Organocatalytic Fluoroalkylation of Polyaromatics
By
Bradley Wilhelmy Jr.
Senior Honors Thesis Department of Chemistry
University of North Carolina at Chapel Hill
2/20/2019
Approved:
Abstract:
Commodity polymers, such as polystyrene and polycarbonate, are ubiquitous in society with 40.4Mt produced in 2015.1 Unfortunately, due to their mass production and longevity in the environment, which stems from their lack of reactivity, these polymers are often found as pollutants. Even when recycled, these materials are difficult to sort and separate leading to heterogenous products with decreased material properties and value. To circumvent this “downcycling” of plastic waste, my research team has studied a post-polymerization modification method to leverage the scale of commodity plastics while imparting value to recycled waste by chemically altering the polymer structure, thus incentivizing recycling. This type of recycling, where value is added to the plastic waste, is called “upcycling.”
demonstrate the range of properties that can be given to a simple polystyrene starting material. The effects of these modifications on physical properties such as contact angle and the glass transition temperature will also be measured in future work. Finally, the substrate scope of our method was expanded to other commodity polymers.
Introduction:
In today’s society, it may be impossible to go a day without coming into contact with some form of plastic material. An estimated 380Mt were produced in 2015 alone, and greater than 7800Mt have been produced since the 1950s.1 Unfortunately, this means a large amount of plastic waste is produced every year. Recycling is a possible solution to this issue, however recycling often creates a low-quality material. Unless recycling can displace plastic production altogether, it only delays waste generation. Upcycling—through post polymerization modification—is a viable alternative that takes post-consumer waste and imparts new value to the material. The challenge in upcycling is that commodity materials are ubiquitous in part due to their chemical stability, making them challenging substrates for further functionalization. Thus, we have utilized a C–H functionalization route that views unreactive C–H bonds as points of diversification.
initiation sites for grafted comb copolymers which have the ability to act as compatibilizing agents with uses in recycling plastic wastes.
Previous, attempts to functionalize polyaromatics have involved Friedel-Crafts chemistry and utilizing perfluoroacyl peroxides (PFAPs) to generate electrophilic radicals. These methodologies are able to impart tunable functionality to the parent material, but at the cost of unwanted chain scission and chain coupling events.3 Additionally, PFAPs in particular are difficult to work with due to their short half-lives and challenging synthesis.
Bypassing the issues previous methods encountered, photoredox catalysis has been shown as a simple, effective method to functionalize aromatic materials.3 Stephenson and coworkers have shown a mechanism that involves the in situ formation of an adduct from pyridine N-oxide and trifluoroacetic acid anhydride (TFAA) which is able to produce an electrophilic radical species upon single electron reduction by a photocatalyst.4 The incorporation of a photocatalyst is an important distinguishing factor between this methodology and those utilizing PFAPs which access electrophilic radicals through heat. Work in the Leibfarth lab has shown that this methodology can also be applied to polymer substrates without significant changes to the molecular weight distribution. It is also able to access many radical species including bromodifluoromethyl groups which can undergo subsequent reactions to modify material properties. This technique is benefitted by its operational simplicity and relatively low-cost reagents; however, it employs a ruthenium catalyst which is more expensive than an organic catalyst and presents the possibility of metal contamination in the final product.
photocatalysts with similar qualities but the advantage of lower operational costs and greater sustainability. Specifically, phenothiazine, phenoxazine, and phenazine derivatives were tested in this work because they are able to replicate the qualities that are desired in an inorganic photocatalyst.5,6 When compared to Ru(bpy)3Cl2, which has a triplet state reduction potential (E0*) of -0.81 V vs. SCE, organic photocatalysts such as 5,10 (2-naphthyl) phenazine and 3,7 biphenyl 1-naphthalene-10-phenoxazine have E0* values of -1.69 and -1.80 V vs. SCE respectively. The reduction potential of these two photocatalysts is attractive as the reduction onset for a pyridine N -oxide-TFAA adduct is -0.86V vs SCE.4 These two catalysts also show promising triplet excited state lifetimes of 4.3 and 480µs compared to Ru(bpy)3Cl2’s 1.1µs.5
Herein, we present a methodology using organic photoredox catalysts to perform post-polymerization modifications on polyaromatic substrates to demonstrate their potential for use in upcycling.
Procedure:
Synthesis of Phenyl Phenothiazine:
mixture was then rinsed twice with deionized water, brine, dried with MgSO4 and concentrated under vacuum. The product was recrystallized from DCM/MeOH at -25℃
and confirmed by 1H NMR matching literature spectra. 6 Synthesis of 1-Napthyl Phenothiazine:
An 8mL septa-capped reaction vial was flame dried under vacuum and charged with phenothiazine (200mg, 1.0mmol, 1.0 equiv.), 1-bromonaphthalene (0.28mL, 2.0mmol, 2.0 equiv.), and sodium tert-butoxide (192mg, 2.0mmol, 2.0 equiv.). The vial was then submitted to a nitrogen filled glovebox where bis(dibenzylideneacetone)palladium(0) (5.77mg, 0.01mmol, 0.01 equiv.), tri-tertbutyl phosphine (6.09mg, 0.03mmol, 0.03 equiv.), and 1.5mL toluene were added. The vial was then sealed, removed from the glovebox, and submitted to a 100˚C heating block for 24 hours. Afterwards the reaction was allowed to cool to room temperature, diluted in DCM, and ran through a short silica plug. The reaction mixture was then rinsed twice with deionized water, brine, dried with MgSO4 and concentrated under vacuum. The product was recrystallized from DCM/MeOH at -25℃
and confirmed by 1H NMR matching literature spectra.6 Synthesis of 5,10 dihydrophenazine:
A 250mL round bottom flask was charged with 75mL deionized water and 18.75mL EtOH then sparged with nitrogen for 30 minutes. Phenazine (750mg, x mmol, 1 equiv.) and Na2S2O4 (8750mg, x mmol, 10 equiv.) were then added and the solution was refluxed under nitrogen for 3 hours. The product was then isolated via cannula filtration and placed under reduced pressure for 48 hours before being stored in a nitrogen filled glovebox.
An 8mL reaction vial was flame dried under vacuum and filled with sodium tert-butoxide (384mg, 4 mmol, 4 equiv.) then submitted to a nitrogen filled glovebox. Inside, 5,10 dihydro phenazine (180 mg, 1 mmol, 1 equiv.), RuPhos Pd G2 (31.1mg, 0.04mmol, 0.04 equiv.), RuPhos (18.6mg, 0.04mmol, 0.04 equiv.) were added and the sealed vial was removed. A nitrogen sparged solution of bromobenzene (0.48mL, 4 mmol, 4 equiv.) and 1.5mL dioxane was then added and the vial submitted to a 110℃ heating block for 24 hours. The reaction mixture was then allowed to cool and rinsed with deionized water three times, once with brine, then dried with MgSO4 before concentrating under reduced pressure. The product was then purified by layering hexanes on top of a solution of product dissolved in hexanes. NMR matched previously reported spectra.7
Synthesis of 5,10 (2-naphthyl) phenazine:
An 8mL reaction vial was flame dried under vacuum and filled with sodium tert-butoxide (384mg, 4 mmol, 4 equiv.) and 2-bromonaphthene (828mg, 4mmol, 4 equiv.) then submitted to a nitrogen filled glovebox. Inside, 5,10 dihydro phenazine (180 mg, 1 mmol, 1 equiv.), RuPhos Pd G2 (31.1mg, 0.04mmol, 0.04 equiv.), RuPhos (18.6mg, 0.04mmol, 0.04 equiv.) were added and the sealed vial was removed. A nitrogen sparged solution of 2.0mL dioxane was then added and the vial submitted to a 110℃ heating block for 24 hours. The reaction mixture was then allowed to cool and deionized water and DCM were added to the reaction mixture, causing the product to precipitate. NMR matched previously reported spectra.7
An 8mL reaction vial was flame dried under vacuum and charged with phenoxazine (183mg, 1mmol, 1 equiv.) and sodium tertbutoxide (192mg, 2 mmol, 2 equiv.) before submitting to a nitrogen filled glovebox. Inside, RuPhos (12mg, 0.03mmol, 0.03 equiv.) and RuPhos Pd G2 (21mg, 0.03 mmol, 0.03 equiv.) were added and the vial was sealed and removed. Bromobenzene (0.11mL, 2 mmol, 2 equiv.) and 1 mL dry dioxane were added and the vial placed in a 130℃ heating block for 48 hours. The reaction was then allowed to room to room temperature, rinsed three times with deionized water, once with brine, dried with MgSO4 and recrystallized in DCM/MeOH. NMR matched previous reports.8 Synthesis of 1-Naphthalene-10-phenoxazine:
An 8mL reaction vial was flame dried under vacuum and charged with phenoxazine (183mg, 1mmol, 1 equiv.) and sodium tertbutoxide (192mg, 2 mmol, 2 equiv.) before submitting to a nitrogen filled glovebox. Inside, RuPhos (12mg, 0.03mmol, 0.03 equiv.) and RuPhos Pd G2 (21mg, 0.03 mmol, 0.03 equiv.) were added and the vial was sealed and removed. 1-bromonaphthalene (0.28mL, 2 mmol, 2 equiv.) and 1 mL dry dioxane were added and the vial submitted to a 130℃ heating block for 48 hours. The reaction was then allowed to room to room temperature, rinsed three times with deionized water, once with brine, dried with MgSO4 and recrystallized in DCM/MeOH. NMR matched previous reports.8
Synthesis of 2-Naphthalene-10-phenoxazine:
Inside, RuPhos (12mg, 0.03mmol, 0.03 equiv.) and RuPhos Pd G2 (21mg, 0.03 mmol, 0.03 equiv.) were added and the vial was sealed and removed. 1 mL dry dioxane was added and the vial submitted to a 130℃ heating block for 48 hours. The reaction was then allowed to room to room temperature, rinsed three times with deionized water, once with brine, dried with MgSO4 and recrystallized in DCM/MeOH. NMR matched previous reports.8
Synthesis of 3,7 dibromo 1-naphthalene-10-phenoxazine:
1-naphthalene-10-phenoxazine (822 mg, 2.65 mmol, 1 equiv.) was dissolved in 82mL chloroform. 82 mL acetic acid was then added to the mixture. The round bottom flask was then wrapped in aluminum foil and N-bromosuccinimide (970 mg, 5.3 mmol, 2.05 equiv.) was added slowly over twenty minutes. The solution was then allowed to stir for two hours and then concentration under vacuum. The concentration solution was then rinsed three times with deionized water, once with brine, then dried with MgSO4 and concentration under vacuum. The product matched previously reported NMR spectra.8
Synthesis of 3,7 biphenyl 1-naphthalene-10-phenoxazine:
dissolved in DCM before passing through a short silica plug. The reaction mixture was then washed three times with deionized water, once with brine, dried with MgSO4 and recrystallized from DCM/MeOH. Peak separation made characterization difficult and is still ongoing with the next step to be 13C NMR.
Synthesis of 3,7 dibromo 2-naphthalene-10-phenoxazine:
2-naphthalene-10-phenoxazine (370 mg, 1.19 mmol, 1 equiv.) was dissolved in 37 mL chloroform. 37 mL acetic acid was then added to the mixture. The round bottom flask was then wrapped in aluminum foil and N-bromosuccinimide (436 mg, 2.45 mmol, 2.05 equiv.) was added slowly over twenty minutes. The solution was then allowed to stir for two hours and then concentration under vacuum. The concentration solution was then rinsed three times with deionized water, once with brine, then dried with MgSO4 and concentration under vacuum. The product matched previously reported NMR spectra.9
Synthesis of 3,7 cyanophenyl 2-naphthalene-10-phenoxazine:
was then washed three times with deionized water, once with brine, dried with MgSO4 and recrystallized from DCM/MeOH. Product was verified with 1HNMR matching reported spectra.9
C–H Functionalization Reaction Procedure:
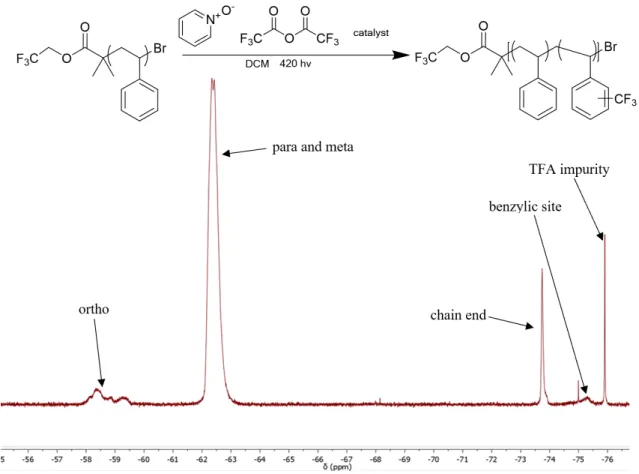
Polystyrene (0.8mmol, 83mg), trifluoroacetic anhydride (0.88mmol, 0.12mL), pyridine N -oxide (0.8mmol, 76.1mg), and photocatalyst were dissolved in 2mL DCM in 1-dram scintillation vials and then submitted to 420nm light while stirring and cooled by a fan. Following the reaction, the contents were concentrated under reduced pressure before precipitating in methanol from DCM. The precipitant was filtered, washed with methanol, then dried before analysis by Gel Permeation Chromatography (GPC) and 19F NMR. Results:
The organic photocatalysts were synthesized according to the procedures described previously, and then used in the post-polymerization modification reaction shown in Figure 1 to determine their efficacy. The reactions were run at 1.0 equivalent of trifluoroacetic anhydride (TFAA) and 1.1 equivalents or pyridine N-oxide per monomer unit, and 0.01 equivalent catalyst to pyridine
regiorandom with functionalization mostly at the para position, with some ortho, double functionalization, and benzylic site functionalization as well.
Figure 1:The general functionalization reaction used in catalyst screening, and example 19F NMR of functionalized polystyrene.
31.5 32 32.5 33 33.5 34 34.5
Retention Time (min)
4 6 7 8
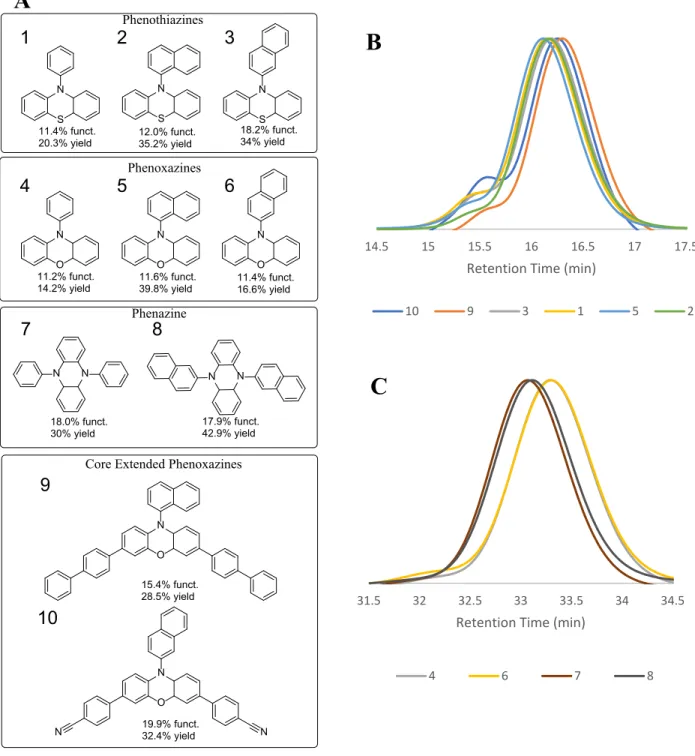
Figure 2. A: Photocatalysts tested in the reaction scheme with functionalization achieved and yield after synthesis, B, C: GPC of the functionalized polystyrene samples.
S N S N S N 11.4% funct.
20.3% yield 12.0% funct.35.2% yield
O N O N O N 11.6% funct.
39.8% yield 11.4% funct.16.6% yield
N N 18.0% funct. 30% yield Phenothiazines Phenazine N N 17.9% funct. 42.9% yield O N O N N N Phenoxazines 15.4% funct. 28.5% yield 19.9% funct. 32.4% yield
Core Extended Phenoxazines
18.2% funct. 34% yield
11.2% funct. 14.2% yield
1 2 3
4 5 6
7 8
9
10
14.5 15 15.5 16 16.5 17 17.5
Retention Time (min)
10 9 3 1 5 2
A
B
This data led to the selection of 5,10-di(2-naphthyl) phenazine as the most efficient catalyst for the reaction scheme described in Figure 1 due to it achieving high functionalization, lack of chain coupling events, and ease of synthesis. Following catalyst
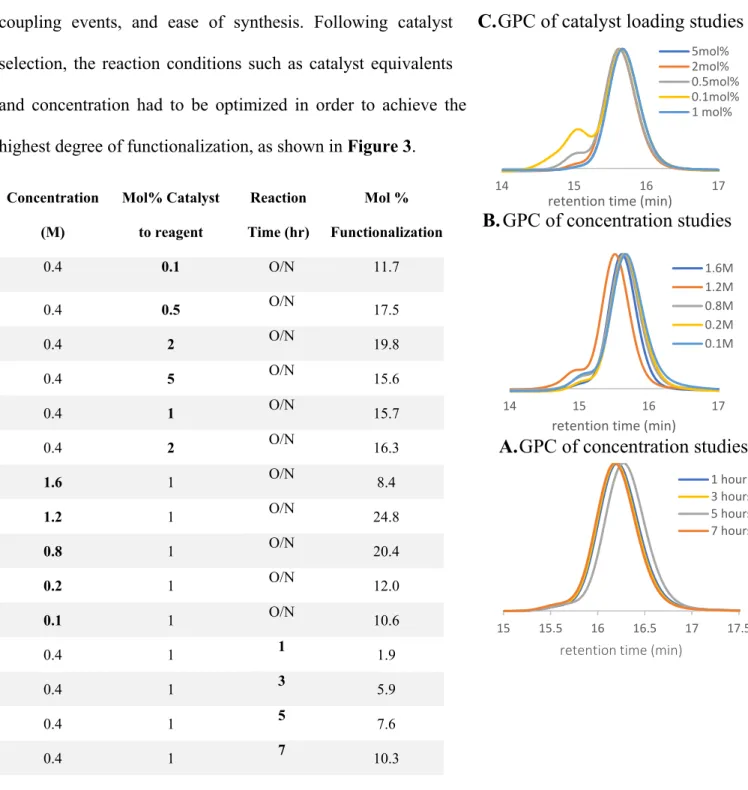
selection, the reaction conditions such as catalyst equivalents and concentration had to be optimized in order to achieve the highest degree of functionalization, as shown in Figure 3.
Concentration (M) Mol% Catalyst to reagent Reaction Time (hr) Mol % Functionalization
0.4 0.1 O/N 11.7
0.4 0.5 O/N 17.5
0.4 2 O/N 19.8
0.4 5 O/N 15.6
0.4 1 O/N 15.7
0.4 2 O/N 16.3
1.6 1 O/N 8.4
1.2 1 O/N 24.8
0.8 1 O/N 20.4
0.2 1 O/N 12.0
0.1 1 O/N 10.6
0.4 1 1 1.9
0.4 1 3 5.9
0.4 1 5 7.6
0.4 1 7 10.3
Figure 3: Optimization of 5,10-di(2-naphthyl) phenazine reaction conditions. Note: The studies in
Figure 3B were done on a different polystyrene sample than Figures 1-3A.
14 15 16 17
retention time (min)
5mol% 2mol% 0.5mol% 0.1mol% 1 mol%
14 15 16 17
retention time (min)
1.6M 1.2M 0.8M 0.2M 0.1M C.GPC of catalyst loading studies
B.GPC of concentration studies
15 15.5 16 16.5 17 17.5
retention time (min)
R O R O O R=
CF2Cl CF2Br
C F2 CF3 C F2 F2 C CF 3 A B C D
Anhydride Equivalents Anhydride
N-oxide to Repeat Unit
Mol% Functionalization
A 2 13.0
B 2 6.8
C 2 39.0
D 2 36.7
Figure 4. Studies of Various Anhydrides as Reagents with 5,10-di(2-naphthyl) phenazine.
DCM 420nm hv
N N
TFAA, Pyridine N-oxide
CF3
Ar Ar
O O
O
Figure 5. Functionalization of Commodity Polymers.
Discussion:
Substrate:
Of the eleven organic photoredox catalysts (PCs) evaluated in this research, three showed the most promising results when comparing their percent functionalization in the reaction shown by Figure 1. PC 7, PC 8, and PC 10 performed the best in the initial screen, imparting 18.0%, 17.8%, and 19.9% functionalization respectively.
The differences in the catalyst’s performance was then gauged based on the GPC trace of the functionalized polymer. PC 10, while achieving the highest degree of functionalization, suffered from chain coupling events, resulting in the formation of a high molecular weight shoulder on the GPC chromatogram seen in Figure 2B. The remaining two phenazine derivatives were nearly equal in their effectiveness and both lacked evidence of chain coupling or degradative effects during the reaction. PC 8 was chosen as the preferred photocatalyst as its purification procedure was simpler, consisting of a precipitation upon the addition of water instead of the recrystallization needed to purify PC 7.
loading have the same efficiency within error. Thus, 1 mol% of catalyst was used for further studies to conserve materials.
The concentration of the solution with respect to monomer units was then manipulated to optimize the reaction conditions. Decreasing the concentration from 0.4M led to a decrease in functionalization, possibly because of a reduced likelihood of the trifluoromethyl radical finding a styrene ring in solution before it encountered another species it could react with such as pyridine or another trifluoromethyl radical. Increasing the concentration led to an optimal point at 1.2M, achieving 24.8% functionalization, before the efficiency began to decrease from further concentrating the reaction. Increasing the concentration too much may have limited the amount of light that was able to penetrate the solution, prohibiting the catalysts within solution from becoming excited and reducing pyridine N-oxide/TFAA adducts. Additionally, solubility became an issue as the amount of good solvent decreased.
Optimization was finished by testing the effects of reaction time on the final functionalization. Previous reactions were run overnight for approximately 16 hours; however, in a laboratory context it would prove beneficial if the reaction could be shortened to occur over the course of a workday and a shortened reaction time would be optimal for an industry scale as well. Reaction times of 1, 3, 5, and 7 hours were conducted and gave functionalizations of 1.9%, 5.9%, 7.6%, and 10.3% respectively (Figure 3). All lesser than 17.8% achieved in a 16-hour overnight reaction, so it was decided to continue to run the reactions overnight. GPC analysis of the samples shows a decrease in retention time as functionalization increased and no shoulder peak formation, indicating that unwanted side reactions were not a factor at these time scales (Figure 3C).
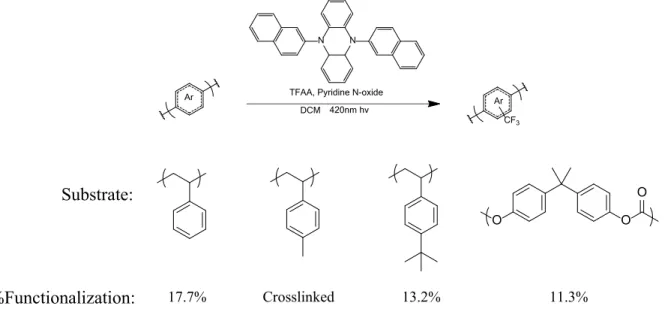
radicals that function as handles for further reactivity, chlorodifluoro- and bromodifluoromethyl radicals were appended to the polymer using their respective acetic acid anhydrides. Bromodifluoromethyl is capable of being hydrolyzed to create difluoromethyl groups on the polymer. The polarity of the fluoride groups draws electron density from the carbon-hydrogen bond, and as a result it mimics that of a phenol. Specifically, this imparts hydrophilic properties onto the polymer as opposed to the hydrophobic properties from trifluoromethyl. Bromodifluoromethyl groups are also unique in that they may be used as an initiator for Atom Transfer Radical Polymerization (ATRP) off of the polymer, creating comb copolymers. Both of the radical species are less electrophilic than trifluoromethyl radicals and as a result achieved less functionalization when compared to trifluoromethylation reactions. Using two equivalents of anhydride and pyridine N-oxide to each repeat unit yielded 13.0% functionalization for chlorodifluoromethyl acetic acid anhydride and 6.8% for the brominated version (Figure 4). Longer perfluoroalkyl chains were also added to the polymer through the use of pentafluoroethyl and heptafluoropropyl radicals. These radical species were similarly reactive as the trifluoromethyls and at two equivalents produced 39.0% and 36.7% functionalization (Figure 4).
The final portion of this study was to determine the reaction’s substrate scope. Commodity polymers such as high molecular weight polystyrene, poly(4-methylstyrene), poly(4-
which the repeat unit has undergone radical addition, upon which its reduced form is capable of forming a bond with another nearby reduced repeat unit. This crosslinking is not seen with
poly(4-tert-butylstyrene) which is functionalized 13.2%, less than polystyrene likely due to steric hindrance. Lastly polycarbonate was also successfully functionalized to 11.3%, with its lower functionalization attributed to the less electron rich nature of the carbonate repeat unit.
References:
1. Geyer R, Jambeck JR, Law KL. Production, use, and fate of all plastics ever made. Sci Adv. 2017;3(7). doi:10.1126/sciadv.1700782
2. Grainger DW, Stewart CW. Fluorinated Coatings and Films: Motivation and Significance. In: Fluorinated Surfaces, Coatings, and Films. ; :1-14. doi:10.1021/bk-2001-0787.ch001 3. Williamson JB, Lewis SE, Johnson III RR, Manning IM, Leibfarth F. C–H
Functionalization of Commodity Polymers. Angew Chemie Int Ed. 0(ja). doi:doi:10.1002/anie.201810970
4. Beatty JW, Douglas JJ, Cole KP, Stephenson CRJ. A scalable and operationally simple radical trifluoromethylation. Nat Commun. 2015;6:7919.
https://doi.org/10.1038/ncomms8919.
5. Du Y, Pearson RM, Lim C-H, et al. Strongly Reducing, Visible-Light Organic Photoredox Catalysts as Sustainable Alternatives to Precious Metals. Chem – A Eur J.
2017;23(46):10962-10968. doi:doi:10.1002/chem.201702926
6. Pan X, Lamson M, Yan J, Matyjaszewski K. Photoinduced Metal-Free Atom Transfer Radical Polymerization of Acrylonitrile. ACS Macro Lett. 2015;4(2):192-196.
doi:10.1021/mz500834g
Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J Am Chem Soc. 2016;138(35):11399-11407. doi:10.1021/jacs.6b08068