Acknowledgments
Thank you to Professor Wei You for your direction, support, and edits.
Thank you to Professors Michael Rubinstein and Jillian Dempsey for being part of my honors thesis committee.
Thank you to Travis LaJoie and Wentao Li for all of your edits, synthesis help, advice, and support; and again to Wentao for conducting the FTAZ functionalization and click reaction.
Thank you to Adam Alman for your CV expertise.
Abstract. Conjugated polymers are promising materials for next generation solar cells.
However, a disadvantage of polymer-based solar cells is that most polymers have a limited light-absorbing range. This research aims to investigate the addition of a side-chain light-light-absorbing molecule to the existing highly efficient polymer PBnDT-FTAZ as a method of increasing the polymer’s light-absorbing range, with the ultimate goal of achieving a higher efficiency. Two chromophores, TT-DPP-TT and T-SBTa-T, were synthesized and investigated as possible candidates to expand the light-absorbing range of PBnDT-FTAZ. Using UV-Vis absorption and electrochemistry, T-SBTa-T was found to be the more suitable chromophore, while TT-DPP-TT’s band gap was found to be too large to effectively expand the absorption range of PBnDT-FTAZ. The polymer PBnDT-FTAZ with a T-SBTa-T side chain is currently being synthesized.
1. Introduction
Polymer-based solar cells are a very promising alternative approach to harvest solar energy, with a projected low cost of fabrication. Inside the solar cell, the conjugated polymer is typically mixed with a fullerene derivative called PCBM (phenyl C60 butyric acid methyl ester)
to form a bulk heterojunction layer (BHJ). Polymers can be tailored to absorb a specific range of light by adjusting their HOMO and LUMO levels.
When a conjugated polymer absorbs light, an electron is promoted from its HOMO to its LUMO, forming an exciton via photoexcitation. The exciton then travels to the interface
between the polymer and the PCBM, where upon exciton dissociation, the electron is transferred to the LUMO of PCBM, leaving a hole in the HOMO of the donor polymer. The now-separated charges move toward electrodes, where they perform work and re-enter the solar cell to continue the process.1
The difference in the energies of the polymer’s HOMO and LUMO energy levels is its band gap, which determines the range of light the polymer can absorb; the smaller the band gap, the less energy the polymer needs to promote an electron from its HOMO to its LUMO, so it can absorb light with higher
wavelengths than a polymer with a larger band gap. Often, the range of light that a polymer can absorb is narrower than many inorganic materials, which is a major disadvantage of polymer-based solar cells.
To increase this light-absorbing range of polymers, three strategies are typically employed. On the device level, the films of two different
polymers with different – ideally complementary – light-absorbing ranges can be deposited on top of
each other to form a tandem cell, although this is difficult to process and requires many
fabrication steps.2 On the material processing level, the two polymers can be mixed together, but
the mixing of polymers in a BHJ is not well understood and the selection rationale is not very clear.3 However,
on the molecular level, a polymer can be synthesized to contain
complementary units that absorb light of wavelengths outside of the main polymer’s light-absorbing range. In this work, we chose to investigate the third approach.
Specifically, we chose to extend the light-absorbing range of the conjugated polymer PBnDT-FTAZ (FTAZ) (Figure 1). FTAZ has a relatively large band gap with a light-absorbing range of 450-650 nm; however, with a high hole mobility and other desirable attributes, FTAZ can achieve excellent solar cell device performance, with a power conversion efficiency (PCE) as high as 7.1%.4 These characteristics
make it an ideal candidate for modification.
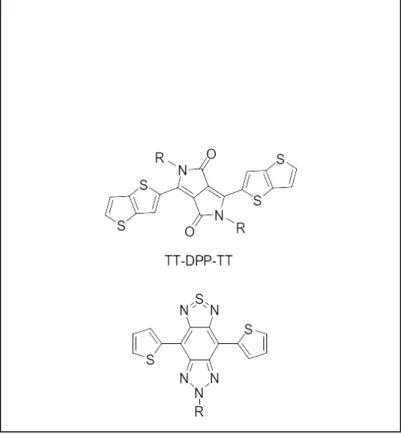
Two chromophores were chosen as candidates for side-chain modifiers on FTAZ: TT-DPP-TT and T-SBTa-T (Figure 2). These chromophores were selected because they form very low-band gap polymers that absorb light of longer wavelengths than FTAZ (Figure 3). As a side chain on FTAZ, they would theoretically absorb light of longer wavelengths than the light absorbed by the FTAZ polymer backbone, thereby extending the polymer’s light-absorbing range, provided that the blue-shift in absorbance from polymer to monomer is not too extreme.
Figure 2. TT-DPP-TT (top) and T-SBTa-T
2. Experimental Details
The synthesis of each chromophore was completed according to literature procedures with some modifications. 1H NMR spectra of the molecules synthesized in each step of the
syntheses can be seen in the Appendix in Figures 1A-8A.
TT-DPP-TT
In Scheme 1, the synthesis of TT-DPP-TT, designed to be attached to the BnDT
monomer which would then be polymerized with FTAZ (see Appendix Scheme 1A), is outlined. Figure 4 shows the proposed polymer P1 with FTAZ backbone and TT-DPP-TT side chain. The synthesis of P1 was abandoned after we finished the synthesis of monoalkylated TT-DPP-TT (F) due to low yields and studies discussed below that showed that TT-DPP-TT was a poor
candidate for a side-chain chromophore on FTAZ.
Scheme 1. Synthesis of TT-DPP-TT
Synthesis of thieno[3,2-b]thiophene-2-carboxylic acid
In a round-bottomed flask with a condenser and stir bar, 18.667 g (84.67 mmol) of ethyl
thieno[3,2-b]thiophene-2-carboxylate (A) was dissolved in DMAc, making an orange solution. An aqueous solution of NaOH was added slowly until the pH reached 10/11, causing an
immediate color change to red. The solution was refluxed for two days, after which the pH was found to be around 6. At 0 °C, HCl (aq) was added until the solution had a pH<1. The solution was then refrigerated overnight. Tan crystals of product (B) precipitated out and were collected by vacuum filtration multiple times. The prodcut weighed 7.4466 g, giving a 47.7% yield. 1H
Synthesis of thieno[3,2-b]thiophene-2-carboxamide
Approximately 30 mL of SOCl2 was
added slowly to 2.8802 g (15.63 mmol) of (B) in a round-bottomed flask with a condenser and stir bar. The solution was heated to reflux for 3.5 hours, resulting in a dark-orange solution. Excess SOCl2 was
evaporated off, leaving a white and orange solid. Next, 72 mL of DCM was added to the solid, which was then added dropwise to a solution of 87 mL of 30% NH4OH(aq) and 117
mL of DCM in an ice/acetone bath. Product (C), a white precipitate, formed. The solution was stirred for 20 minutes. The precipitate was filtered, washed with water, and heated on vacuum overnight. It had a mass of 2.0912 g, giving a yield of 73.1%. The product was not further purified. The product was characterized by 1H NMR, but the J coupling constants were not calculated because δ values
are rough estimates. 1H NMR (CDCl
3, 600 MHz): δ 8.10 (b, 1H), 8.07 (s, 1H), 7.80 (d, 1 H),
7.53 (b, 1H), 7.45 (d, 1H).
Synthesis ofthieno[3,2-b]thiophene-2-carbonitrile
In a round-bottomed flask with a condenser and stir bar, 3.111 g (16.98 mmol) of (C) and 43 mL of POCl3 were refluxed for two hours. The cloudy tan solution became dark red/brown and
clear. The reaction mixture was added dropwise to 100 mL of ice water, turning a cloudy brown. This aqueous solution was extracted with chloroform and the organic layer was dried with MgSO4. The resulting red solution was run through a short silica gel plug with chloroform as the
mobile phase. A yellow oil was obtained, which when dried became an off-white solid (D) weighing 2.5 g, giving a yield of 89%. 1H NMR (CDCl
3, 400 MHz): δ 7.798 (s, 1H), 7.711 (d,
1H, J = 5.2), 7.298 (d, 1H, J = 5.2).
Cyclization: Synthesis of TT-DPP-TT
To a dry, argon-filled 100-mL 2-necked round-bottomed flask with a stir bar, 1.827 g (16.29 mmol, 1.5 equiv.) of t-BuOK and 18 mL of t-BuOH were added. The solution was heated to 60 °C to dissolve the base. Then, 1.7941 g (10.86 mmol, 1.0 equiv.) of (D) was dissolved in 9 mL of t-BuOH using heat and sonication. This solution was added to the 2-necked flask, forming a cloudy tan precipitate. The solution was warmed to 85 °C for 20 minutes and 5 mL of t-BuOH was added to dissolve the precipitate. In 4 mL of t-BuOH, 1.11 mL (5.429 mmol, 0.5 equiv.) of diisopropyl succinate was added. The solution was stirred at 100 °C overnight. The next day, the solution was viscous and dark purple. To the reaction mixture, 36 mL of methanol and 9 mL of acetic acid were added. The solution was then filtered. The precipitate was washed with
water and hot methanol. The final product (E) was a dark purple solid that weighed 1.062 g, giving a yield of 47.6%. (E) was not characterized by NMR due to poor solubility.
Monoalkylation of TT-DPP-TT
In a 25-mL 2-necked flask with a stir bar, 0.2146 g (0.523 mmol, 1.0 equiv.) of (E), 0.4449 g (1.60 mmol, 3.06 equiv.) of 5-(bromomethyl)tridecane, 0.2045 g (0.628 mmol, 1.2 equiv.) of CsCO3 and 12 mL of DMF were added. The solution was purged with argon for 10 minutes.
The solution was a deep turquoise color. It was heated at 60 °C for 36 hours, after which the solution was dark gray-purple. The solution was cooled to room temperature. Chloroform was added to the solution, which was then washed with water three times, dried with MgSO4, and
evaporated. However, a crude NMR revealed a lot of remaining DMF in the product, so it was precipitated out of methanol. The crude product, a very dark purple solid, was purified with a silica gel column with a 1:1 hexanes:CHCl3 by volume mobile phase. The recovered product
was shown by NMR to be dialkylated TT-DPP-TT, a blue solid, so the column was flushed with CHCl3 to recover the desired monoalkylated product (F). (F) was a purple solid with a mass of
0.0408 g, giving a yield of 12.8%, but was not characterized by NMR due to poor solubility. Dialkylated product, however, was characterized. 1H NMR (CDCl
3, 400 MHz): δ 9.277 (s, 2H),
7.597 (d, 2 H, J = 3.6), 7.346 (d, 2H, J = 3.6), 4.068 (d, 4H, J = 5.2), 1.989 (b, 2H), 1.228 (m, 32H), 0.834 (m, 20H).
(Note: multiple reaction conditions were explored for this reaction once the first attempt gave a low yield. Different equivalents of bromide and different bases [CsCO3 and KtBuO-] were added
at different times after heating the reaction mixture for different durations. It was found that dissolving TT-DPP-TT in DMF results in a purple solution, which after adding the base becomes turquoise. A column in 100% chloroform mobile phase was established to best separate di- and monoalkylaed TT-DPP-TT. However, no other reaction conditions gave higher yields.)
T-SBTa-T
Multiple routes were explored to synthesize the other side-chain chromophore, T-SBTa-T. Ultimately, the route outlined in Scheme 2 was chosen, in which T-SBTa-T was
functionalized with a terminal alkene so that it could be attached to the FTAZ monomer using the robust thiol-ene click reaction, and then polymerized with BnDT . Figure 5 shows the proposed polymer P2 with FTAZ backbone and T-SBTa-T side chain. The synthesis of P2 was continued after studies discussed below that showed that T-SBTa-T was a good candidate for a side-chain chromophore on FTAZ.
Nitration: Synthesis of 4,7-dibromo-5,6-dinitrobenzo[c][1,2,5]thiadiazole To two 2-necked 25-mL round-bottomed flask with medium-sized stir bars, 0.70 mL (15.1 mmol, 2.8 equiv.) of fuming HNO3 was added to 5.50 mL (62.5
mmol, 11.6 equiv.) of CF3SO3H at 0 °C.
A white solid formed in both flasks. The reactions were taken off of ice, and 1.6 g (5.4 mmol, 1 equiv.) of
4,7-dibromobenzo[c][1,2,5]thiadiazole (A) was added to each reaction flask in four portions over 20 minutes. The solutions became viscous, mustard-yellow liquids. The reactions were stirred at 50 °C for four days. The reactions were then taken off heat and added dropwise to an
aqueous NaOH solution on ice. The final solution was orange and had a pH of between 3 and 5. The crude product was vacuum filtered from the solution and recrystallized from 160 mL of hot ethanol. The pure product was found to be a yellow crystal with a mass of 3.008 g, giving a yield of 72.0%. The product was
characterized by 1H NMR, which contains no peaks to report.
Stille Coupling: Synthesis of 5,6-dinitro-4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole To a flame-dried, 100-mL 2-necked flask with a condenser and stir bar under argon, 4.16 g (16.8 mmol, 2.20 equiv.) of trimethyl(thiophen-2-yl)stannane was added by syringe. This was
degassed under vacuum. Under argon, 2.9342 g (7.64 mmol, 1.0 equiv.) of (B), 0.269 g (0.383 mmol, 0.05 equiv.) of Pd(PPh3)2, and 50 mL of anhydrous toluene were added. The solution was
purged with argon for ten minutes. The solution was then heated to reflux over two nights. A color change from cloudy yellow-orange to dark red shortly after heating. The reaction was worked up with brine and DCM, dried under MgSO4, and purified using a silica gel column with
a mobile phase of 1:1 DCM/hexanes by volume. The product (C) was a red-orange solid with a mass of 2.1 g, giving a 70.4% yield. 1H NMR (CDCl
3, 400 MHz): δ 7.749 (dd, 2H, J = 4.8, 0.8),
7.524 (dd, 2H, J = 3.6, 1.2), 7.243 (dd, 2H, J = 5.2, 3.6).
Reduction: Synthesis of 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole-5,6-diamine To a 500-mL flask with a stir bar, 2.1 g (5.38 mmol, 1.0 equiv.) of (C), 6.0 g (107 mmol, 19.9 equiv.) of spherical iron powder, and 80 mL of acetic acid were added. The solution was stirred at 70 °C for 4.5 hours. A color change from brown-orange to lime green occurred shortly after heating. The reaction was poured into an aqueous solution of NaOH over ice. The final solution had a pH of around 10. The product was partially extracted with ethyl acetate, but the excess iron powder made this work-up extremely difficult. DCM was then poured into the solution and the organic layer was collected through vacuum filtration to remove the iron. The organic phase was washed with NaHCO3 and brine and dried over MgSO4. The crude product (D) consisted of
purified. 1H NMR (CDCl
3, 400 MHz): δ 7.568 (d, 2H, J =5.2), 7.371 (d, 2H, J = 3.6), 7.267 (dd,
2H, J = 8.0, 2.8), 4.439 (b, 4H).
Ring Closing: Synthesis of T-SBTa-T
In a 250-mL round-bottomed flask with a condenser and stir bar, 1.7 g (5.15 mmol, 1.0 equiv.) of (D) was dissolved in 40 mL of THF and 25 mL of acetic acid and heated to 80 °C. In portions, 0.7135 g (10.3 mmmol, 2.0 equiv.) of NaNO2 was added. The solution boiled and underwent a
color change from dark brown to red to dark pink as more NaNO2 was added. The solution was
stirred at 80 °C overnight. Without neutralizing, the reaction was worked up with water and ether, and the organic layer was then washed with NaHCO3 and brine. The crude product (E)
was a dark blue-purple solid (dark pink in solution) with a mass of 1.1 g and was not further purified, giving a crude yield of 62.5%. The product was not characterized by NMR due to poor solubility. (Note: I have also done this reaction on a smaller, 1.1-mmol scale, and gotten a crude yield of 97%.)
Alkylation: Synthesis of T-SBTa-T with alkene chain
In a 250-mL round-bottomed flask with a stir bar, 1.1 g (3.22 mmol, 1.0 equiv.) of (E) was dissolved in about 80 mL of DMF. 0.89 g (6.44 mmol, 2.0 equiv.) of solid K2CO3 and 0.52 mL
(3.89 mmol, 1.21 equiv.) of 6-bromo-1-hexene were added. The solution underwent a color change from dark purple to dark turquoise upon addition of the base. The solution was stirred at room temperature overnight. The next day, the reaction was determined by TLC to be
incomplete and low yielding. Theorizing that the base had been ineffective, 0.90 mL (6.45 mmol, 2.0 equiv.) of TEA was added. An hour and a half later, the reaction was determined by TLC to have remained unchanged, and 0.63 g (5.61 mmol, 1.74 equiv.) of stronger base,
potassium tert-butoxide was added. The solution immediately turned forest green. The solution was stirred over the weekend at 45 °C. When next tested by TLC, the reaction had not gone any farther toward completion. Therefore, 1.00 mL (7.48 mmol, 2.32 equiv.) of 6-bromo-1-hexene was added and 45 minutes later, the reaction had started producing product again. To ensure a high yield,1.5 mL (11.22 mmol, 3.48 equiv.) more of the bromide was added, and within an hour, the reaction was deemed to have gone to completion by TLC. The reaction was worked up using brine and ether. The ether layer was rinsed with brine twice to remove as much excess DMF as possible and condensed. DMF still remained in the crude product, which was dried with a stream of air for two days. The crude product was purified using column chromatography with a mobile phase of 1:7 ethyl acetate:hexanes by volume. The pure product (F) was found to be a dark blue solid (blue in solution) with a mass of 0.11 g, giving a yield of 7.7 %. 1H NMR
(CDCl3, 400 MHz): δ 8.743 (d, 2H, J = 2.8), 7.609 (d, 2H, J = 4.4), 7.288 (dd, 2H, J = 5.2, 3.6),
Scheme 3. Functionalization of FTAZ and click reaction with T-SBTa-T
As seen above in Scheme 3, FTAZ was functionalized by adding a thiol chain to the triazole ring. This thiol chain was then reacted with the alkene chain on the T-SBTa-T monomer synthesized in Scheme 2. The results of this reaction are discussed below. The proposed final polymerization reaction to form P2 can be seen in the Appendix (Scheme 2A).
3. Results and Discussion
UV-2600 UV/Vis spectrophotometer. Electrochemical properties of the same compounds were measured using cyclic voltammetry (CV) on a BASI E2 potentiostat. Dialkylated TT-DPP-TT and T-SBTa-T with an alkene chain were the molecules used in these studies.
Optical measurements allowed for an estimation of the increase in FTAZ’s
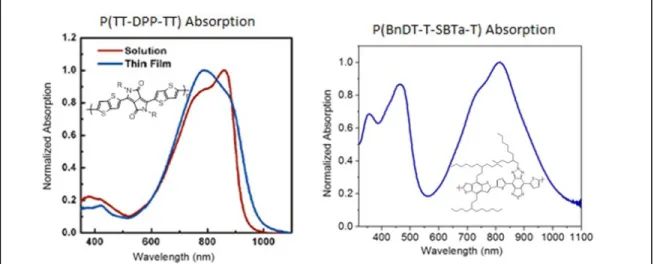
light-absorbing range from adding each chromophore as a side chain. Figure 6 compares the film absorption spectra of FTAZ and TT-DPP-TT. TT-DPP-TT’s absorption spectrum was much more blue-shifted than would be expected from its polymer’s spectrum. The difference between light-absorbing ranges of TT-DPP-TT and its polymer are most likely due to
conjugations effects seen in P(TT-DPP-TT) that are not present in the monomer, and adding TT-DPP-TT as a monomer side chain to FTAZ is not expected to significantly red-shift its absorption. Figure 6 clearly shows that adding TT-DPP-TT as a side chain to FTAZ will not significantly extend the light range of the polymer.
Figure 7 compares the film absorption spectra of FTAZ and T-SBTa-T. The film absorption of T-SBTa-T is about 200 nm more red-shifted than FTAZ’s, and it is therefore likely that as a side-chain chromophore, it will extend the polymer’s light absorbance from 450-650 nm to 450-800 nm, a significant increase. Therefore, T-SBTa-T was deemed to be the most likely candidate for a useful side-chain chromophore on FTAZ.
The results of both the optical and electrochemistry
measurements of FTAZ and the chromophores are summarized Figure 6. Absorption spectra of FTAZ and TT-DPP-TT
below in Tables 1-3. TT-DPP-TT’s LUMO level could not be determined from CV measurements, so it was calculated from the optical band gap.
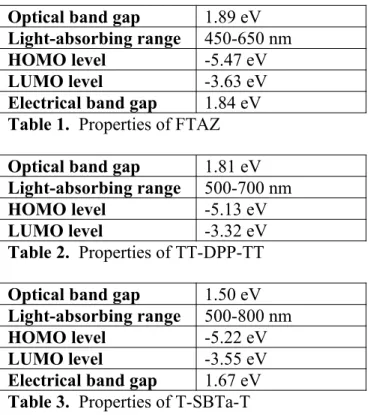
Optical band gap 1.89 eV Light-absorbing range 450-650 nm
HOMO level -5.47 eV
LUMO level -3.63 eV
Electrical band gap 1.84 eV Table 1. Properties of FTAZ Optical band gap 1.81 eV Light-absorbing range 500-700 nm
HOMO level -5.13 eV
LUMO level -3.32 eV
Table 2. Properties of TT-DPP-TT Optical band gap 1.50 eV Light-absorbing range 500-800 nm
HOMO level -5.22 eV
LUMO level -3.55 eV
Electrical band gap 1.67 eV Table 3. Properties of T-SBTa-T
The CV data also supports the selection of T-SBTa-T as the more promising side-chain chromophore. The electrical band gap of T-SBTa-T was found to be significantly smaller than both the electrical band gap of FTAZ and the optical band gap of TT-DPP-TT. This suggests that it would significantly extend the light-absorbing range of FTAZ as a side chain.
The results of the click reaction between FTAZ and T-SBTa-T of Scheme 3 above are not entirely clear. The reaction mixture was purified using column chromatography, and the product was identified using solution UV-Vis spectrometry on the same instrument mentioned above; because of the small scale of the reaction, there was not enough product for NMR characterization.
The product was identified by comparing the absorption spectra of the staring materials of the click reaction, FTAZ and T-SBTa-T, with that of the product. As can be seen in Figures 8 and 9, the product’s absorption spectrum has similar features to both starting materials’ spectra. There is a peak around 600 nm, indicating the presence of the T-SBTa-T monomer in the product. There is also a peak between 300 and 400 nm that looks more similar to the shape of FTAZ’s peak than T-SBTa-T’s 350-nm peak, seen by the presence of the shoulder on the product’s peak. This indicates that the product also contains the FTAZ monomer. Therefore, the tentative conclusion of this analysis is that the click reaction was successful, although the reaction must be redone on a larger scale in order to characterize the product definitively using NMR.
4. Conclusions
Due to its smaller band gap and more red-shifted light-absorbing range, T-SBTa-T was deemed to be a good candidate for a side-chain chromophore on the polymer FTAZ, while TT-DPP-TT’s band gap is too large and its light-absorbing range is too similar to FTAZ’s to be useful. Therefore, the synthesis of the proposed polymer P2, consisting of FTAZ with a T-SBTa-T side chain, was selected to be continued. Future work on this project includes polymerizing the thiol-ene click reaction product with BnDT to form P2 and measuring the properties of the final polymer in a solar cell. A few of the steps in the synthesis of T-SBTa-T with an alkene chain and in the FTAZ functionalization also need to be reexamined and modified for higher yields.
This research explored the effectiveness of attaching a low band-gap side-chain
chromophore to a polymer to increase its light-absorbing range in a solar cell, with final results pending. It also developed new methods of synthesizing a chromophore functionalized for side-chain attachment to a polymer backbone, which can be extended to other chromophores and polymer backbones in future work. The thiol-ene click chemistry reaction in particular has potential to be useful in future attachment of chromophore to polymer due to its robust nature and the relatively simple functionalization it requires of the reactants involved. Further projects along this same line of work include those that experiment with conjugated side chains, other side-chain chromophores, and other polymer backbones.
5. References
2. You, J.; Dou, L.; Yoshimura, K.; Kato, T.; Ohya, K.; Moriarty, T.; Emery, K.; Chen, C.-C.; Gao, J.; Li, G.; Yang, Y. Nat. Commun. 2013, 4, 1446−55.
3. Yang, L.; Yan, L.; You, W. J. Phys. Chem. Lett. 2013, 4, 1802−1810.
4. Price, S. C.; Stuart, A. C.; Yang, L.; Zhau, H.; You, W. J. Am. Chem. Soc. 2011, 133 (12), 4625-4631.
Appendix




![Figure 1A. 1 H NMR spectrum of thieno[3,2-b]thiophene-2-carboxylic acid](https://thumb-us.123doks.com/thumbv2/123dok_us/8331983.2210573/17.918.141.799.114.578/figure-a-nmr-spectrum-thieno-thiophene-carboxylic-acid.webp)
![Figure 2A. 1 H NMR spectrum of thieno[3,2-b]thiophene-2-carboxamide](https://thumb-us.123doks.com/thumbv2/123dok_us/8331983.2210573/18.918.136.793.104.595/figure-a-h-nmr-spectrum-thieno-thiophene-carboxamide.webp)
