0095-1137/97/$04.0010
Copyright © 1997, American Society for Microbiology
Detection of Vancomycin-Resistant Enterococci in Fecal
Samples by PCR
SACHIKO SATAKE,1† NANCYE CLARK,1DAVID RIMLAND,2FREDERICK S. NOLTE,3 ANDFRED C. TENOVER1*
Hospital Infections Program, National Center for Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia 303331; Medical Service, Veterans Affairs Medical Center,
Atlanta, Georgia 303232; and Department of Pathology, Emory University Hospital, Atlanta, Georgia 303223
Received 24 January 1997/Returned for modification 4 March 1997/Accepted 20 March 1997
Surveillance cultures for vancomycin-resistant enterococci (VRE) are time-consuming and expensive for the laboratory to perform. Therefore, we investigated the use of PCR as an alternative method of detecting and identifying VRE directly in fecal samples. PCR primers directed tovanA,vanB,vanC1,vanC2, and enterococcal ligase genes were used to detect and identify VRE in fecal material obtained by rectal or perirectal swabbing. Although PCR-inhibitory substances were present in DNA prepared directly from the swabs, the inhibitory substances could be reduced by processing the nucleic acid with two commercially available DNA preparation columns. Fecal material from 333 swabs was cultured on several selective agar media before and after broth enrichment. DNA was extracted from the fecal material and was analyzed by PCR. By using all four primer sets, only 59 (67.8%) of the samples were positive forvanA. However, after retesting the negative samples with only the vanA primer set, 77 (88.5%) of 87 specimens that were culture positive for Enterococcus faecium
containingvanAwere positive by PCR. One specimen was PCR positive for thevanAgene but culture negative for enterococci. The specificity of thevanAassay was 99.6%. PCR analysis of enrichment broth samples with all four primers sets after 15 to 18 h of incubation detected 74 (85.1%) of the 87 culture-positive specimens. The specificity of the vanA assay after the enrichment step was 100%. No vanB-containing enterococci were recovered by culture. Since 16 samples can be tested by PCR in 4 h (including electrophoresis), identification of VRE is possible within 8 h of specimen submission at a cost of approximately $10.12/assay. Thus, PCR may be a cost-effective alternative to culture for surveillance of VRE in some hospitals.
Vancomycin-resistant enterococci (VRE) have emerged as important nosocomial pathogens in the United States and else-where (2, 7, 15, 19, 21, 27). These organisms are often resistant to multiple antimicrobial agents limiting the number of thera-peutic options available to the physician (10, 20). Many hos-pitals have initiated surveillance programs for VRE; however, these programs have proven to be time-consuming for infec-tion control personnel and expensive for the microbiology lab-oratory to conduct (6, 21). Several different selective media for enhancing the recovery of VRE from stool or rectal samples have been proposed, but no single medium has proven to be universally acceptable (6, 12, 17, 21, 27, 31). Nonetheless, sur-veys of stools or rectal swabs are recommended by the Hospital Infection Control Practices Advisory Committee to allow for the early identification of colonized patients so that infection control measures to prevent person-to-person transmission of VRE can be instituted (11).
Vancomycin is a member of a broader class of antimicrobial agents referred to as glycopeptides (1). Glycopeptide resis-tance genotypes in enterococci includevanA(high-level resis-tance);vanB,vanB2, andvanD(moderate to high-level resis-tance); and vanC1, vanC2, and vanC3 (intrinsic low-level resistance) (1, 5, 24–26). While thevanAresistance gene has been detected in a wide variety of enterococcal species (1, 3, 4),
vanC1,vanC2, andvanC3 have been recognized only in En-terococcus gallinarum, Enterococcus casseliflavus, and Entero-coccus flavescens, respectively (1, 18, 24). There is little infor-mation in the literature regarding the occurrence of vanB outside ofEnterococcus faecalisandEnterococcus faecium(1, 13, 26), and only a single isolate of vanDhas been reported (25). PCR assays have been devised to recognize the vanA, vanB, andvanC1genotypes (3, 5) and have demonstrated that enterococci may contain more than one of these determinants (4).
Detection of glycopeptide resistance in enterococci by either disk diffusion or automated susceptibility testing methods has been shown to be less sensitive than that by broth microdilu-tion methods (13, 28, 30). However, the broth methods require a full 24 h of incubation to detect strains with low-level resis-tance (23). Thus, the detection of VRE, particularly in rectal surveillance cultures where vancomycin-susceptible entero-cocci are often present, has been a challenge. In this study, we compared the sensitivities and specificities of PCR assays for vanA,vanB,vanC1,vanC2, and the enterococcal ligase primers (5) with the sensitivity and specificity of culture with and with-out broth enrichment, for the detection of VRE in clinical specimens.
MATERIALS AND METHODS
Selective media and culture condition.This study was carried out in three
phases, each of which used a slightly different set of selective media. Five different selective agars and three different selective broths were used to screen for growth of VRE (Table 1). Rectal or perirectal swabs were collected from 333 patients from two hospitals in the metropolitan Atlanta area, placed in Amies transport medium, and processed within 8 h of collection. The fecal material from the swabs was suspended in 350ml of sterile water, and the mixture was * Corresponding author. Mailing address: Nosocomial Pathogens
Laboratory Branch (G-08), Centers for Disease Control and Preven-tion, 1600 Clifton Rd., NE, Atlanta, GA 30333. Phone: (404) 639-3246. Fax: (404) 639-1381.
† Present address: College of Medical Care and Technology, Gunma University, Maebashi, Japan.
2325
on May 15, 2020 by guest
http://jcm.asm.org/
vortexed vigorously for 5 s. One hundred microliters of this suspension was inoculated into 3 ml of enrichment broth, and two 50-ml samples of the suspen-sion were inoculated onto each of two selective agar plates. The agar plates were incubated at 35°C, while the broth was incubated at 35 to 45°C, depending on the phase of the study. Ten-microliter samples from the enrichment broths were subcultured onto selective agar plates after 15 to 18 h of incubation. A smaller inoculum was used to avoid overgrowth of the organisms on the plates. The agar plates were examined after 24 h, 48 h, and 5 days of incubation. Colonies resembling enterococci by colonial morphology were subcultured onto Trypti-case soy agar with 5% sheep blood (Becton Dickinson Microbiology Systems, Cockeysville, Md.) and were identified by standard laboratory methods (8, 9). Brain heart infusion agar containing 6mg of vancomycin per ml also was inoc-ulated as described by the National Committee for Clinical Laboratory Stan-dards (NCCLS) (23, 29) to enhance the detection of VRE. The MICs of van-comycin and teicoplanin were determined by the NCCLS broth microdilution method with cation-adjusted Mueller-Hinton broth (Difco) (22, 23). Quality control strains for susceptibility testing included Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 29213, E. faecalis ATCC 29212, and E. faecalis ATCC 51299 (22).
DNA extraction from rectal swab specimens.Fifty-microliter samples of stool suspension were centrifuged at 16,0003g for 5 min. The pellet was resuspended in 180ml of distilled water, and 4 mg of lysozyme perml was added to the suspension, which was then incubated at 37°C for 30 min. The lysate was then treated with 0.5 mg of proteinase K at 70°C for 30 min and then at 95°C for an additional 30 min. Two volumes of ethanol were added to precipitate the nucleic acids. The ethanol mixture was applied directly to a QIAamp Tissue Kit column (QIAGEN Inc., Chatsworth, Calif.) and washed twice in buffer as described by the manufacturer, and the nucleic acid was eluted with 50ml of 10 mM Tris-HCl (pH 9.0). Twenty microliters of the eluate was applied to a Centrisep gel filtra-tion column (Princeton Separafiltra-tion, Inc., Adelphia, N.J.), and the eluate con-taining purified nucleic acid was used in the PCR assays. The procedure is outlined in Fig. 1.
Amplification of vanA, vanB, vanC1, vanC2, ddlE. faecalis, and ddlE. faeciumgenes
by PCR.Oligonucleotide primers directed to the vanA, vanB, vanC1, ddlE. faecalis, and ddlE. faeciumgenes (5) and novel vanC2 primers (primer vanC2-1, 59-CGG GGA AGA TGG CAG TAT-39; primer vanC2-2, 59-CGC AGG GAC GGT GAT TTT-39) were used in conjunction with PCR to amplify DNA from fecal material, isolated colonies, or enrichment broth after inoculation and incubation for 15 to 18 h. The respective PCR product sizes were as follows: vanA, 732 bp; vanB, 635 bp; vanC1, 822 bp; and vanC2, 484 bp (see Fig. 2A). The vanC2 primer also detected strains of E. flavescens that carry the vanC3 gene. The ddl primers yielded a product of 941 bp for E. faecalis and 550 bp for E. faecium (5). Two-or 4-ml samples of DNA purified directly from stool as described above, 2 or 4
ml of uncentrifuged enrichment broth, or a loopful of bacterial cell suspension taken from isolated colonies was added to 23 or 21ml of a chilled PCR mixture containing 10 mM Tris (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.2 mM
de-oxynucleotide triphosphates (dATP, dCTP, dGTP, and dTTP), 0.5 mM (each) primer, and 2.5 U of Taq DNA polymerase. A Perkin-Elmer Cetus model 9600 DNA thermocycler (Norwalk, Conn.) was programmed as follows: 10 min at 95°C; 30 cycles of 30 s at 94°C, 30 s at 58°C, and 30 s at 72°C; and 10 min at 72°C. Samples were held at 4°C until the products could be analyzed. Sixteen-micro-liter samples of the PCR products were electrophoresed through a 1.5% agarose gel for 45 min at 150 V. The gels were stained with ethidium bromide and photographed under UV light. PCR was simultaneously performed with four
pairs of primers for vanA, vanB, vanC1, and vanC2 in one reaction tube to identify vancomycin resistance determinants and two pairs of primers for ddlE. faecalisand ddlE. faeciumin another reaction tube for organism identification. In later experiments, only vanA primers were used. Quality control organisms for PCR included E. faecium NJ1 (vanA), E. faecalis NJ3 (vanB), E. gallinarum NJ4 (vanC1) (30), E. casseliflavus ATCC 25788 (vanC2), and E. faecalis ATCC 29212 (negative control).
RESULTS
Detection of VRE by culture methods. Ninety-nine VRE isolates were recovered from 97 of 333 rectal specimens by at least one culture method (i.e., direct plating on agar or after broth enrichment; Table 2). Of the 99 isolates, 87 were iden-tified biochemically as E. faecium and were shown by PCR to contain vanA, 6 were identified as E. gallinarum and contained
vanC1, and another 6 were identified as E. casseliflavus and
contained vanC2. One specimen contained both E. gallinarum and vanA-containing E. faecium, and another specimen con-tained both E. gallinarum and E. casseliflavus. No vanB-con-taining enterococci were isolated. VRE were detected in 6 specimens by direct plating only (vanA-containing enterococci plus vanC-containing enterococci; Table 2), in 16 specimens by the use of enrichment broth only, and in 76 specimens by both techniques. Many of the specimens positive only by broth
[image:2.612.58.556.83.207.2]en-FIG. 1. Method for isolating and purifying nucleic acid from fecal material using QIAamp and Centrisep column for testing by PCR.
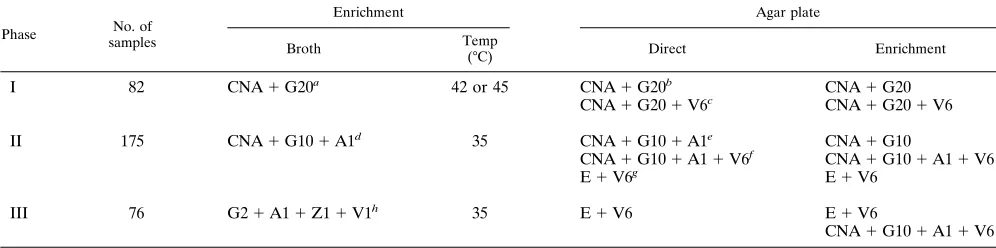
TABLE 1. Selective media and enrichment broths used in this study
Phase No. of samples
Enrichment Agar plate
Broth Temp
(°C) Direct Enrichment
I 82 CNA1G20a 42 or 45 CNA1G20b CNA1G20
CNA1G201V6c CNA1G201V6
II 175 CNA1G101A1d 35 CNA1G101A1e CNA1G10
CNA1G101A11V6f CNA1G101A11V6
E1V6g E1V6
III 76 G21A11Z11V1h 35 E1V6 E1V6
CNA1G101A11V6
a
Trypticase soy broth with 15mg of colistin per ml, 15mg of nalidixic acid per ml, and 20mg of gentamicin per ml. b
Columbia CNA agar with 20mg of gentamicin per ml. c
Columbia CNA agar with 20mg of gentamicin per ml and 6mg of vancomycin per ml. d
Trypticase soy broth with 15mg of colistin per ml, 15mg of nalidixic acid per ml, 10mg of gentamicin per ml, and 1mg of amphotericin B per ml. e
Columbia CNA agar with 10mg of gentamicin per ml and 1mg of amphotericin B per ml. f
Columbia CNA agar with 10mg of gentamicin per ml, 1mg of amphotericin B per ml, and 6mg of vancomycin per ml. g
Enterococcosel agar with 6mg of vancomycin per ml. h
Trypticase soy broth with 2mg of gentamicin per ml, 1mg of amphotericin B per ml, 1mg of aztreonam per ml, and 1mg of vancomycin per ml.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:2.612.321.553.559.705.2]richment showed,10 colonies of VRE on subculture, suggest-ing that these samples contained low numbers of VRE.
The sensitivities of two types of selective media, Enterococ-cosel agar and Columbia CNA agar, were compared for recov-ery of VRE. Because the CNA formulation with 20 mg of gentamicin per ml proved to be too inhibitory for some en-terococci, only the results obtained with the formulation with 10mg of gentamicin per ml will be discussed. Enterococcosel agar with 6mg of vancomycin per ml (Enterococcosel-V) and Columbia CNA agar with 10mg of gentamicin per ml, 6mg of vancomycin per ml, and 1mg of amphotericin B per ml (CNA-VGA agar) were tested in parallel. Forty-twovanA-containing E. faeciumisolates were recovered from the 142 samples inoc-ulated onto both types of media. Of the 42, 41 were detected on Enterococcosel-V agar and 39 were detected by CNA-VGA. The sensitivities of Enterococcosel-V agar and Colum-bia CNA-VGA agar for the recovery of organisms containing high-level vancomycin resistance were 97.6 and 92.9%, respec-tively. Eight samples in the comparison study were found to harborvanC-containing enterococci (i.e.,E. gallinarumand/or E. casseliflavus) by at least one selective medium; of the eight samples, Enterococcosel-V agar detected enterococci in six and CNA-VGA detected enterococci in four. The sensitivities of Enterococcosel-V agar and Columbia CNA-VGA agar for the recovery ofvanC-containing enterococci were 75 and 50%, respectively. ThevanBcontrol strainE. faeciumATCC 51299 did not grow on the Columbia CNA-VGA agar used in phase I, which contained 20mg of gentamicin per ml, or in phase II, in which the amount of gentamicin was reduced to 10mg/ml. The control strain did grow on the Columbia CNA agar used in phase III, which contained only 1mg of gentamicin per ml and 1mg of aztreonam per ml, the latter of which was added to help suppress gram-negative flora. However, because of the expense of this agar formulation, the supplemented Columbia CNA medium was not used for direct inoculation of specimens in phase III but was used only for comparison of the recovery of VRE from broth.
Detection of VRE in broth enrichment culture and by PCR. A slightly different enrichment broth was used in each phase of the study (Table 1). In phase I several of theE. faeciumisolates did not grow when the broth was incubated at 45°C, even though this species should thrive at this temperature (9). Sub-cultures of the organisms which were recovered by direct plat-ing into the same selective broth incubated at 42°C also failed
to grow. In addition, agar plates containing specimens from immunocompromised patients often showed growth of yeasts. Thus, in phases II and III, the temperature of the broth was reduced to 35°C and 1mg of amphotericin B per ml was added to both the CNA broth and agar.
The sensitivities of PCR assays with 2ml of the enrichment broth after overnight incubation for the detection of vanA-containing enterococci in phases I, II, and III were 88.5, 83.3, and 84.0%, respectively. Overall, 74 (85.1%) of 87 vanA-con-tainingE. faeciumculture-positive samples were detected (Ta-ble 3). The specificities of the assays were 100% in all three phases. The sensitivities of the assays were not significantly improved by repeating the assays with 4ml of broth.
Identification of VRE strains by PCR.Of 327 enterococci isolated during the course of the study from all selective media (which included multiple isolates of the same organism from agar and broth), 325 (99.4%) could be unambiguously identi-fied by PCR with the ligase gene primers. This included 240E. faeciumand 61E. faecalisisolates. In addition, using thevanC1 and vanC2primers, we confirmed the biochemical identifica-tions for 11E. gallinarumand 13E. casseliflavusisolates. Two of 63 strains identified biochemically asE. faecalisconsistently failed to produce a PCR product withddlE. faecalisgene primers.
Two additional strains of vancomycin-susceptible E. faecalis, both isolated from the same specimen but on different selective media, were positive for thevanAgene by PCR. The presence ofvanA gene sequences was confirmed by Southern blotting and DNA hybridization with avanA-specific DNA probe (3). In addition, two strains of vancomycin-resistant E. faecium isolated from the same specimen were positive for both the vanA and vanBgenes by PCR, while a third strain, this one vancomycin susceptible, also was positive for vanB by PCR (data not shown). The presence of vanB sequences in the isolates was confirmed by using a vanB-specific DNA probe and an alternate set of PCR primers (3). All 206 strains show-ing high-level resistance to vancomycin by broth microdilution producedvanAproducts by PCR. The sensitivities of the assays withvanA,vanC1, andvanC2primer sets compared with those of the reference broth microdilution MIC method were all 100%, while the sensitivities of the assays withddlE. faecalisand
ddlE. faecium primer sets compared with the sensitivity of
bio-chemical identification were 96.8 and 100%, respectively. The specificities were 98.4% forvanA, 99.1% forvanB, and 100% forvanC1,vanC2,ddlE. faecalis, andddlE. faecium.
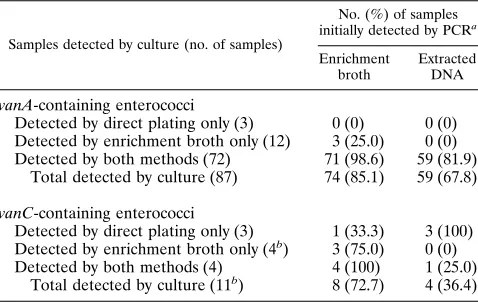
[image:3.612.59.298.82.233.2]Detection of thevanAandvanCgenes in fecal material by PCR.The sensitivity of the PCR assay for detecting thevanA determinant in organisms grown on agar plates by using only thevanAprimer pair was determined to be between 1 and 10 TABLE 2. Detection of VRE in 333 fecal samples by PCR
Samples detected by culture (no. of samples)
No. (%) of samples initially detected by PCRa
Enrichment broth
Extracted DNA vanA-containing enterococci
Detected by direct plating only (3) 0 (0) 0 (0) Detected by enrichment broth only (12) 3 (25.0) 0 (0) Detected by both methods (72) 71 (98.6) 59 (81.9)
Total detected by culture (87) 74 (85.1) 59 (67.8)
vanC-containing enterococci
Detected by direct plating only (3) 1 (33.3) 3 (100) Detected by enrichment broth only (4b) 3 (75.0) 0 (0)
Detected by both methods (4) 4 (100) 1 (25.0) Total detected by culture (11b) 8 (72.7) 4 (36.4)
aDoes not include repeat testing of negative samples withvanAprimers only. bOne sample contained bothE. gallinarumandE. casseliflavus, so the total
number ofvanC-containing organisms isolated by culture was 12.
TABLE 3. Adjusted sensitivity and specificity of PCR assay with
vanAprimers compared with those of culture
PCR result in assays with the indicated source of DNA
No. of samples with following result by culture: Positive
(n587)
Negative (n5246) Enrichment broth culturea
Positive 74 0
Negative 13 246
DNA extracted from rectal swabb
Positive 77 1
Negative 10 245
aSensitivity, 74 (85.1%) of 87 samples; specificity, 246 (100%) of 246 samples. bSensitivity, 77 (88.5%) of 87 samples; specificity, 245 (99.6%) of 246 samples.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.612.315.556.90.206.2]CFU (data not shown). When combined with the other three primer sets in the multiplex assay, the sensitivity of thevanA primer set dropped to approximately 10 to 100 CFU (data not shown). This change in sensitivity was also reflected in the results obtained with clinical samples.
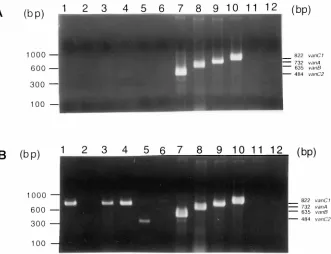
The sensitivity of PCR for detecting the vanA and vanC genes in nucleic acid extracted directly from the fecal suspen-sions varied according to the extent to which the nucleic acid was purified. For example, Fig. 2 demonstrates the differences invanAdetection by PCR for three samples in which the DNA was purified only with a QIAamp column (Fig. 2A) and the results obtained after the nucleic acid underwent an additional purification step using Centrisep columns (Fig. 2B). The re-sults indicate that three samples would have been classified as negative if only the QIAamp column had been used. Thus, inhibitors can reduce the sensitivity of the direct PCR assays. When a 2-ml sample was used with all four primer sets, the vanA PCR assay detected 59 (67.8%) of 87 specimens that were positive for E. faeciumcontainingvanA by at least one culture method. Of 28 false-negative specimens, 3 were posi-tive forE. faeciumcontainingvanAby direct plating, 12 were positive by broth enrichment, and 13 were positive by both direct plating and broth enrichment. One specimen that was PCR positive for vanA was negative by all culture methods. Thus, the specificity of the PCR assay for detection of thevanA gene DNA prepared directly from specimens was 99.6%. All 28 PCR-negative samples were retested with 2 and 4 ml of sample and only thevanAprimer set. Ten specimens remained negative with both 2- and 4-ml samples, 14 were positive with both 2- and 4-ml samples, 2 were positive with only 2-ml sam-ples, and 2 were positive with only 4-ml samples. Thus, after retesting, an additional 18 samples were positive for thevanA
gene. Sixteen of the samples that were initially PCR negative showed,10 colonies on selective medium. Thus, after repeat testing, 88.5% (77 of 87) of culture-positive specimens were positive for thevanAgene by PCR (Table 3).
ThevanC1assay detected 4 of 11 specimens that were cul-ture positive forE. gallinarum. Two specimens that were PCR positive forvanC1were negative by all culture methods. The negative samples were not retested.
Cost of performing the PCR tests.The total cost of the PCR assay for VRE by the two-column method depicted in Fig. 1 including four primer sets (vanA, vanB, vanC1, vanC2) was $10.12 per reaction. The cost of the reagents ($1.62/reaction including the primers), the columns (QIAamp $1.10 and Cen-trisep $2.40), and labor ($15.00/h320 min or $5.00 for tech-nologist time to process 16 samples plus 4 controls) will vary significantly on the basis of the volume of assays performed and the salary of the individual performing the test. By con-trast, the cost of the culture method for VRE with broth enrichment ($0.75), a single selective agar plate and a nonse-lective CNA agar plate ($1.05), biochemicals for identification ($2.30), an antimicrobial susceptibility test ($7.42), and labor ($15.00/h317 min5$4.25) was $15.77.
DISCUSSION
[image:4.612.144.475.69.323.2]This study focused on whether PCR could be used in the laboratory to replace more traditional surveillance culture methods for the detection of VRE in colonized patients. PCR has the potential to reduce both the time and cost of detecting VRE and can provide information on the vancomycin resis-tance genotype, which may be useful for epidemiologic studies (3). The cost of the PCR assay was less than the cost of the FIG. 2. Comparison of PCR results after purification of DNA by a single or two consecutive nucleic acid purification procedures. An ethidium bromide-stained agarose gel (1.5%) containing the PCR products produced by amplification of DNA extracted from rectal swabs is shown. PCR was performed with four pairs of oligodeoxynucleotide primers, i.e., primers forvanA,vanB,vanC1, andvanC2, simultaneously. (A) Lanes 1 to 6, PCR products obtained from specimens 289 to 294, respectively, after purifying DNA only with QIAamp spin columns; lane 7,E. casseliflavus(vanC2); lane 8,E. faecalisNJ3 (vanB); lane 9,E. faeciumNJ1 (vanA); lane 10,E. gallinarumNJ4 (vanC1); lane 11,E. faecalisATCC 29212; lane 12, no sample DNA. (B) Lanes 1 to 6, PCR products obtained from specimens 289 to 294, respectively, after purifying DNA with QIAamp and Centrisep spin columns; lanes 7 to 12, as described above for panel A. The faint band in lane 5 did not match the size of any of the controls and was not reproducible. It was assumed to be nonspecific.
on May 15, 2020 by guest
http://jcm.asm.org/
culture method used in the study. A survey of three microbi-ology laboratories in the Atlanta area suggested that the cost of VRE screening per specimen ranged from $9.55 to $19.80, depending on the degree to which the organisms were identi-fied and whether an MIC test or simply a vancomycin agar screening plate was used to confirm the resistance profile. While the PCR assay may be cost-effective, one potential draw-back to this approach is that it precludes strain typing studies, which are integral to many infection control investigations. While it would be possible, especially if an enrichment broth was used, to culture those specimens that are positive by PCR to recover enterococci for strain typing, this would increase the overall cost of the surveillance system.
Stool is the most difficult specimen on which to perform PCR due to the presence of multiple substances that inhibit the polymerase enzyme (14, 16). Because of this inhibition, the PCR assays initially proved to be less sensitive than the culture techniques for the detection of VRE. However, the sensitivity of the assay increased when only thevanA primer was used with an increased amount of target DNA. Although the primer sequences were screened for the presence of possible hairpin and dimer formation, it is likely that there are unforeseen interactions among the eight primers when used together in a single tube. The amount of fecal material used for culture also was 1 to 2 orders of magnitude higher than that used in the PCR assays, which probably contributed to the overall lower sensitivity of the PCR assay. Increasing the amount of target DNA did not significantly increase the sensitivity of the assay, probably because too much target nucleic acid is also known to inhibit PCR.
The results of our PCR assays for VRE were compared with the results of culture tests with several different selective media with and without the aid of broth enrichment. Several studies have advocated the use of broth enrichment for surveillance of VRE (12, 31). Enterococcosel-V agar and CNA-VGA agar showed equivalent sensitivities for the recovery of vanA-con-taining enterococci from rectal and perirectal samples (no at-tempt was made in this study to assess the differences between the recovery of VRE from perirectal versus rectal swabs); however, the Enterococcosel-V agar required at least 48 h of incubation to optimize recovery of VRE. Colonies were often only pinpoints at 24 h, and additional colonies were frequently recognized at 48 h; however, prolonged incubation for up to 5 days yielded relatively few additional isolates. While colonies on CNA-VGA were often large enough to be subcultured at 24 h, this medium was more likely to show growth of yeast and occasional gram-negative organisms than was Enterococ-cosel-V agar. Two formulations of CNA-VGA agar, i.e., those containing 20 or 10 mg of gentamicin per ml, inhibited the growth of bothE. faecalisNJ3, which harbors avanBgene, and the vanC2-containing E. casseliflavus strain; however, these strains grew well on Enterococcosel-V agar. Vancomycin and gentamicin probably have a synergistic effect that inhibits the growth of bothvanB- and somevanC-containing enterococci. Because of the additional cost of the CNA-VGA medium and its suppression of some vanB and vanCisolates, Enterococ-cosel-V agar or similar bile-esculin azide agar formulations may be a better choice for surveillance cultures. We did not evaluate Enterococcosel broth; however, the CNA-based en-richment broths used in this study, which were incubated for 15 to 18 h before subculture, did enhance the recovery of VRE, although it added approximately $1.50 to the cost of culture due to the additional supplies and technologist time required for processing.
PCR assays were performed with samples taken from the enrichment broth after 15 to 18 h of incubation and with
nucleic acid extracted from fecal material from rectal swabs suspended in distilled water. We had presumed that the dilu-tion of the inhibitory substances in water would allow us to use a simple and inexpensive column purification system for the isolation of nucleic acid. However, a single column did not yield sufficiently pure DNA for amplification. Inhibition was consistently eliminated only by processing the nucleic acid with an additional commercially available DNA preparation col-umn. Nonetheless, the cost of processing samples through the two columns at $10.12 per specimen, including all PCR-related reagents and labor, was still less than the cost of culture. Sixteen samples could be completed in 8 h from the time of receipt of the specimens. If PCR reagents are already in use in the laboratory, this cost could be further reduced.
We examined the effect of using all four primer sets (vanA, vanB,vanC1, andvanC2) versus just the singlevanAprimer set in the PCR assays. The use of four primer sets clearly reduced the sensitivity of the PCR assay for detectingvanA-containing enterococcal strains both with purified organisms and with stool specimens. Thus, laboratories should consider using only vanAprimers, or perhaps the combination ofvanAandvanB primers, and forego the detection of the vanC isolates. The significance of detecting vanC-containing VRE from rectal samples remains unclear; however, nosocomial infections caused byE. gallinarumandE. casseliflavusappear to be rare (3, 15, 21). We could not adequately evaluate the detection of vanB-containing enterococci because they occurred infre-quently in the hospitals participating in this study. Given that a non-vancomycin-containing CNA agar medium was included in the protocol and all enterococci including vancomycin-sus-ceptible organisms were identified, we feel confident that vanB-containing strains would have been recovered had they been present in the patient population sampled. In addition, the vanB primer set used has successfully been used in our laboratory and other laboratories for the past 2 years (5).
A 15- to 20-h enrichment step in selective broth incubated at 35°C enhanced the sensitivity of the culture method. Incuba-tion of the broth at 45°C, which is considered a differential test for enterococci, inhibited the growth of many of theE. galli-narum and vanA-containing E. faecium strains recovered in this study. The enrichment broth with 20mg of gentamicin per ml and 6 mg of vancomycin per ml was too inhibitory for vanC-containing species. In phase II, a different enrichment broth, in which the concentration of gentamicin was reduced to 10 mg/ml and to which 1 mg of amphotericin B per ml was added, incubated at 35°C was used. This improved the recovery ofvanC-containing species, but some VRE were still inhibited. In phase III, the concentrations of both gentamicin and van-comycin were reduced to 1mg/ml and 1mg of aztreonam per ml was added. This change allowed for the growth of thevanB andvanCcontrol organisms but did not significantly improve the overall recovery of VRE and was very expensive. Given the results of this study and those of several other investigators (12, 31), Enterococcosel broth containing 6mg of vancomycin per ml or a similar bile-esculin azide formulation may be the best enrichment broth for use in maximizing the detection of VRE from fecal samples.
Identification of VRE by PCR worked well. Only two iso-lates of E. faecalis failed to produce a PCR product of the appropriate size. Biochemically, these two strains were typical ofE. faecalis. Of the three vancomycin-susceptible strains that were positive by PCR for vanA orvanB, all three were also positive in assays with different PCR primers, and each hybrid-ized with avanAorvanBgene probe, suggesting that the genes were present in the isolates but were nonfunctional (1). (We did not test PCR primer sets to the other genes in the
on May 15, 2020 by guest
http://jcm.asm.org/
mycin operon to determine whether each component of the vangene cluster was present.) This suggests that the PCR assay does have sufficiently high specificity for use with fecal samples, although some vancomycin-susceptible organisms (3 [0.9%] of 333) will be falsely classified as VRE.
In summary, culture of rectal or perirectal samples with Enterococcosel-V agar after enrichment in supplemented CNA broth at 35°C followed by biochemical species identifi-cation and broth microdilution susceptibility testing was the most sensitive method in this study for detecting all types of VRE. However, completion of this method required at least 5 days, and additional tests were needed to distinguish VRE fromLeuconostocandPediococcus, which can be vancomycin resistant and esculin hydrolysis positive. Since 16 samples can be tested by PCR in 4 h (including electrophoresis), identifi-cation of VRE is possible within 8 h of specimen submission by PCR of DNA extracted from a clinical specimen. If enrichment broth is used, the costly step of DNA extraction is eliminated but the time to the final results is extended to approximately 30 to 36 h, although this may make it easier for laboratories to batch process specimens that arrive in the laboratory through-out the day. Thus, PCR may be an attractive alternative to culture for surveillance of VRE in some hospitals. However, in doing so, the laboratory may lose the ability to perform strain typing studies with the organisms in their institutions.
ACKNOWLEDGMENTS
We thank Elaine Miller and Ronda Sinkowitz for collecting the samples and Jerome Tokars for help with data analysis.
REFERENCES
1.Arthur, M., and P. Courvalin.1993. Genetics and mechanisms of
glycopep-tide resistance in enterococci. Antimicrob. Agents Chemother.37:1563– 1571.
2.Centers for Disease Control and Prevention.1993. Nosocomial enterococci
resistant to vancomycin—United States, 1989–1993. Morbid. Mortal. Weekly Rep.42:597–599.
3.Clark, N. C., R. C. Cooksey, B. C. Hill, J. M. Swenson, and F. C. Tenover.
1993. Characterization of glycopeptide-resistant enterococci from U.S. hos-pitals. Antimicrob. Agents Chemother.37:2311–2317.
4.Dutka-Malen, S., B. Blaimont, G. Wauters, and P. Courvalin.1994.
Emer-gence of high level resistance to glycopeptides inEnterococcus gallinarum
andEnterococcus casseliflavus. Antimicrob. Agents Chemother. 38:1675– 1677.
5.Dutka-Malen, S., S. Evers, and P. Courvalin.1995. Detection of
glycopep-tide resistance genotypes and identification to the species level of clinically relevant enterococci by PCR. J. Clin. Microbiol.33:24–27.
6.Edberg, S. C., C. J. Hardalo, C. Kontnick, and S. Campbell.1994. Rapid
detection of vancomycin-resistant enterococci. J. Clin. Microbiol.32:2182– 2184.
7.Emori, T. G., and R. P. Gaynes.1993. An overview of nosocomial infections,
including the role of the microbiology laboratory. Clin. Microbiol. Rev.
6:428–442.
8.Facklam, R., and J. A. Elliott.1995. Identification, classification, and clinical
relevance of catalase-negative, gram-positive cocci, excluding the strepto-cocci and enterostrepto-cocci. Clin. Microbiol. Rev.8:479–495.
9.Facklam, R., and D. F. Sahm.1995.Enterococcus, p. 308–314.InP. R.
Murray, E. J. Baron, M. A. Pfaller, F. C. Tenover, and R. H. Yolken (ed.), Manual of clinical microbiology, 6th ed. American Society for Microbiology, Washington, D.C.
10. Handwerger, S., B. Raucher, D. Altarac, J. Monka, S. Marchinone, K. V.
Singh, B. E. Murray, J. Wolff, and B. Walters.1993. Nosocomial outbreak
due toEnterococcus faeciumhighly resistant to vancomycin, penicillin and gentamicin. Clin. Infect. Dis.16:750–755.
11. Hospital Infection Control Practices Advisory Committee.1995.
Recom-mendations for preventing the spread of vancomycin resistance. Infect. Con-trol Hosp. Epidemiol.16:105–113.
12. Jensen, B. J.1996. Screening specimens for vancomycin-resistant
enterococ-cus. Lab. Med.27:53–56.
13. Jett, B., L. Free, and D. F. Sahm.1996. Factors influencing the Vitek
Gram-Positive Susceptibility system’s detection ofvanB-encoded vancomy-cin resistance among enterococci. J. Clin. Microbiol.34:701–706.
14. Johnson, S. R., D. H. Martin, C. Cammarata, and S. A. Morse.1995.
Alterations in sample preparation increase sensitivity of PCR assay for diagnosis of chancroid. J. Clin. Microbiol.33:1036–1038.
15. Kaplan, A. H., P. H. Gilligan, and R. R. Facklam.1988. Recovery of resistant
enterococci during vancomycin prophylaxis. J. Clin. Microbiol.26:1216– 1218.
16. Kato, N., C. Ou, H. Kato, S. L. Bartley, C. Luo, G. E. Killgore, and K. Ueno.
1993. Detection of toxigenicClostridium difficilein stool specimens by the polymerase chain reaction. J. Infect. Dis.167:455–458.
17. Landman, D., J. M. Quale, E. Oydna, B. Willey, V. Ditore, M. Zaman, K.
Patel, G. Saurina, and W. Huang.1996. Comparison of five selective media
for identifying fecal carriage of vancomycin-resistant enterococci. J. Clin. Microbiol.34:751–752.
18. LeClercq, R., S. Dutka-Malen, J. Duval, and P. Courvalin.1992.
Vancomy-cin resistance genevanCis specific toEnterococcus gallinarum. Antimicrob. Agents Chemother.36:2005–2008.
19. Livornese, L. L., S. Dias, C. Samel, B. Romanowski, S. Taylor, P. May, P.
Pisakis, G. Woods, D. Kaye, M. E. Levision, and C. C. Johnson.1992.
Hospital-acquired infection with vancomycin-resistantEnterococcus faecium
transmitted by electronic thermometers. Ann. Intern. Med.117:112–116.
20. Montecalvo, M. A., H. Horowitz, C. Gedris, C. Carbonaro, F. C. Tenover, A.
Issah, P. Cook, and G. P. Wormser.1994. Outbreak of vancomycin-,
ampi-cillin-, and aminoglycoside-resistantEnterococcus faeciumbacteremia in an adult oncology unit. Antimicrob. Agents Chemother.38:1363–1367.
21. Morris, J. G., Jr., D. K. Shay, J. N. Hebden, R. J. McCarter, Jr., B. E.
Perdue, W. Jarvis, J. A. Johnson, T. C. Dowling, L. B. Polish, and R. S.
Schwalbe.1995. Enterococci resistant to multiple antimicrobial agents,
in-cluding vancomycin. Ann. Intern. Med.123:250–259.
22. National Committee for Clinical Laboratory Standards.1995. Performance
standards for antimicrobial susceptibility testing. M100-S6. National Com-mittee for Clinical Laboratory Standards, Wayne, Pa.
23. National Committee for Clinical Laboratory Standards.1997. Methods for
dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 4th ed. Approved standard. M7-A4. National Committee for Clinical Lab-oratory Standards, Wayne, Pa.
24. Navarro, F., and P. Courvalin.1994. Analysis of genes encodingD
-alanine-D-alanine ligase-related enzymes inEnterococcus casseliflavusand Entero-coccus flavescens. Antimicrob. Agents Chemother.38:1788–1793.
25. Perichon, B., P. E. Reynolds, and P. Courvalin.1996. AvanD-type
glyco-peptide-resistantEnterococcus faecium, abstr. LB-12, p. 9.InProgram and abstracts of the 36th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, D.C.
26. Quintiliani, R., Jr., and P. Courvalin.1994. Conjugal transfer of the
vanco-mycin resistance determinantvanBbetween enterococci involves the move-ment of large genetic elemove-ments from chromosome to chromosome. FEMS Microbiol. Lett.119:359–364.
27. Rubin, L. G., V. Tucci, E. Cerenado, G. Eliopoulos, and H. D. Isenberg.1992.
Vancomycin-resistantEnterococcus faeciumin hospitalized children. Infect. Control Hosp. Epidemiol.13:700–705.
28. Sahm, D. F., and L. Olsen.1990. In vitro detection of enterococcal
vanco-mycin resistance. Antimicrob. Agents Chemother.34:1846–1848.
29. Swenson, J. M., N. C. Clark, M. J. Ferraro, D. F. Sahm, G. Doern, M. A.
Pfaller, L. B. Reller, M. P. Weinstein, R. J. Zabransky, and F. C. Tenover.
1994. Development of a standardized screening method for detection of vancomycin-resistant enterococci. J. Clin. Microbiol.32:1700–1704.
30. Tenover, F. C., J. Tokars, J. Swenson, S. Paul, K. Spitalny, and W. Jarvis.
1993. Ability of clinical laboratories to detect antimicrobial agent-resistant enterococci. J. Clin. Microbiol.31:1695–1699.
31. VanHorn, K. G., C. A. Gedris, and K. M. Rodney.1996. Selective isolation of
vancomycin-resistant enterococci. J. Clin. Microbiol.34:924–927.