Copyright © 1999, American Society for Microbiology. All Rights Reserved.
GB Virus C/Hepatitis G Virus Groups and Subgroups: Classification
by a Restriction Fragment Length Polymorphism Method Based
on Phylogenetic Analysis of the 59
Untranslated Region
J. F. QUARLERI,1,2V. L. MATHET,1,2M. FELD,1D. FERRARIO,1M. P. DELLA LATTA,1 R. VERDUN,3D. O. SA´ NCHEZ,3ANDJ. R. OUBIN˜ A1,2*
Laboratorio de Hepatitis Virales, Departamento Microbiologı´a, Facultad de Medicina, Universidad de Buenos Aires,1
Facultad de Medicina, Universidad del Salvador,2and Instituto de Investigaciones Biotecnolo´gicas,
Universidad de San Martı´n,3Buenos Aires, Argentina
Received 28 August 1998/Returned for modification 19 November 1998/Accepted 27 January 1999
A phylogenetic tree based on 150 5*untranslated region sequences deposited in GenBank database allowed segregation of the sequences into three major groups, including two subgroups, i.e., 1, 2a, 2b, and 3, supported by bootstrap analysis. Restriction site analysis of these sequences predicted that HinfI and either AatII or AciI could be used for genomic typing with 99.4% accuracy. cDNA sequencing and subsequent alignment of 21 Argentine GB virus C/hepatitis G virus strains confirmed restriction fragment length polymorphism patterns theoretically predicted. This method may be useful for a rapid screening of samples when either epidemiolog-ical or transmission studies of this agent are carried out.
GB virus C (GBV-C)/hepatitis G virus (HGV) is a recently described agent which has been proposed as a member of the
Flaviviridae family, distantly related to GBV-A and even more
related to HCV (18, 33). The virus is able to infect humans parenterally and vertically, although its true pathogenicity is under extensive study (2, 22). Its viral genome is a positive-strand RNA that appears to be approximately 9.4 kb long and that includes a long open reading frame. Based on sequence homology with HCV, it has been proposed that the predicted HGV polyprotein contains two putative envelope proteins (E1 and E2), an RNA helicase, a serine protease, and an RNA-dependent RNA polymerase (18, 27). Initial suggestions of an apparent lack of core coding region have very recently been challenged, when the expression of sequences upstream of the E1 coding region was demonstrated (41).
Sequence analysis of the most 59-terminal region shows a certain degree of heterogeneity among different isolates. Based on a study of 44 sequences from around the world, Muerhoff et al. (24) initially proposed the existence of three genotypes, i.e., 1, 2, and 3, the first two including two subtypes each. When such analysis was extended to a fragment of 374 nucleotides (nt) of the 59untranslated region (UTR) from 83 isolates, four groups, namely, 1, 2a, 2b, and 3, were finally established (23). Up to now, at least two attempts have been made in order to provide a rapid method for HGV typing (25, 29). However, the growing number of HGV sequences deposited in the GenBank database and the changing view regarding types and subtypes led us to improve our initial method. To reach this goal, all so-called complete HGV sequences deposited in GenBank at the time of submission of this manuscript (n522) and partial sequences from the 59UTR were used for phylogenetic anal-ysis and predictive restriction fragment length polymorphism (RFLP) patterns in this study. Since a consensus nomenclature about GBV-C/HGV grouping and subgrouping is still lacking and because the observed degree of genomic diversity at 59
UTR is not similarly represented within other genomic regions (34), we will use the terms group and subgroup instead of type and subtype. In this study, we propose a rapid method for GBV-C/HGV classification by 59UTR cDNA amplification followed by enzymatic cleavage to obtain group- and subgroup-specific RFLP profiles.
MATERIALS AND METHODS
RNA extraction and cDNA synthesis.RNA was initially obtained from 200ml of a reference plasma, PNF2161 (18), kindly provided by Margaret Gallagher (Centers for Disease Control and Prevention, Atlanta, Ga.). After reverse tran-scription (RT) and amplification conditions were settled, serum samples from 100 parenterally HIV-infected patients were studied. RNA was obtained from serum by guanidinium isothiocyanate-acidic phenol extraction (5), as previously described (28). RT was performed by incubating the template (equivalent to 100 ml of serum) in the presence of 200 pM outer antisense primer (GOA) 59CCC GGC CCC CAC TGG TCC TTG 39and 200 U of Moloney murine leukemia virus for 1 h at 37°C. Reverse transcriptase was inactivated by being heated to 95°C for 10 min, and cDNA was kept cold until PCR amplification, which was performed immediately.
PCR amplification.An aliquot of cDNA was amplified in a volume of 50ml by using GOA and an outer sense primer (GOS) 59CGG CAC TGG GTG CAA GCC CCA 39. After a denaturation step (2 min 30 s at 94°C), 35 cycles of PCR, with 1 cycle consisting of denaturation (30 s at 94°C), annealing (30 s at 55°C), and primer extension (45 s at 72°C), followed. Nested PCR was performed in a volume of 50ml after transfer of 5ml from the first round of PCR to a mix containing 200 pM inner sense primer (GIS, 59AGC CCC AGA AAC CGA CGC CTA 39) and an inner antisense primer (GIA, 59TAT TGG TCA AGA GAG ACA TTG 39). This second PCR was performed after a denaturation step as given above, followed by 40 cycles of PCR, with 1 cycle consisting of dena-turation (30 s at 94°C), annealing (30 s at 53°C), and extension (45 s at 72°C).
Amplicons of 325 to 329 bp were expected. Amplified sequences corresponded to positions located from nt 10 to 335 (with eventual insertions of deletions) from the most extreme 59UTR of GBV-C. Since this prototype strain appears to be 19 nt shorter than other isolates, another useful numbering method has been recently proposed by Smith et al. (34), who propose decreasing negative values from the most 59UTR nucleotide up to the last base before the first (putatively assigned) coding AUG.
cDNA sequencing.The specificity of the reaction was assessed by direct se-quencing of PCR amplicons. Briefly, 3 nested PCR mixtures were pooled, puri-fied in 7% polyacrylamide gels, eluted in 0.5 M NH4acetate, 0.01 M EDTA, and 0.1% sodium dodecyl sulfate, phenol-chloroform extracted, ethanol precipitated, and resuspended in 10ml of sterile distilled water. Manual and/or automatic sequencing by the chain termination method (31) was performed alternately with [g-32P]dATP-labelled GIS or GIA primer or with either primer in the presence of fluorescent dye-labelled chain terminators, respectively.
Molecular evolutionary genomic analysis.Considering the location and length (325 to 329 bp) of the above-mentioned predicted amplicons, both reported * Corresponding author. Mailing address: Departamento
Microbio-logı´a, Facultad de Medicina, Universidad de Buenos Aires, Paraguay 2155, Piso 11, (1121) Buenos Aires, Argentina. Phone: 54-11-4508-3689. Fax: 54-11-4508-3705. E-mail: [email protected].
1340
on May 15, 2020 by guest
http://jcm.asm.org/
complete (n5120) and partial 59UTR GBV-C/HGV sequences (n530) from GenBank, as well as from virus-infected Argentine patients (n521), were included for alignment and phylogenetic analysis. An initial phylogenetic analysis was performed on downloaded GenBank sequences. DNA alignments were generated with the Clustal X program (11, 13, 38). Evolutionary distances be-tween sequences were determined with the DNADIST program (Kimura two-parameter method) of the PHYLIP package version 3.5c (8). The computed distances were used for the construction of phylogenetic trees by the neighbor-joining method of NEIGHBOR program. The robustness of the trees was as-sessed by bootstrap resampling with the programs SEQBOOT (to generate 1,000 reshuffled sequences), DNADIST, and NEIGHBOR. The consensus tree was calculated with CONSENSE. Bootstrap values of less than 70% were regarded as not providing strong support for the phylogenetic grouping. The tree was ob-tained from RETREE program (PHYLIP package) with the midpoint rooting option. Final graphical output was created with the program TREEVIEW. The MapDraw program (DNASTAR Package, Lasergene for Windows) was selected to detect group- and subgroup-specific restriction sites.
Sequences deposited in GenBank database and used in the analysis are shown in Table 1.
Nucleotide sequence accession numbers.The following sequences from GBV-C/HGV Argentine strains were deposited in GenBank: AF081562, AF081564, AF116325, AF116326, AF116327, AF116328, AF116329, AF116334, AF116335, AF116338, AF116339, AF116340, AF081561, AF116330, AF116332, AF116333, AF081563, AF116324, AF116331, AF116336, and AF116337.
RESULTS
Molecular genomic GBV-C/HGV analysis and predicted RFLP patterns.In order to determine whether restriction sites of GBV-C/HGV sequences deposited in the GenBank database might be useful in distinguishing groups and subgroups, 150 59
[image:2.612.57.552.81.563.2]UTR sequences discussed above were selected. Of the 150 TABLE 1. GBV-C/HGV sequences deposited in GenBank
Isolate accession no.GenBank Reference Isolate accession no.GenBank Reference Isolate accession no.GenBank Reference
1 AF031828 4 51 U59521 24 101 Y15258 19
2 AB008335 14 52 U59522 24 102 Y15259 19
3 AB008342 1 53 U59523 24 103 Y15263 19
4 AF006957 17 54 U59524 24 104 Y15264 19
5 AF006958 17 55 U59525 24 105 Y15265 19
6 AF006959 17 56 U59526 24 106 Y15266 19
7 AF006960 17 57 U59527 24 107 AF038796 20
8 AF006961 17 58 U59528 24 108 AF038797 20
9 AF006962 17 59 U59529 24 109 AF038798 20
10 AF006963 17 60 U59530 24 110 AF038799 20
11 AF006964 17 61 U59531 24 111 AF038800 20
12 AF006965 17 62 U59532 24 112 AF038801 20
13 AF006966 17 63 U59533 24 113 AF038802 20
14 AF006968 17 64 U59534 24 114 AF038803 20
15 AF006969 17 65 U59535 24 115 AF038804 20
16 AF006970 17 66 U59536 24 116 AF038805 20
17 AF006971 17 67 U59537 24 117 AF038806 20
18 AF006972 17 68 U59538 24 118 AF038807 20
19 AF006973 17 69 U59539 24 119 AF038808 20
20 AF006974 17 70 U59540 24 120 AF038809 20
21 AF006975 17 71 U59541 24 121 AF038810 20
22 AF006976 17 72 U59542 24 122 AF038811 20
23 AF006977 17 73 U59543 24 123 AF038812 20
24 AF006978 17 74 U59544 24 124 AF038813 20
25 AF006979 17 75 U59545 24 125 AF038814 20
26 AF006980 17 76 U59546 24 126 AF078047 40
27 AF006981 17 77 U59547 24 127 AF078049 40
28 AF006982 17 78 U59548 24 128 AF078050 40
29 AF006983 17 79 U59549 24 129 AF078054 40
30 AF006984 17 80 U59550 24 130 AF078055 40
31 AF006985 17 81 U59551 24 131 AF078057 40
32 AF006986 17 82 U59552 24 132 AF078058 40
33 AF031827 4 83 U59553 24 133 AF078059 40
34 AF031829 4 84 U59554 24 134 AF078060 40
35 D87249 9 85 U59555 24 135 AF078062 40
36 D87250 9 86 U59556 24 136 AF078063 40
37 D87251 9 87 U59557 24 137 AF078064 40
38 D87252 9 88 U59558 24 138 AF078065 40
39 D87253 9 89 U63715 7 139 AF078799 40
40 D87254 9 90 U76892 12 140 AF081782 45
41 D87255 32 91 U76893 12 141 D87262 26
42 D87708 14 92 U76894 12 142 D87263 26
43 D87715 14 93 U94695 39 143 D87709 14
44 D90600 27 94 AF015870 43 144 D87710 14
45 U36380 9 95 AF015871 43 145 D87711 14
46 U44402 18 96 AF015872 43 146 D87712 14
47 U45966 18 97 AF015876 43 147 D87713 14
48 U59518 24 98 AF015877 43 148 D87714 14
49 U59519 24 99 AF015878 43 149 AF078048 40
50 U59520 24 100 Y15257 19 150 AF006967 17
on May 15, 2020 by guest
http://jcm.asm.org/
sequences, 120 encompassed the complete length of 325 to 329 nt extended between the 59ends of GIS and GIA prim-ers; 22 of them were regarded as full-length genomic se-quences. Another group of 30 sequences (AF006957 to AF006986) did not have the first 37 nt (positions 10 to 46 according to the GBV-C numbering system) of our amplified products. Consequently, our sequence alignment used a frag-ment corresponding to positions 47 to 335, following the GBV-C numbering system.
After sequence alignment, a phylogenetic tree was con-structed (Fig. 1). Three major groups, one including two sub-groups, were evident, namely, 1, 2a, 2b, and 3.
Analysis of restriction sites of all the sequences showed that endonucleases could be used in genomic typing and subtyping. Initial cleavage with HinfI produces at least 10 different pat-terns, which we named H1, H2, H3, H4, H5, H6, H7, H8, H9, and H10 (Fig. 2). Partial sequences (about 290 bp long) were also included and classified as compatible with a given pattern (the pattern number followed by an asterisk). Group 1 was as-sociated with H1, H2, and H5 patterns; group 2 was asas-sociated with H1, H3, or H3* (strong association), H4 or H4*, H6*, H8, H9, and H10 patterns; and group 3 was associated with H1 or H1* or infrequently with H7 (Fig. 2).
Since H1 and H1* patterns were observed almost exclusively with either group 1 or 3, sequences with these profiles were resolved by means of AatII, taking into account that all such group 1 sequences (n518) were not cut, in contrast to all H1 and H1* group 3 sequences (n529) which could be cleaved at position 231 of the fragment (nt 241 from the most 59terminal base) (Fig. 2). Thus, computer-predicted RFLP with AatII of H1 or H1*, H2, H5, or H7 sequences (whose HinfI RFLP profile was compatible with group 1 and/or 3) allowed their segregation into either group: 22 sequences were assigned to group 1 and the other 18 were assigned to group 3 (Fig. 2). AciI cleavage allowed subgroup 2a to be segregated from subgroup 2b; while all 2b sequences (n 5 12) were not cut, all 2a se-quences (n584) could be cleaved at several restriction sites (Fig. 2).
RT-PCR amplification.As a positive control, the specificity of the designed primers was assessed when only products of the expected size (no spurious bands) were observed after RT and subsequent amplification of the RNA obtained from the ref-erence plasma PNF2161 (U44402). This system also allowed the detection of GBV-C/HGV RNA in 21 of 100 parenterally HIV-infected patients.
RFLP and cDNA sequencing.All Argentine isolates (n 5
21) were grouped by the RFLP method described in this study. GBV-C/HGV strains were classified as group 1, 2a, 2b, and 3. The accuracy of the proposed methodology was subsequently substantiated by cDNA sequencing, since the phylogenetic analysis of the sequences obtained showed complete agree-ment with the observed RFLP patterns from each sample (Fig. 3 and 4). GBV-C/HGV sequences from Argentine strains deposited in GenBank, belonged to the following groups: group 1, AF081562; subgroup 2a, AF081564, AF116325, AF116326, AF116327, AF116328, AF116329, AF116334, AF116335, AF116338, AF116339, and AF116340; subgroup 2b, AF081561, AF116330, AF116332, and AF116333; and group 3, AF081563, AF116324, AF116331, AF116336, and AF116337.
In agreement with a previous study (23), reference sample PNF2161 was classified as a member of group 2a by phyloge-netic analysis. Its predicted group 2a RFLP profile was also con-firmed by gel electrophoresis.
Within group 2, two unusual sequences were observed among Argentine samples with GenBank accession nos. AF116327 and AF116340, which showed a G at position 166, leading to
the loss of a HinfI restriction site. These isolates were classified as group 2a by both RFLP and sequence alignment with pre-viously characterized isolates. Their unusual sequences showed 100% homology with a rare GBV-C/HGV variant downloaded from GenBank (sequence AF006961).
Phylogenetic analysis.Three major groups, i.e., 1, 2 (includ-ing two subgroups) and 3 were observed (Fig. 1). This result was substantiated after 1,000 replicates for bootstrap analysis. Grouping into subgroups 2a and 2b was undoubtedly support-ed by 70 and 93% of all trees, respectively. In contrast, no suf-ficient support was observed for subgroups within group 1 (45 and 86% for the two observed branches) with the sole excep-tion of sequence AF078048 from Singapore (40) which might represent a different (sub)group. RFLP profile from this se-quence is shown among AatII restriction patterns in Fig. 2, fol-lowed by a “?” and in the phylogenetic tree from Fig. 1 as se-quence 149.
DISCUSSION
Our sequence alignment confirmed isolate grouping of GBV-C/HGV strains included in a recently reported study by anal-ysis of a partially overlapping 374-nt region (positions 79 to 447).
Considering all 150 sequences, three major groups (i.e., 1, 2 [including two subgroups], and 3) were observed as depicted in Fig. 1.
The initial visual and computational analyses, including both their alignment and phylogenetic relatedness, of 94 sequences allowed the detection of conserved regions to design universal primers, as well as group- and subgroup-specific restriction sites. When extended to additional 56 sequences (total num-ber, 150) the same clustering was observed, although a few more RFLP patterns were evident. Although not closely re-lated to other group 2a strains, sequence analysis of the refer-ence isolate PNF2161 also supported the proposed methodol-ogy. Moreover, cDNA sequencing of 21 Argentine isolates confirmed GBV-C/HGV typing based on 59UTR RFLP (data not shown).
Interestingly, sequence U75356 (Chinese isolate HGVC964 [44]) was putatively assigned to group 2a or 3 according to the length and subregion location analyzed at 59 UTR (23) (not included in the phylogenetic analysis shown). It was classified into a separate group by another phylogenetic tree when we analyzed its sequence, associated with the observed insertion of three consecutive nucleotides (data not shown). This se-quence exhibited the predicted group 3 RFLP pattern, also in agreement with an analysis of its artificially combined E2-NS3-NS5b genome (23). Subsequently, sequence U75356 was de-scribed as the unique example of a putative genotype 4 by Smith et al. (34).
At least three items deserve special comments. First, al-though to a lesser extent than with 59UTR HCV isolates (since genetic distances between GBV-C/HGV groups are most sim-ilar to those observed between HCV subtypes) GBV-C/HGV strains show sufficient heterogeneity to allow their clustering in at least three major groups, group 2 including two subgroups and group 1 possibly including at least one sequence (sequence 149 [Fig. 1]) clustered separately. Considering that such low level of heterogeneity might signify minor changes at the amino acid level in coding regions, antigenic diversity would seem to be unlikely reflected in clinical or biological differ-ences of this viral infection. However, the effects of mutations at 59UTR should be thoroughly investigated, since significant differences of biological properties could be associated with them, as has been shown with other viruses, i.e. poliovirus,
on May 15, 2020 by guest
http://jcm.asm.org/
FIG. 1. Phylogenetic tree of 290-nt 59UTR sequences deposited in GenBank. The GenBank accession numbers of the isolates are given in Table 1. A distance scale (in nucleotide substitutions per position) is shown.
on May 15, 2020 by guest
http://jcm.asm.org/
HCV, etc. Second, of 150 previously reported sequences and of the 21 Argentine isolates shown here, almost no discrepancies between phylogenetic and RFLP analyses of the 59UTR were observed with the unique exception of sequence AF078048 (40) which clustered with genogroup 1 but exhibited a group 3 RFLP profile (Fig. 1 and 2). As stated above, this isolate might represent a new (sub)group. Third, regarding a putative geo-graphical distribution of genogroups, it should be noted that all groups and subgroups were observed in Argentine patients. These results extend the initial observations of Muerhoff et al, who documented the existence of two 2a and one 2b isolate from Argentina (23). GBV-C/HGV genotypic prevalence in Ar-gentine patients and blood donors will be published elsewhere.
Based on a recent report showing that a 374-nt sequence from the 59 UTR truly represents differences in the whole genome (23), the methodology described here might be useful for a rapid screening of GBV-C/HGV isolates when epidemi-ological studies are undertaken. Indeed, selected 59UTR re-gions from both studies partially overlap. Furthermore,
[image:5.612.59.547.87.483.2]an-other recent study also supports the use of the 59 UTR for GBV-C/HGV grouping (34). Mukaide et al. have also pro-posed RFLP typing of GBV-C/HGV variants based on the phylogenetic study of shorter 59UTR sequences (183 nt). How-ever, their analysis of 33 sequences was unable to substantiate group 2 subgrouping (25). Moreover, when we analyzed Argen-tine sequence AF116331, its phylogenetic grouping showed that it belongs to group 3, as also exhibited by our RFLP method (restriction patterns H1 and Aa2). However, accord-ing to the previously mentioned procedure (25), it should have been classified as HG type, assumed to correspond to group 2 following Muerhoff’s nomenclature (23). Likewise, recently available sequences downloaded from GenBank (i.e., AF038810 to AF038814 and Y15266) exhibit RFLP patterns not initially described by Mukaide et al. In this regard, it should be stressed that other researchers have demonstrated that only sequences that are long enough could be used to properly perform phylogenetic analysis to obtain reliable data (23, 34). In our proposed method, the use of GIS primer allowed the
FIG. 2. Predicted RFLP patterns obtained by computer analysis of 150 59 UTR GBV-C/HGV sequences downloaded from GenBank. The 21 Argentine
GBV-C/HGV sequences in this study also supported the proposed RFLP method. RFLP patterns with an asterisk belong to 290-bp partial sequences starting at position 47 according to the GBV-C numbering system. The numbers under restriction fragment length polymorphism indicate the expected sizes (in base pairs) of the fragments, and the numbers of sequences with a given pattern in group or subgroup 1, 2a, 2b, and 3 and the total number (n) are shown.
on May 15, 2020 by guest
http://jcm.asm.org/
eventual detection of a HinfI cleavage site at position 38 of a given amplicon (nt 48 according to the GBV-C numbering system). Such a site appeared to be important to detect a high percentage of genogroup 2 sequences, while group 2 sub-grouping is consistently determined by AciI digestion (Fig. 2). However, among 84 group 2a sequences downloaded from GenBank and characterized by phylogenetic relationships sup-ported by bootstrap analysis, two (accession nos. AF038812 and AF038813; [20]) deserve special comments. These two sequences are shown with a dagger symbol in Fig. 2 among sequences producing H1 (HinfI) and Aa1 (AatII) restriction patterns. Therefore, such a profile is compatible with genotype 1. For properly assigning genogroup to these two unusual se-quences, it was necessary to perform also AciI digestion, where an Ac2 pattern was observed, in contrast to all genuine group 1 sequences where this profile was not documented (n522). A proposed algorithm of enzyme digestions is shown in Fig. 5. We suggest initially cleaving amplicons with HinfI. Accord-ing to the pattern observed (H1 to H10), subsequent diges-tions could be performed either with AatII (usually to distin-guish between groups 1 and 3) and AciI to differentiate subgroups 2a and 2b. From a practical point of view, the two subgroup 2a sequences discussed above, AF038812 and AF038813, appeared to add a third step in the algorithm of digestions to distinguish between group 1 and group 3 [i.e., (i)
HinfI, (ii) AatII, and (iii) AciI]. However, it is worth
mention-ing that these two sequences belong to a smention-ingle isolate (called S4) from which five clones were analyzed, only two (clones 4 and 5) showing the loss of a HinfI site due to the presence of a C at position 157, instead of G (20). Therefore, the possibility of picking an RT-PCR amplification error or the detection of nonpredominant quasispecies cannot be ruled out. Whether such sequences truly represent a feature from a proportion of Spanish patients remains to be determined. This methodology allowed proper assignment of 149 of 150 sequences
down-loaded from GenBank and 21 of 21 Argentine strains (n5171; 99.4% accuracy).
With regard to other sequences not included in this study, it should be mentioned that those entire polyprotein open read-ing frame sequences (AB003291 and AB003292) proposed to be new genotypes (36) could not be analyzed, since infor-mation about their sequence at 59UTR does not show 111 nt from the most 59 end of the corresponding amplicon of our study. A similar exclusion criteria—with variable lengths in the region to be analyzed—was used for other several sequences (1, 21, 44).
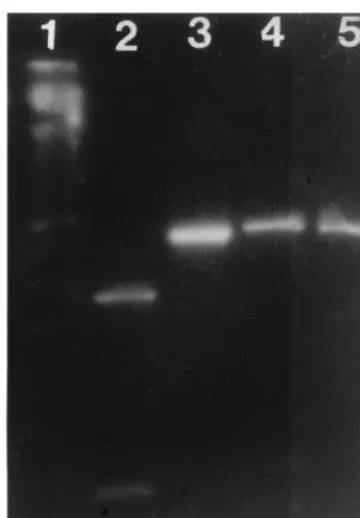
[image:6.612.84.264.71.330.2]To be useful, a typing method based initially on RT-PCR detection of genomic sequences must be able to detect most, if not all, strains. Therefore, universal primers are usually select-ed from conservselect-ed regions. This was the case when we initially designed primers used in this method. However, the increasing number of GBV-C/HGV sequences deposited in GenBank demonstrated that few mismatches were evident with some sequences. This appeared to be a problem with sequences like FIG. 4. RFLP characterization of GBV-C/HGV 2a (lanes 4 and 5) and 2b (lanes 2 and 3) subgroups. AciI (lanes 2 and 4) and HinfI were used. Lane 1 contains 50-bp ladder.
FIG. 5. Proposed algorithm for GBV-C/HGV typing by enzymatic digestion. FIG. 3. RFLP characterization of GBV-C/HGV group 1 (lanes 4 and 5) and
group 3 (lanes 2 and 3). AatII (lanes 2 and 4) or HinfI (lanes 3 and 5) were used. Lane 1 contains 50-bp ladder.
on May 15, 2020 by guest
http://jcm.asm.org/
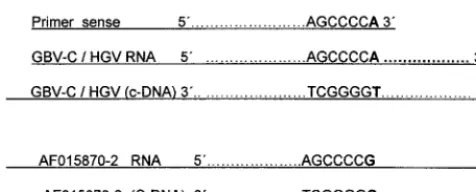
AF015870 to AF015872 and AF015876 to AF015878 (43) for which GOS primer exhibits a mismatch at its 39end. Potential inefficacy in the RT-PCR amplification might be overcome by using primers with ambiguities at the critical nucleotide posi-tions. As can be observed by visual and computer analyses, the mismatch at the most 39end of the outer sense primer would be crucial to hybridize the cDNA synthesized by extension from the 39end of the antisense primer, as shown in Fig. 6.
At first glance, it would appear that sense primer cannot efficiently amplify sequences like AF015870 to AF015872 and other related sequences. However, Kwok et al. (15) have ele-gantly demonstrated that this is not the case. They unambigu-ously demonstrated by experiments done in triplicate that de-spite an A at the 39end of a primer, efficiency of amplification is 100% when the corresponding position at the template is occupied by a C. This effect is reciprocal (either the mismatch is in the template or in the primer). This research group, among many others, have also observed that a single internal mismatch (as very unfrequently observed with few GBV-C/ HGV sequences) does not significantly affect yield of amplifi-cation (15, 35).
So far, whether GBV-C/HGV sequence variability influ-ences the course of infection is unknown. However, such vari-ation has been associated with a different outcome with the related HCV (3). In this regard, Japanese (42) and German (10) researchers have reported strain-specific association with fulminant hepatitis. Although such findings were not subse-quently extended, the recent discovery of GBV-C/HGV in bone marrow, spleen, and liver (16) deserves thorough studies regarding cell tropism and potential influence of viral genomic variations in the infection of such tissues. While newly discov-ered sequences will probably warrant periodical updating of the proposed methodology, this study also contributes to the knowledge of the molecular epidemiology of viral hepatitis in Argentina (28, 30, 37).
ACKNOWLEDGMENTS
This study was partly supported by grants from University of Buenos Aires (ME/009) and CONICET (PIP 6554), Argentina.
We are truly indebted to Betty H. Robertson and Margaret Gal-lagher (Centers for Disease Control and Prevention) for providing an aliquot of the reference plasma PNF2161 and to Osvaldo Libonatti and Mirna Biglione for kindly providing most of the HIV-infected sera.
ADDENDUM IN PROOF
While this article was in press, a related paper was published by J. M. Lo´pez-Alcorocho, I. Castillo, J. F. Tomas, and I. Carren˜o (J. Med. Virol. 57:80–84, 1999).
REFERENCES
1. Abe, K., and T. Kaneko. 1997. Unpublished data.
2. Alter, H. 1996. The cloning and clinical implications of HGV and HGBV-C. N. Engl. J. Med. 334:1536–1537.
3. Amoroso, P., M. Rapicetta, M. E. Tosti, A. Mele, E. Spada, S. Buonocore, G.
Lettieri, P. Pierri, P. Chionne, A. R. Ciccaglione, and L. Sagliocca.1998. Correlation between virus genotype and chronicity rate in acute hepatitis. C. J. Hepatol. 28:939–944.
4. Bukh, J., J. P. Kim, S. Govindarajan, C. L. Apgar, S. K. H. Foung, J. Wages,
J. A. Yun, M. Shapiro, S. U. Emerson, and R. H. Purcell.1998. Experimental infection of chimpanzees with hepatitis G virus and genetic analysis of the virus. J. Infect. Dis. 177:855–862.
5. Chomczynski, P., and N. Sacchi. 1987. Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Bio-chem. 162:156–159.
6. Christopherson, C., J. Sninsky, and S. Kwok. 1997. The effects of internal primer-template mismatches on RT-PCR: HIV-1 model studies. Nucleic Acids Res. 25:654–658.
7. Erker, J. C., J. N. Simons, A. S. Muerhoff, T. P. Leary, M. L. Chalmers, S. M.
Desai, and I. K. Mushahwar.1996. Molecular cloning and characterization of a GB virus C isolate from a patient with non-A-E hepatitis. J. Gen. Virol.
77:2713–2720.
8. Felsenstein, J. 1993. PHYLIP inference package, version 3.5c. University of Washington, Seattle.
9. Fukushi, S., C. Kurihara, N. Ishiyama, H. Okamura, F. B. Hoshino, A. Oya,
and K. Katayama.1996. Nucleotide sequence of the 59non-coding region of hepatitis G virus isolated from Japanese patients: comparison with reported isolates. Biochem. Biophys. Res. Commun. 226:314–318.
10. Heringlake, S., S. Osterkamp, C. Trautwein, H. L. Tillmann, K. Bo¨ker, S.
Muerhoff, I. K. Mushahwar, G. Hunsmann, and M. P. Manns.1996. Asso-ciation between fulminant hepatic failure and a strain of GBV virus C. Lancet 348:1626–1629.
11. Higgins, D. G., A. J. Bleasby, and R. Fuchs. 1992. Clustal V: improved software for multiple sequence alignment. Comput. Appl. Biosci. 8:189–191. 12. Hsieh, S. Y., P. Y. Yang, H. C. Chen, and Y. F. Liaw. 1997. Cloning and characterization of the extreme 59-terminal sequences of the RNA genomes of GB virus C/hepatitis G virus. Proc. Natl. Acad. Sci. USA 94:3206–3210. 13. Jeanmougin, F., J. D. Thompson, M. Gouy, D. G. Higgins, and T. J. Gibson. 1998. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 23: 403–405.
14. Katayama, K., S. Fukushi, C. Kurihara, N. Ishiyama, H. Okamura, F. B.
Hoshino, and A. Oya.1998. Full-length GBV-C/HGV genomes from nine Japanese isolates: characterization by comparative analyses. Arch. Virol.
143:1–13.
15. Kwok, S., D. E. Kellogg, N. McKinney, D. Spasic, L. Goda, C. Levenson, and
J. J. Sninsky.1990. Effects of primer-template mismatches on the polymer-ase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 18:999–1005.
16. Laskus, T., M. Radkowski, L.-F. Wang, H. Vargas, and J. Rakela. 1998. Detection of hepatitis G virus replication sites by using highly strand-specific
Tth-based reverse transcriptase PCR. J. Virol. 72:3072–3075.
17. Linnen, J. M., K. Fung, K. E. Fry, M. Mizokami, K. Ohba, J. M. Wages, Jr.,
Z.-Y. Zhang-Keck, K. Song, and J. P. Kim.1997. Sequence variation and phylogenetic analysis of the 59terminus of hepatitis G virus. J. Viral Hepa-titis 4:293–302.
18. Linnen, J., J. Wages, Jr., Z.-Y. Zhang-Keck, K. E. Fry, K. Z. Krawczynski,
H. Alter, E. Koonin, M. Gallagher, M. Alter, S. Hadziyannis, P. Karayiannis, K. Fung, Y. Nakatsuji, J. W.-K. Shih, L. Young, M. Piatak, Jr., C. Hoover, J. Ferna´ndez, S. Chen, J.-C. Zou, T. Morris, K. C. Hyams, S. Ismay, J. D. Lifson, G. Hess, S. K. H. Foung, H. Thomas, D. Bradley, H. Margolis, and J. P. Kim.1996. Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science 271:505–508.
19. Liu, H. F., C. Cornu, M. Jadoul, K. Dahan, G. Loute, and P. Goubau. 1998. Molecular analysis of GB virus C isolates in Belgian hemodialysis patients. J. Med. Virol. 55:118–122.
20. Lo´pez-Alcorocho, J. M., I. Castillo, J. F. Tomas, and V. Carren˜o. Unpub-lished data.
21. Lu, L., S. W. K. Im, M. H. Ng, and Y. S. Fu. Unpublished data. 22. Miyakawa, Y., and M. Mayumi. 1997. Hepatitis G virus: a true hepatitis virus
or an accidental tourist? N. Engl. J. Med. 336:795–796.
23. Muerhoff, A. S., D. B. Smith, T. P. Leary, J. C. Erker, S. M. Desai, and I. K.
Mushahwar.1997. Identification of GB virus C variants by phylogenetic analysis of 59untranslated and coding region sequences. J. Virol. 71:6501–6508. 24. Muerhoff, A. S., J. N. Simons, T. P. Leary, J. C. Erker, M. L. Chalmers, T. J.
Pilot-Matı´as, G. J. Dawson, S. M. Desai, and I. K. Mushahwar.1996. Sequence heterogeneity within the 59-terminal region of the hepatitis GB virus C genome and evidence for genotypes. J. Hepatol. 25:379–384. 25. Mukaide, M., M. Mizokami, E. Orito, K. Ohba, T. Nakano, R. Ueda, K.
Hikiji, S. Iino, S. Shapiro, N. Lahat, Y.-M. Park, B.-S. Kim, T. Oyunsuren, M. Rezieg, M. N. Al-Ahdal, and J. Y. N. Lau.1997. Three different GB virus C/hepatitis G virus genotypes. Phylogenetic analysis and a genotyping assay based on restriction fragment length polymorphism. FEBS Lett. 407:51–58. 26. Nakao, H., H. Okamoto, M. Fukuda, F. Tsuda, T. Mitsui, K. Masuko, H.
[image:7.612.56.294.70.166.2]Iizuka, Y. Miyakawa, and M. Mayumi.1997. Mutation rate of GB virus C/ hepatitis G virus over the entire genome and in subgenomic regions. Virol-ogy 233:43–50.
FIG. 6. Sequence complementarity between GOS primer and GBV-C/HGV RNA and cDNA. AF015870-2, AF015870 to AF015872.
on May 15, 2020 by guest
http://jcm.asm.org/
27. Okamoto, H., H. Nakao, T. Inoue, M. Fukuda, J. Kishimoto, H. Iizuka, F.
Tsuda, Y. Miyakawa, and M. Mayumi.1997. The entire nucleotide sequence of two GB virus C/hepatitis G virus isolates of distinct genotypes from Japan. J. Gen. Virol. 78:737–745.
28. Oubin˜a, J. R., J. F. Quarleri, M. Rudzinski, C. Parks, I. Badı´a, and S. M.
Gonza´lez Cappa.1995. Genomic characterization of hepatitis C virus isolates from Argentina. J. Med. Virol. 47:97–104.
29. Quarleri, J. F., and J. R. Oubin˜a. 1997. A proposed rapid method for ge-nomic characterization of GBV-C/hepatitis G virus (HGV). Medicina (Bue-nos Aires) 57:717–719.
30. Quarleri, J. F., B. H. Robertson, V. Mathet, S. D. Sinha, I. Badı´a, B. Frider,
A. Ferro, C. Galoppo, S. Sookoian, G. Castan˜o, and J. R. Oubin˜a.1998. Ge-nomic and phylogenetic analysis of hepatitis C virus strains from Argentina. Medicina (Buenos Aires) 58:153–159.
31. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463–5467. 32. Shao, L., H. Shinzawa, K. Ishikawa, X. Zhang, M. Ishibashi, H. Misawa, N.
Yamada, H. Togashi, and T. Takahashi.1996. Sequence of hepatitis G virus genome isolated from a Japanese patient with non-A-E-hepatitis: amplifica-tion and cloning by long reverse transcripamplifica-tion-PCR. Biochem. Biophys. Res. Commun. 228:785–791.
33. Simons, J. N., T. P. Leary, G. J. Dawson, T. J. Pilot-Matı´as, A. S. Muerhoff,
G. G. Schlauder, S. M. Desai, and I. K. Mushahwar.1995. Isolation of novel virus-like sequences associated with human hepatitis. Nature Med. 1:564– 569.
34. Smith, D. B., N. Cuceanu, F. Davidson, L. M. Jarvis, J. L. K. Mokili, S.
Hamid, C. A. Ludlam, and P. Simmonds.1997. Discrimination of hepatitis G virus/GBV-C geographical variants by analysis of the 59non-coding region. J. Gen. Virol. 78:1533–1542.
35. Sommer, R., and D. Tautz. 1989. Minimal homology requirements for PCR
primers. Nucleic Acids Res. 17:6749.
36. Takahashi, K., M. Hijikata, K. Hino, and S. Mishiro. 1997. Entire polypro-tein-ORF sequences of Japanese GBV-C/HGV isolates: implications for new genotypes. Hepatol. Res. 8:139–148.
37. Telenta, P. F. S., G. Palacios Poggio, J. L. Lo´pez, J. Gonza´lez, A. Lemberg,
and R. H. Campos.1997. Increased prevalence of genotype F hepatitis B virus isolates in Buenos Aires, Argentina. J. Clin. Microbiol. 35:1873–1875. 38. Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G.
Higgins.1997. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882.
39. Wang, H. L., and D.-Y. Jin. 1997. Prevalence and genotype of hepatitis G virus in Chinese professional blood donors and hepatitis patients. J. Infect. Dis. 175:1229–1233.
40. Wong, J., S. H. Chan, and E. C. Ren. Unpublished data.
41. Xiang, J., D. Klinzman, J. McLinden, W. N. Schmidt, D. R. LaBrecque, R.
Gish, and J. T. Stapleton.1998. Characterization of hepatitis G virus (GB-C virus) particles: evidence for a nucleocapsid and expression of sequences upstream of the E1 protein. J. Virol. 72:2738–2744.
42. Yoshiba, M., H. Okamoto, and S. Mishiro. 1995. Detection of the GBV-C hepatitis virus genome in serum from patients with fulminant hepatitis of unknown aetiology. Lancet 346:1131–1132.
43. Zanella, I., G. Fiordalisi, A. Bettinardi, M. Roncini, R. Stellini, and D.
Primi.Unpublished data.
44. Zhou, Y. S., W. Chen, Q. M. Zhao, H. L. Zhao, J. S. Zhang, J. Xu, and H. T.
Wang.1996. cDNA cloning and sequencing of HGV genome from Chinese. Bull. Acad. Mil. Med. Sci. 20:249–253.
45. Zhu, F. L., Z. T. Qi, and L. Shao. 1998. Splicing and cloning of the full length genomic cDNA of GB virus C/hepatitis G virus. Ti Erh Chun Ta Hsueh Hsueh Pao 19:301–306.