, protec-tive antibodies, define independently segregating serotype specificities (18), and are implicated in virus cell entry, al-though the role of VP7 may be minor (14, 31). The spike protein VP4 is important in determination of host cell tropism (19, 25), virulence (21, 24, 36), and viral attachment and pen-etration (6, 20, 22, 31). VP4 is a hemagglutinin in some animal strains (25, 33) and contains a putative fusion region (34), and its cleavage by trypsin into subunits VP5* and VP8* (12, 17, 19) enhances virus infectivity by permitting virus penetration (2, 17, 19) rather than by increasing virus binding (26).
Rotaviruses infect the mature epithelial cells lining the villi of the small intestine. Infection can be asymptomatic or cause symptoms varying from loose stools to copious watery life-threatening diarrhea. Symptoms of rotavirus infection reflect the balance between host resistance factors and viral virulence. Virulence has been associated with genes coding for structural proteins VP3 (24), VP4 (7, 21, 24, 36), and VP7 (24) and nonstructural proteins NSP1 (9), NSP2 (9, 24), and NSP4 (3, 24). The existence of bovine (brv P7[5], G6) and porcine (prv 4F, prv 4S) rotavirus strains of varying virulence has been clearly demonstrated in animal models (7, 8) and has been linked to differences in the ability to replicate in vivo and/or in vitro (7, 23, 39).
Two human rotavirus strains, RV-3 and S12/85, of identical P2A[6],G3 serotype and subgroup II specificity have been identified in newborn babies in Melbourne, Australia, as causes of asymptomatic nosocomial infection in a neonatal nursery (RV-3) or severe community-acquired diarrhea after discharge from hospital (S12/85). RV-3 was isolated from a stool specimen containing rotavirus of the R electropherotype obtained in 1977 (1, 38), and S12/85 was isolated from a stool specimen obtained in 1985 (28). These strains have very similar amino acid sequences in genes coding for VP4, VP7, and NSP4 and have identical VP6 sequences (28). However, conserved amino acid differences between these two strains were identi-fied at, or near, sites on VP4 that are implicated in antigenicity,
recognition of a2b1 integrin (14), and cell fusion and at an antigenic site on VP7 that is possibly involved in recognition of a4b1 integrin (14) and that is subject to changes in glycosyla-tion. Any, or all, of these changes have the potential to influ-ence viral growth in vivo and in vitro.
We now report the growth characteristics of these and other human rotaviruses in vitro. The abilities of these viruses and their reassortants, bearing mixtures of genes from RV-3 and S12/85, to bind and grow in an African green monkey kidney epithelial cell line (MA104) and in a human colonic adenocar-cinoma cell line showing characteristics typical of small intes-tinal epithelial cells (Caco-2 [40]) were studied in order to identify the human rotavirus cell attachment protein and spe-cific amino acids that may influence this process.
Rotavirus S12/85 was adapted to growth in MA104 cells as described previously (1). Human rotavirus strains previously isolated from asymptomatic neonates were NnB1, a P2A[6],G3 serotype and subgroup II strain of S electropherotype (38) obtained from a stool specimen collected in 1978 (1), M37 (P2A[6],G1), 1076 (P2A[6],G2), and RV-3. An escape mutant of RV-3, vRV-3:3, with a single amino acid mutation at posi-tion 383 (Asn-Lys), which we had derived by selecposi-tion with the neutralizing monoclonal antibody (MAb) RV-3:3 directed to VP4 (13, 27), was also studied.
Reassortants of RV-3 and S12/85 were derived by coinfec-tion of MA104 cells at a multiplicity of infeccoinfec-tion (MOI) of 1 fluorescent cell-forming unit (FCFU) as described previously (29). Selection was carried out with antirotavirus MAbs RV3:3 (to neutralize viruses with VP4 derived from RV-3 [13, 27]) and A10/N3 (to neutralize viruses with VP7 derived from S12/85 [16, 30]). MAb RV-3:3 neutralized RV-3 and S12/85 to reciprocal titers of 820,000 and 4,000, respectively. MAb A10/N3 neutralized RV-3 and S12/85 to reciprocal titers of ,100 and 1,300, respectively. The parental origin of each gene segment of all reassortants was determined by electrophoresis in 7.5, 10, and 15% polyacrylamide gels (data not shown). The origin of VP4 and VP7 was confirmed by sequence analysis of amino acids (aa) 235 to 242 of VP7 and aa 330 to 380 of VP4, which was performed as described previously (28). The origin of VP4 and VP7 also was determined by the virus tion titer measured by fluorescent-focus reduction neutraliza-tion assay (13) with MAbs RV-3:3 and A10/N3, the VP4-specific MAb HS11 (37), and rabbit hyperimmune antisera raised to RV-3 and S12/85 (data not shown). MAb HS11 neu-tralized S12/85 to low titer but did not neutralize RV-3 (data not shown). As shown in Table 1, all reassortants contained single or multiple segments from S12/85 in the genetic back-* Corresponding author. Mailing address: Department of
Microbi-ology and ImmunMicrobi-ology, The University of Melbourne, Royal Parade, Parkville 3052, Victoria, Australia. Phone: 61 3 9344 8823. Fax: 61 3 9347 1540. E-mail: [email protected].
† Present address: Viral Gastroenteritis Section, Division of Viral and Rickettsial Diseases, National Center for Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA 30333.
9348
on November 9, 2019 by guest
http://jvi.asm.org/
ground of RV-3. These reassortants also all contained gene segment 9 of RV-3, which we have shown by Northern blot hybridization of a digoxigenin-labelled RV-3 VP7 probe to encode VP7 (data not shown). We were unable to generate reassortants containing gene 4 of RV-3 in the genetic back-ground of S12/85, probably because we had no MAb suitable for selection against VP7 of RV-3.
All viruses were propagated in MA104 and Caco-2 cells after activation with 10mg of porcine trypsin (Sigma) per ml, fol-lowed by incorporation of 1mg of trypsin per ml in the main-tenance medium (15). Prior to infection, Caco-2 cells were incubated overnight in medium without fetal calf serum. All viruses grew to titers of 43105to 13106when inoculated at a MOI of 2 in MA104 cells. As shown in Table 1, Caco-2 cells infected with S12/85, M37, 1076, or NnB1 (MOI, 2) produced a moderate titer of infectious virus (105to 106 FCFU/ml) in each of the three to six passages tested. Conversely, RV-3 and vRV-3:3 at the same MOI were unable to establish productive infection in Caco-2 cells. Low levels of virus were detected in the first two passages only, which may have been due to the virus binding without penetration of the cell or penetration occurring by a nonproductive pathway. It was not possible to achieve higher MOI with RV-3 or vRV-3:3. All reassortants that contained gene segment 4 (encoding VP4) of S12/85 were able to infect and adapt to Caco-2 cells. The two reassortants unable to infect and adapt to Caco-2 cells (r34 and r214.2) contained gene segment 4 of RV-3.
To determine whether the inability of RV-3 to adapt to growth in Caco-2 cells was due to lack of suitable receptors, the binding of infectious rotaviruses to cells was measured essen-tially as described previously (31), by using 10mg of trypsin per ml to activate virus, a MOI of 1 to 3, and an infectivity assay developed previously (13). The extent of rotavirus binding to Caco-2 cells varied among the viruses tested (Table 1). Viruses S12/85, NnB1, M37, and 1076, which were able to infect and adapt to growth in Caco-2 cells, showed high levels of binding (13.3, 16.8, 11, and 13.8%, respectively). Reassortants that were able to infect and adapt to Caco-2 cells also exhibited high levels of attachment ranging from 11.5 to 17.1%. In con-trast, viruses which contained gene 4 of RV-3 and were unable to adapt to growth in Caco-2 cells (RV-3, r34, and r214.2) showed low binding levels (4.8, 4.6, and 2.7%, respectively). The MOI did not influence the level of binding of RV-3 to
Caco-2 cells, since 4.8% of virus bound at a MOI of 1 and 4.9% bound at a MOI of 10. Variant vRV-3:3, which also was unable to adapt to growth in Caco-2 cells, also showed a low level of binding (3.6%). Thus, aa 383 of RV-3 VP4 is not involved in the inability of RV-3 to bind to and grow in Caco-2 cells.
[image:2.612.51.548.82.225.2]As shown in Fig. 1, for the 11 serotype P2A[6] viruses tested, there was a highly statistically significant correlation at the 95% confidence level between the amount of infectious virus able to bind to Caco-2 cells and the titer of virus produced after one passage in these cells (r250.93; P,0.0001). The four virus strains with no more than a single amino acid change from the RV-3 VP4 sequence and a VP7 sequence identical to that of RV-3 (RV-3, vRV-3, r34, and r214.2) showed signifi-cantly lower levels of binding and replication than did the
[image:2.612.315.541.469.655.2]FIG. 1. Correlation of binding of infectious, MA104 cell-adapted, serotype P2A[6] rotaviruses to Caco-2 cells with the titer of virus produced after a single passage in these cells. Viruses with similar binding and growth characteristics as determined by statistical analysis were grouped as described in the text. A, viruses with VP4 derived from RV-3 (RV-3, r34, and r214.2) or with a single mutation of the RV-3 VP4 amino acid sequence (vRV-3:3); B, viruses with VP4 sequences showing at least three amino acid differences from the sequence of RV-3 (S12/85, r6, r26, r31, NnB1, M37, and 1076).
TABLE 1. Attachment and growth of neonatal, reassortant, and variant rotaviruses in Caco-2 cellsa
Virus Serotype Gene segmentsfrom S12/85 FCFU/ml at passage
b:
% of infectious virus bound to cellsc
1 2 3
RV-3 P2A,G3 1.13104 2.23103 0 4.8
vRV-3:3 P2A,G3 1.03104 1.43103 0 3.6
S12/85 P2A,G3 1–11 3.43105 6.13105 7.73105 13.3*
M37 P2A,G1 5.83105 9.03105 1.53106 11.0*
1076 P2A,G2 1.83105 4.13105 5.93105 13.8*
NnB1 P2A,G3 6.03105 1.53105 1.83105 16.8§
r6 P2A,G3 4 1.43106 1.03106 1.63106 16.8§
r26 P2A,G3 4 1.23106 1.03106 1.63106 17.1§
r223 P2A,G3 4 1.53106 1.53106 1.93106 Not done
r31 P2A,G3 4, 10 1.53106 1.53106 1.53106 16.2§
r34 P2A,G3 1 1.23104 0 0 4.6
r214.2 P2A,G3 6, 8, 10 5.23103 900 0 2.7
aAll viruses produced titers between 53105and 13107FCFU/ml after three passages in MA104 cells.
bRV-3 and S12/85 were passaged six times in Caco-2 cells. RV-3 produced no infectious virus in passages 4 to 6. Titers for S12/85 were 7.73105, 4.43105, and
5.93105FCFU/ml in passages 4 to 6, respectively.
cResults are representative of two experiments. Standard errors ranged from 0.9 to 1.8%. Significant differences in binding from that of RV-3 at the 95% level in
the unpaired, two-tailed t test are indicated as follows: *, P50.04; §, P50.02.
on November 9, 2019 by guest
http://jvi.asm.org/
remaining seven strains, which had at least three amino acid differences in VP4 from RV-3, and were of identical or differ-ent G serotype (unpaired, two-tailed t test, 95% confidence interval, 0.025#P#0.047 for binding and 0.0025#P#0.048 for virus replication).
These results show that the ability of a range of cultivable serotype P2A rotaviruses to infect MA104 and Caco-2 cells correlated with the ability of at least 11% of infectious virus present to bind to the cells, and VP4 was critical for S12/85 to bind and infect Caco-2 cells. VP7 did not appear to influence Caco-2 binding and replication. In previous studies, Bass et al. (5) showed that radiolabelled rotaviruses bound to similar ex-tents to both permissive (MA104) and nonpermissive (mouse L) cells, as measured by virus escape from cell surface protease treatment. This difference from our findings may be due to the different viruses, cell lines, and virus detection methods em-ployed. Our results suggest either that RV-3 cannot identify a necessary receptor on Caco-2 cells or that the level or affinity of binding to this receptor is too low for productive infection. This may be because the binding sites necessary on the VP4 protein are altered compared with strain S12/85. It is also possible that RV-3 does not efficiently penetrate or uncoat in Caco-2 cells. However, it is unlikely that the inability of RV-3 and vRV-3:3 to grow in Caco-2 cells was due to a postentry block in virus replication. Firstly, the monoreassortants, con-taining a single gene from S12/85 and the remaining 10 gene segments from RV-3, grew to a titer indistinguishable from that of S12/85 itself, suggesting that RV-3 would replicate adequately in Caco-2 cells if the binding and entry steps are bypassed. Secondly, it is very unlikely that gene 4, encoding the cell attachment protein VP4, which is also required for entry, would also on its own control any later growth restriction. Thirdly, both of the two rotaviruses (rhesus rotavirus RRV and bovine rotavirus UK) tested previously (5) were able to repli-cate in both permissive (MA104 and HT29) and nonpermissive (L and HEp 2) cell lines after lipofection of noninfectious, double-layered virus particles.
In contrast to their binding to Caco-2 cells, RV-3 and S12/85 showed similar abilities to bind to MA104 cells, with 15.7% of input infectious RV3 virus and 13.7% of input infectious S12/85 virus binding to MA104 cells (P50.52). Hence, the cell receptors utilized by these viruses may vary with cell line, as was found for murine rotavirus EHP. The infectivity of EHP in
Caco-2 cells was dependent on the presence of sialic acid on the cell surface but was independent of sialic acid in MA104 cells (31).
The VP4 proteins of fecally derived RV-3 and S12/85 rota-viruses showed 99.1% amino acid identity (28). Upon adaption to growth in MA104 cells, both RV-3 and S12/85 sustained mutations in VP4. The changes in RV-3 were at aa 439 (Leu to Ser) and at aa 466 (Arg to Gly). In S12/85, aa 388 (Ile to Leu) and aa 479 (Tyr to His) were altered (Table 2). Sequence comparisons of the entire VP4 of MA104 cell-adapted S12/85 and NnB1 viruses with RV-3 revealed three conserved amino acid differences at aa 30 (Asp to Ser), 241 (Ser to Ala), and 388 (Leu to Ile). Similar comparisons with M37 and 1076 rotavi-ruses showed conservation of Ser at aa 241 and either Leu (M37) or Met (1076) at aa 388, whereas Ser was present at aa 30 in 1076.
Thus, conserved differences in sequence at a site of trypsin cleavage (aa 241) and in the putative fusion region at aa 388 (34) were consistent with differences in the ability of neonatal rotaviruses to replicate in Caco-2 cells. It is possible that dif-ferences in VP7 sequence between some of these viruses con-tribute to this replication property in some cases. Differences at these positions in VP4 do not correspond to those at aa 133, 303, and 380 in P2A[6],G3 rotaviruses, including stool RV-3 and S12/85, which were implicated in viral virulence in our earlier study (28). Thus, results from in vitro cell culture stud-ies may not necessarily provide information to help explain differences in virulence between rotavirus strains.
[image:3.612.54.547.89.220.2]The efficiency of virus attachment after activation at differ-ent trypsin levels was studied for RV-3 and S12/85 in MA104 and Caco-2 cells. Stocks of RV-3 and S12/85 with uncleaved VP4 were produced by inoculation of MA104 cells with tryp-sin-activated virus for 1 h. The inoculum was removed, and monolayers were washed twice with phosphate-buffered saline before Dulbecco modified Eagle medium containing the pro-tease inhibitors leupeptin and pepstatin at 10 mg/ml and 0.5% (vol/vol) fetal calf serum was added (32). Virus was harvested at 20 h postinfection. Lack of VP4 cleavage was confirmed by polyacrylamide gel electrophoresis and Western blotting with the appropriate hyperimmune antiserum. Virus with uncleaved VP4 was treated with concentrations of trypsin from 0 to 100 mg/ml and then used in the virus-cell binding assay (Fig. 2). RV-3 and S12/85 with uncleaved VP4 bound to MA104 cells TABLE 2. Amino acid sequence differences in the gene encoding VP4 between neonatal rotaviruses with different abilities to attach to and
grow in Caco-2 cells
Straina VP4 sequenceb Ability to grow in
Caco-2 cellsc
RV-3
MA104 cell culture 25IGSEKSQNVTI35 235VALSSRAVTYQRAQVN250 380SGGNYDFQIPVGA392 2
Stool --- --- --- Not done
vRV-3:3 --- --- ---K--- 2
S12/85
MA104 cell culture ---N--- ---S--- I---L---- 1
Stool ---N--- ---S--- I--- Not done
NnB1 ---N--- ---S--- ---L---- 1
M37 ---T--- ---S--- ---N--L---- 1
1076 --- ---S--- ---N--M---- 1
ST-3 --- --- ---N--L---- 1
aFrom MA104 cell culture unless indicated otherwise. The complete nucleotide and deduced amino acid sequences of S12/85 and NnB1 were determined in this study. The complete nucleotide and amino acid sequences of the VP4 of MA104 cell-adapted RV-3, stool-derived RV-3, and stool-derived S12/85 were determined previously (23). The sequence of culture-adapted RV-3 VP4 has GenBank accession no. U16299. VP4 sequences of M37 and 1076 were determined by Gorziglia et al. (21).
bAmino acids conserved between NnB1 and S12/85, but which differ from RV-3, are boldfaced.
cVirus growth (1) was defined as the ability to replicate and produce progeny virus for at least three passages.
on November 9, 2019 by guest
http://jvi.asm.org/
with an efficiency similar to that of virus treated with 10 to 100 mg of trypsin per ml (t test, 0.10 # P # 0.36). The level of S12/85 attachment to Caco-2 cells and to MA104 cells was similar to that of RV-3 to MA104 cells over the range of trypsin levels tested (0.15 # P # 0.83), and a significantly greater percentage of S12/85 virus not treated with trypsin bound to Caco-2 cells than that of S12/85 treated with 50 to 100 mg of trypsin per ml (0.026# P# 0.037). Of most interest, levels of binding of RV-3 to Caco-2 cells were significantly lower than those to MA104 cells at 0 to 50mg of trypsin per ml (0.012#P#0.05) but not at 100mg of trypsin per ml (P5 0.059).
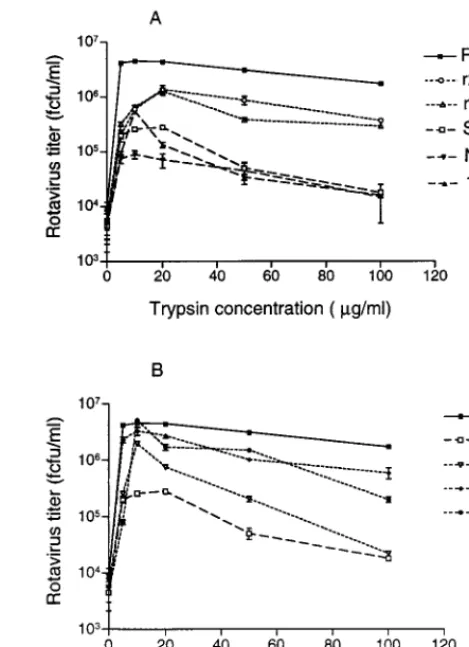
To determine the effect of trypsin on rotavirus growth, virus stocks with intact VP4 were activated with a range of trypsin concentrations at 37°C for 20 min. Confluent monolayers of MA104 cells in 24-well trays were inoculated with activated virus at a MOI of 1, and virus was harvested at 20 h postin-fection. The titer of rotaviruses produced was influenced by the trypsin concentration used for virus activation. The maximum titer of RV-3 was obtained after activation with 10 mg of trypsin per ml (Fig. 3). At 100mg of trypsin per ml, virus yield was reduced to 37.6% of that at 10mg/ml. In contrast, S12/85 showed maximal virus production after activation at a trypsin concentration of 20mg/ml, and at a trypsin concentration of 100mg/ml the S12/85 virus titer was only 1% of the titer at 20 mg/ml. Thus, RV-3 tolerated a much wider range of trypsin concentrations than S12/85, which was sensitive to high levels of trypsin. As shown in Fig. 3A, RV-3 titers at 10 to 100mg of trypsin per ml were significantly different from all the other rotaviruses tested, at the 99% level (t test, 0.0006#P#0.008), whereas S12/85 titers were not significantly different from NnB1 titers (0.01 # P # 0.83) or 1076 titers (0.018# P #
0.31). Thus, like S12/85, NnB1 and 1076 were also very sensi-tive to trypsin levels above 10mg/ml. Reassortants r26 and r223 showed titers indistinguishable from each other at 10 to 100mg trypsin per ml (0.04#P#0.62). The titers of both reassor-tants were significantly different from that of RV-3 (0.001#
P#0.008) but not from that of S12/85 (0.012#P#0.084) at 10 to 50mg of trypsin per ml. At 100mg of trypsin per ml, the titers of r223 (P50.0004) and r26 (P50.0018) were signifi-cantly different from that of S12/85 and from that of RV-3 (0.004#P#0.005). This shows that, with the exception of the highest trypsin level tested, the sensitivities of r223 and r26 to trypsin are similar to that of S12/85 and that this sensitivity is
a property of S12/85 VP4. These results support previous find-ings that cleavage of VP4 with trypsin concentrations up to 25 mg/ml correlated with increasing rotavirus infectivity (2). The reassortant titers also suggest that genes other than VP4 may affect trypsin sensitivity. It is likely that VP4-VP7 interactions, previously reported to affect antigenicity (11), may also affect trypsin sensitivity. Additionally, at elevated trypsin levels, other viral proteins may be trypsin sensitive. For example, solubilized VP7 is cleaved by trypsin and induces permeabilization of cell vesicles (10). We did not see evidence of VP7 cleavage by Western blot at 10mg of trypsin per ml (data not shown).
Community viruses P (P1A[8],G3), RV-4 (P1A[8],G1), and RV-5 (P1B[4],G2) grew to high titer in Caco-2 cells (data not shown). In MA104 cells, all showed an increase in titer with concentrations of trypsin up to 10 mg/ml (Fig. 3B). Further increases in trypsin concentration to 100mg/ml produced virus titer reductions of 98.9% 4), 96.1% (P), and 82.1% (RV-5). P and RV-4 showed significantly lower titers than RV-3 at 20 to 100mg of trypsin per ml (0.0008#P#0.007), and RV-5 showed significantly lower titers at 50mg of trypsin per ml (P5 0.004). Thus, the trypsin resistance and high titers of RV-3 in MA104 cells are unusual among human rotaviruses.
[image:4.612.309.544.71.395.2]Assuming that the VP4 amino acid sequence changes which occurred on adaption of RV-3 and S12/85 to culture, and which were not at or near sites of trypsin cleavage, do not affect the trypsin sensitivity of these viruses, this result may have implications for virus growth in the intestine. Theoretically, it
FIG. 2. Binding of RV-3 and S12/85 rotaviruses activated with various con-centrations of trypsin to MA104 and Caco-2 cell monolayers. Results, expressed as means, are representative of two experiments. Standard errors of the means are indicated.
FIG. 3. Effect of the concentration of trypsin used to activate infectivity on replication of human rotaviruses and their reassortant viruses. Results, expressed as means, are representative of two independent experiments. Standard error of the means are indicated, except for those too small to be visible on the graphs.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.64.273.72.221.2]would be advantageous for a virus to be able to replicate over a wide range of trypsin concentrations, particularly in neonates whose intestinal trypsin levels can vary greatly during the first few weeks of life (4, 35). P2A[6],G3 rotaviruses, of which RV-3 and NnB1 are representative, have been shown to be endemic in obstetric hospital nurseries in Melbourne, Australia, without causing disease for at least 10 years (1). Those rotaviruses like RV-3 may be particularly well suited to growth in the neonatal gut.
Nucleotide sequence accession numbers.The sequences of
S12/85 and NnB1, determined in this study, have been depos-ited in GenBank under accession no. AF076925 and AF076926, respectively.
This study was supported by project grants 940302 and 940315 from the National Health and Medical Research Council of Australia and by the Royal Children’s Hospital Research Foundation.
We are grateful to Jon Gentsch and Luis Padilla-Noriega for the HS11 MAb.
REFERENCES
1. Albert, M. J., L. E. Unicomb, G. L. Barnes, and R. F. Bishop. 1987. Culti-vation and characterization of rotavirus strains infecting newborn babies in Melbourne, Australia, from 1975 to 1979. J. Clin. Microbiol. 25:1635–1640. 2. Arias, C. F., P. Romero, V. Alvarez, and S. Lopez. 1996. Trypsin activation
pathway of rotavirus infectivity. J. Virol. 70:5832–5839.
3. Ball, J. M., P. Tian, C. Q. Zeng, A. P. Morris, and M. K. Estes. 1996. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 272:101–104.
4. Barnes, G. L. 1991. Intestinal viral infections, p. 538–546. In Pediatric gas-trointestinal disease. Marcel Dekker, Inc., New York, N.Y.
5. Bass, D. M., M. R. Baylor, C. Chen, E. M. Mackow, M. Bremont, and H. B.
Greenberg.1992. Liposome-mediated transfection of intact viral particles reveals that plasma membrane penetration determines permissivity of tissue culture cells to rotavirus. J. Clin. Investig. 90:2313–2320.
6. Bass, D. M., E. R. Mackow, and H. B. Greenberg. 1991. Identification and partial characterization of a rhesus rotavirus binding glycoprotein on murine enterocytes. Virology 183:602–610.
7. Bridger, J. C., B. Burke, G. M. Beards, and U. Desselberger. 1992. The pathogenicity of two porcine rotaviruses differing in their in vitro growth characteristics and genes 4. J. Gen. Virol. 73:3011–3015.
8. Bridger, J. C., and D. H. Pocock. 1986. Variation in virulence of bovine rotaviruses. J. Hyg. 96:257–264.
9. Broome, R. L., P. T. Vo, R. L. Ward, H. F. Clark, and H. B. Greenberg. 1993. Murine rotavirus genes encoding outer capsid proteins VP4 and VP7 are not major determinants of host range restriction and virulence. J. Virol. 67:2448– 2455.
10. Charpilienne, A., M. J. Abad, F. Michelangeli, F. Alvarado, M. Vasseur, J.
Cohen, and M. C. Ruiz.1997. Solubilized and cleaved VP7, the outer gly-coprotein of rotavirus, induces permeabilization of cell membrane vesicles. J. Gen. Virol. 78:1367–1371.
11. Chen, D. Y., M. K. Estes, and R. F. Ramig. 1992. Specific interactions between rotavirus outer capsid proteins VP4 and VP7 determine expression of a cross-reactive, neutralizing VP4-specific epitope. J. Virol. 66:432–439. 12. Clark, S. M., J. R. Roth, M. L. Clark, B. B. Barnett, and R. S. Spendlove.
1981. Trypsin enhancement of rotavirus infectivity: mechanism of enhance-ment. J. Virol. 39:816–822.
13. Coulson, B. S., K. J. Fowler, R. F. Bishop, and R. G. Cotton. 1985. Neutral-izing monoclonal antibodies to human rotavirus and indications of antigenic drift among strains from neonates. J. Virol. 54:14–20.
14. Coulson, B. S., S. L. Londrigan, and D. J. Lee. 1997. Rotavirus contains integrin ligand sequences and a disintegrin-like domain that are implicated in virus entry into cells. Proc. Natl. Acad. Sci. USA 94:5389–5394. 15. Coulson, B. S., L. E. Unicomb, G. A. Pitson, and R. F. Bishop. 1987. Simple
and specific enzyme immunoassay using monoclonal antibodies for serotyp-ing human rotaviruses. J. Clin. Microbiol. 25:509–515.
16. Dyall-Smith, M. L., I. Lazdins, G. W. Tregear, and I. H. Holmes. 1986. Location of the major antigenic sites involved in rotavirus serotype-specific neutralization. Proc. Natl. Acad. Sci. USA 83:3465–3468.
17. Espejo, R. T., S. Lopez, and C. Arias. 1981. Structural polypeptides of simian rotavirus SA11 and the effect of trypsin. J. Virol. 37:156–160.
18. Estes, M. K. 1996. Rotaviruses and their replication, p. 1625–1655. In B. N.
Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa.
19. Estes, M. K., D. Y. Graham, and B. B. Mason. 1981. Proteolytic enhance-ment of rotavirus infectivity: molecular mechanisms. J. Virol. 39:879–888. 20. Fukudome, K., O. Yoshie, and T. Konno. 1989. Comparison of human,
simian, and bovine rotaviruses for requirement of sialic acid in hemaggluti-nation and cell adsorption. Virology 172:196–205.
21. Gorziglia, M., K. Green, K. Nishikawa, K. Taniguchi, R. Jones, A. Z.
Kapikian, and R. M. Chanock.1988. Sequence of the fourth gene of human rotaviruses recovered from asymptomatic or symptomatic infections. J. Vi-rol. 62:2978–2984.
22. Greenberg, H. B., J. Flores, A. R. Kalica, R. G. Wyatt, and R. Jones. 1983. Gene coding assignments for growth restriction, neutralization and subgroup specificities of the W and DS-1 strains of human rotavirus. J. Gen. Virol.
64:313–320.
23. Hall, G. A., J. C. Bridger, K. R. Parsons, and R. Cook. 1993. Variation in rotavirus virulence: a comparison of pathogenesis in calves between two rotaviruses of different virulence. Vet. Pathol. 30:223–233.
24. Hoshino, Y., L. J. Saif, S. Y. Kang, M. M. Sereno, W. K. Chen, and A. Z.
Kapikian.1995. Identification of group A rotavirus genes associated with virulence of a porcine rotavirus and host range restriction of a human rotavirus in the gnotobiotic piglet model. Virology 209:274–280.
25. Kalica, A. R., J. Flores, and H. B. Greenberg. 1983. Identification of the rotaviral gene that codes for hemagglutination and protease-enhanced plaque formation. Virology 125:194–205.
26. Keljo, D. J., and A. K. Smith. 1988. Characterization of binding of simian rotavirus SA-11 to cultured epithelial cells. J. Pediatr. Gastroenterol. Nutr.
7:249–256.
27. Kirkwood, C. D., R. F. Bishop, and B. S. Coulson. 1996. Human rotavirus VP4 contains strain-specific, serotype-specific and cross-reactive neutraliza-tion sites. Arch Virol. 141:587–600.
28. Kirkwood, C. D., B. S. Coulson, and R. F. Bishop. 1996. G3P2 rotaviruses causing diarrhoeal disease in neonates differ in VP4, VP7 and NSP4 se-quence from G3P2 strains causing asymptomatic neonatal infection. Arch. Virol. 141:1661–1676.
29. Kobayashi, N., K. Kojima, K. Taniguchi, T. Urasawa, and S. Urasawa. 1994. Genotypic diversity of reassortants between simian rotavirus SA11 and hu-man rotaviruses having different antigenic specificities and RNA patterns. Res. Virol. 145:303–311.
30. Lazdins, I., S. Sonza, M. L. Dyall-Smith, B. S. Coulson, and I. H. Holmes. 1985. Demonstration of an immunodominant neutralization site by analysis of antigenic variants of SA11 rotavirus. J. Virol. 56:317–319.
31. Ludert, J. E., N. Feng, J. H. Yu, R. L. Broome, Y. Hoshino, and H. B.
Greenberg.1996. Genetic mapping indicates that VP4 is the rotavirus cell attachment protein in vitro and in vivo. J. Virol. 70:487–493.
32. Ludert, J. E., A. A. Krishnaney, J. W. Burns, P. T. Vo, and H. B. Greenberg. 1996. Cleavage of rotavirus VP4 in vivo. J. Gen. Virol. 77:391–395. 33. Mackow, E. R., J. W. Barnett, H. Chan, and H. B. Greenberg. 1989. The
rhesus rotavirus outer capsid protein VP4 functions as a hemagglutinin and is antigenically conserved when expressed by a baculovirus recombinant. J. Virol. 63:1661–1668.
34. Mackow, E. R., R. D. Shaw, S. M. Matsui, P. T. Vo, M. N. Dang, and H. B.
Greenberg.1988. The rhesus rotavirus gene encoding protein VP3: location of amino acids involved in homologous and heterologous rotavirus neutral-ization and identification of a putative fusion region. Proc. Natl. Acad. Sci. USA 85:645–649.
35. McLean, B. S., and I. H. Holmes. 1981. Effects of antibodies, trypsin, and trypsin inhibitors on susceptibility of neonates to rotavirus infection. J. Clin. Microbiol. 13:22–29.
36. Offit, P. A., G. Blavat, H. B. Greenberg, and H. F. Clark. 1986. Molecular basis of rotavirus virulence: role of gene segment 4. J. Virol. 57:46–49. 37. Padilla Noriega, L., R. Werner Eckert, E. R. Mackow, M. Gorziglia, G.
Larralde, K. Taniguchi, and H. B. Greenberg.1993. Serologic analysis of human rotavirus serotypes P1A and P2 by using monoclonal antibodies. J. Clin. Microbiol. 31:622–628.
38. Rodger, S. M., R. F. Bishop, C. Birch, B. McLean, and I. H. Holmes. 1981. Molecular epidemiology of human rotaviruses in Melbourne, Australia, from 1973 to 1979, as determined by electrophoresis of genome ribonucleic acid. J. Clin. Microbiol. 13:272–278.
39. Tzipori, S., L. Unicomb, R. Bishop, J. Montenaro, and L. M. Vaelioja. 1989. Studies on attenuation of rotavirus. A comparison in piglets between virulent virus and its attenuated derivative. Arch. Virol. 109:197–205.
40. Vachon, P. H., and J. F. Beaulieu. 1992. Transient mosaic patterns of mor-phological and functional differentiation in the Caco-2 cell line. Gastroen-terology 103:414–423.
![FIG. 1. Correlation of binding of infectious, MA104 cell-adapted, serotypeP2A[6] rotaviruses to Caco-2 cells with the titer of virus produced after a single](https://thumb-us.123doks.com/thumbv2/123dok_us/1772.75131/2.612.51.548.82.225/correlation-binding-infectious-adapted-serotypep-rotaviruses-produced-single.webp)