University of Pennsylvania
ScholarlyCommons
Publicly Accessible Penn Dissertations
1-1-2014
Telomere and Proximal Sequence Analysis Using
High-Throughput Sequencing Reads
Nick Stong
University of Pennsylvania, [email protected]
Follow this and additional works at:
http://repository.upenn.edu/edissertations
Part of the
Bioinformatics Commons
,
Genetics Commons
, and the
Molecular Biology Commons
This paper is posted at ScholarlyCommons.http://repository.upenn.edu/edissertations/1460 For more information, please [email protected].
Recommended Citation
Stong, Nick, "Telomere and Proximal Sequence Analysis Using High-Throughput Sequencing Reads" (2014).Publicly Accessible Penn Dissertations. 1460.
Telomere and Proximal Sequence Analysis Using High-Throughput
Sequencing Reads
Abstract
The telomere is a specialized simple sequence repeat found at the end of all linear chromosomes. It acts as a
substrate for telomere binding factors that in coordination with other interacting elements form what is
known as the shelterin complex to protect the end of the chromosome from the DNA damage repair
machinery. The telomere shortens with each cell division, and once critically short is no longer able to
perform this role. Short dysfunctional telomeres result in cellular senescence, apoptosis, or genome instability.
Telomere length is regulated by many factors including cis-acting elements in the proximal sequence which is
known as the subtelomere. The Riethman lab played a pivotal role in generating the reference sequence of the
subtelomere in both the human and mouse genomes, providing an essential resource for this work. Short high
throughput sequencing (HTS) reads generated from the simple repeat containing telomere or the segmental
duplication rich subtelomere cannot be aligned to a reference genome uniquely. They are filtered and excluded
from many HTS analysis methods. A ChIP-Seq analysis pipeline was developed to incorporate these
multimapping reads to study DNA-protein interactions in the subtelomere. This pipeline was employed to
search for factors regulating the expression TERRA, an essential long non-coding RNA, and to better
characterize their transcription start sites. ChIP-seq analysis in the human subtelomere found colocalization of
CTCF and Cohesin directly adjacent to the telomere and throughout the subtelomere specific repeats. Follow
up functional studies showed this binding regulated TERRA transcription at these sites. Extending these
analyses in the mouse genome showed very different patterns of CTCF and cohesin binding, with no evidence
of binding at apparent sites of TERRA transcription. Mouse subtelomere sequence analysis showed the
co-occurence of two repeats at sites of putative TERRA expression, MurSatRep1 and MMSAT4, one of which
was previously shown to be expressed in lincRNAs. The Telomere Analysis from SEquencing Reads(TASER)
pipeline was developed to capture telomere information from HTS data sets and used to investigate telomere
changes that occur in prostate cancer. TASER analysis of 53 paired prostate tumor and normal samples
revealed an overall decrease in telomere length in tumor samples relative to matched paired normal tissue,
especially sequence containing the exact canonical telomere repeat. Multimapping reads contain important
information, that when used properly, help elucidate understanding of telomere biology, cancer biology, and
genome regulation and stability.
Degree Type
Dissertation
Degree Name
Doctor of Philosophy (PhD)
Graduate Group
Genomics & Computational Biology
First Advisor
Keywords
Cancer Biology, ChIP-Seq, High Throughput, Next Generation, Sequencing, Telomere Biology
Subject Categories
Bioinformatics | Genetics | Molecular Biology
TELOMERE AND PROXIMAL SEQUENCE
ANALYSIS USING HIGH-THROUGHPUT
SEQUENCING READS
Nicholas Stong A DISSERTATION
in
Genomics and Computational Biology
Presented to the Faculties of the University of Pennsylvania in
Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
2014
Supervisor of Dissertation _____________________
Harold Riethman, Ph.D. Associate Professor,
Molecular and Cellular Oncogenesis Program, The Wistar Institute
Graduate Group Chairperson ______________________ Li-San Wang, Ph.D
Associate Professor of Pathology and Laboratory Medicine, University of Pennsylvania Dissertation Committee:
Junhyong Kim, Ph.D.
Endowed Professor, Department of Biology, University of Pennsylvania Lyle Ungar, Ph.D.
Associate Professor, CIS, University of Pennsylvania Bradley Johnson, M.D. Ph.D.
Associate Professor of Pathology and Laboratory Medicine, University of Pennsylvania Hongzhe Li, Ph.D.
ii
TELOMERE AND PROXIMAL SEQUENCE
ANALYSIS USING HIGH-THROUGHPUT
SEQUENCING READS
COPYRIGHT 2014
Nicholas Edward Stong
This work is licensed under the Creative Commons Attribution- NonCommercial-ShareAlike 3.0
License
To view a copy of this license, visit
iv
ACKNOWLEDGMENT
The Riethman lab was a wonderful environment for me to be exposed to the day to day wet lab work used to study telomere biology and have the computational resrouces to gain experience in bioinformatics. Harold has always been a kind mentor and thought of my personal achievments and development as a scientist. The Lieberman lab provided much of the support I needed to complete the biological story surrounding my computational findings. Particularly Zhong Deng who always had the right questions that would help me frame what I was working on along with helping me to validate many of my computational predictions. Amber Weiner was a fantastic undergrad that was always willing to work hard and learn everything I gave her.
ABSTRACT
TELOMERE AND PROXIMAL SEQUENCE ANALYSIS USING HIGH-THROUGHPUT
SEQUENCING READS
Nicholas Stong Harold Riethman, Ph.D.
The telomere is a specialized simple sequence repeat found at the end of all linear chromosomes. It acts as a substrate for telomere binding factors that in coordination with other interacting elements form what is known as the shelterin complex to protect the end of the chromosome from the DNA damage repair machinery. The telomere shortens with each cell division, and once critically short is no longer able to perform this role. Short dysfunctional telomeres result in cellular senescence, apoptosis, or genome instability. Telomere length is regulated by many factors including cis-acting elements in the proximal sequence which is known as the subtelomere. The Riethman lab played a pivotal role in generating the reference
vi
be expressed in lincRNAs. The Telomere Analysis from SEquencing Reads(TASER) pipeline was developed to capture telomere information from HTS data sets and used to investigate telomere changes that occur in prostate cancer. TASER analysis of 53 paired prostate tumor and normal samples revealed an overall decrease in telomere length in tumor samples relative to matched paired normal tissue, especially sequence containing the exact canonical telomere repeat. Multimapping reads contain important information, that when used properly, help
TABLE OF CONTENTS
ACKNOWLEDGMENT ... IV
ABSTRACT ... V
TABLE OF CONTENTS ... VII
LIST OF TABLES ... X
LIST OF ILLUSTRATIONS ... XI
CHAPTER 1: BACKGROUND ... 1
1.1 Telomere Biology ... 1
1.1.1 Telomere Structure ... 1
1.1.2 Telomere DNA Interactions ... 3
1.1.3 Telomeres and Disease ... 6
1.1.4 Subtelomere Structure ... 10
1.1.5 Subtelomere Transcripts ... 11
1.2 Cohesin and CTCF ... 12
1.3 High Throughput Sequencing ... 13
1.3.1 Technology ... 13
1.3.2 Alignment ... 15
1.3.3 Applications ... 16
1.4 Outline of Dissertation ... 18
CHAPTER 2: PROXIMAL 15KB ANALYSIS ... 20
2.1 Introduction ... 20
2.2 Methods ... 22
2.2.1 ChIP-‐Seq data ... 22
2.2.2 Mapping ChIP-‐Seq data to human subtelomeres ... 23
2.3 Results ... 24
2.3.1 CTCF, cohesin, and RNAPII binding to the CpG-‐island promoters in human subtelomeres ... 24
2.3.2 CTCF binds directly upstream of the CpG-‐island and 29 repeat element found in subtelomeres .. 27
viii
2.4 Discussion ... 30
2.4.1 A foundation for a chromatin atlas of the human subtelomeres ... 30
2.4.2 Supplemental Figures ... 31
CHAPTER 3: HUMAN SUBTELOMERE ANALYSIS ... 33
3.1 ABSTRACT ... 33
3.2 INTRODUCTION ... 34
3.3 METHODS ... 36
3.3.1 Fosmid library screening, ... 36
3.3.2 Updated subtelomere assemblies. ... 36
3.3.3 Sequence Feature Annotation. ... 37
3.3.4 Short-‐read-‐based Annotation Pipeline. ... 37
3.3.5 Subtelomere Browser. ... 39
3.3.6 Peak/boundary association enrichment calculation. ... 39
3.3.7 Chromatin Immunoprecipitation (ChIP) assay. ... 39
3.4 RESULTS ... 40
3.4.1 Gap-‐filling and detection of distal telomeric structural variants ... 40
3.4.2 Updated Subtelomere Assemblies ... 41
3.4.3 Subtelomere Annotation ... 45
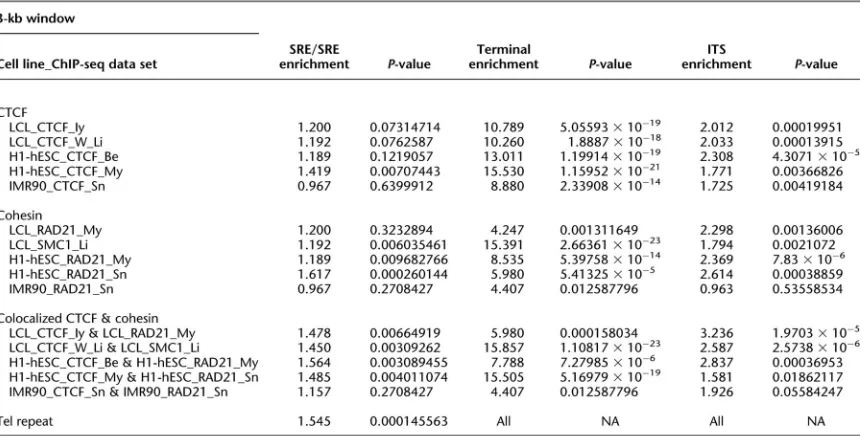
3.4.4 SRE Boundary Enrichments ... 48
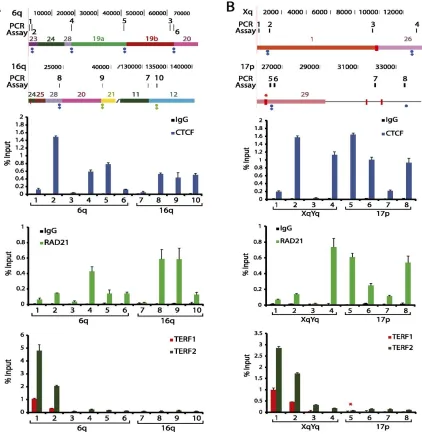
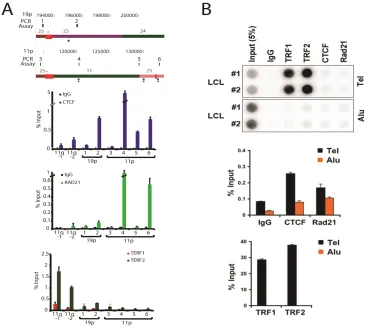
3.4.5 Experimental validation of ChIP-‐seq peaks by ChIP-‐qPCR ... 52
3.4.6 CTCF datasets from additional primary and cancer cell lines ... 56
3.5 DISCUSSION ... 56
3.6 DATA ACCESS ... 59
3.7 ACKNOWLEDGEMENTS ... 60
3.8 Supplementary Information ... 61
3.8.1 Supplementary Figures ... 61
CHAPTER 4: MOUSE SUBTELOMERE ANALYSIS ... 83
4.1 Abstract ... 83
4.2 Introduction ... 84
4.3 Methods ... 86
4.3.1 Sequence Feature Annotation ... 86
4.3.2 Subtelomere Sequence Characterization ... 87
4.3.3 Subtelomere Browser ... 88
4.3.4 Peak/boundary association enrichment calculation ... 88
4.4 Results ... 88
4.4.2 Subtelomere Annotation ... 90
4.4.3 Annotation of subtelomeric CTCF and cohesin binding sites using ChIPseq datasets. ... 96
4.5 Discussion ... 101
4.6 Supplemental Figures ... 103
CHAPTER 5: TELOMERE ANALYSIS USING TASER ... 108
5.1 Abstract ... 108
5.2 Introduction ... 109
5.3 Methods ... 111
5.3.1 Use and Optimization of RepeatMasker ... 111
5.3.2 Summary Statistics ... 112
5.3.3 Dataset ... 112
5.4 Results ... 114
5.4.1 Motivation ... 114
5.4.2 Prostate Cancer has short dysfunctional telomeres ... 116
5.4.3 Telomere metrics can be used to identify cancer ... 122
... 124
5.5 Discussion ... 125
5.6 Supplemental Figures ... 125
CHAPTER 6: CONCLUSION ... 135
BIBLIOGRAPHY ... 138
x
LIST OF TABLES
Table 3.1 − Subtelomeric sequences from telomeric clones ... 41
Table 3.2 −SRE boundary enrichments ... 51
Table 3.3 − Telomere sequence screen of end-sequences from the G248 and ABC7 fosmid libraries ... 70
Table 3.4 − Mate-pair mappings of telomere fosmid end sequences from G248 and ABC7 to the human reference assembly ... 72
Table 3.5 − (CCCTAA)4 - containing end sequences in Structural Variation Fosmid Libraries .... 73
Table 3.6 − Subtelomere mapping distribution of mate-pairs of (CCCTAA)n reads from ABC7, ABC8, and ABC14 libraries ... 76
Table 3.7 − Clone-based Subtelomere Assemblies ... 76
Table 3.8 − Hybrid genome joining coordinates of hg19 ... 77
Table 3.9 − Datasets used in this study and quality metrics ... 77
Table 3.10 − SRE boundary enrichment statistics ... 78
Table 3.11 − CTCF boundary analysis for 4 primary and 4 immortalized cell lines ... 79
Table 3.12 − ChIP-qPCR primers used ... 82
Table 4.1 − Segmental Duplication Content of the Mouse Subtelomere ... 93
Table 4.2 − Mouse Subtelomeric Clones ... 107
Table 4.3 − Datasets used in this study and quality metrics ... 107
LIST OF ILLUSTRATIONS
Figure 2.1 − Enrichment profiles for ChIP-Seq analysis of CTCF, cohesin, and RNAPII binding to
human subtelomeres ... 26
Figure 2.2 − Identification of CTCF-binding site elements in the 61-bp element of human subtelomeres ... 29
Figure 2.3 − Summary of ChIP-Seq analysis of CTCF, cohesin, and RNAPII binding to type I human subtelomeres ... 31
Figure 2.4 − Summary of ChIP-Seq analysis on type II human subtelomeres ... 31
Figure 2.5 − Validation of CTCF binding site at 10q human subtelomeres in BCBL1 cells ... 32
Figure 3.1 − Sequence organization of updated subtelomere sequence assemblies ... 45
Figure 3.2 − Subtelomere annotation features ... 46
Figure 3.3 − Example of an annotated subtelomere with CTCF and cohesin binding enrichment peaks from multiple cell types ... 50
Figure 3.4 − qPCR analysis of subtelomeric DNA protein binding sites predicted by ChIP-seq data set mappings ... 55
Figure 3.5 − G248 fosmid coverage of 2p subtelomere ... 64
Figure 3.6 − Stability of Subterminal DNA in Fosmids ... 65
Figure 3.7 − TERF1 and TERF2 ChIP-seq peak analysis ... 68
Figure 3.8 − Annotated Subtelomeres (screen shots of all subtelomeres) ... 69
Figure 3.9 − ChIP analysis of CTCF, RAD21, TERF1, and TERF2 binding at subtelomeric candidate sites predicted by ChIP-seq dataset mappings ... 69
Figure 4.1 − Subtelomere Annotation Features ... 91
Figure 4.2 − Examples of annotated subtelomeres 2q and 17q ... 92
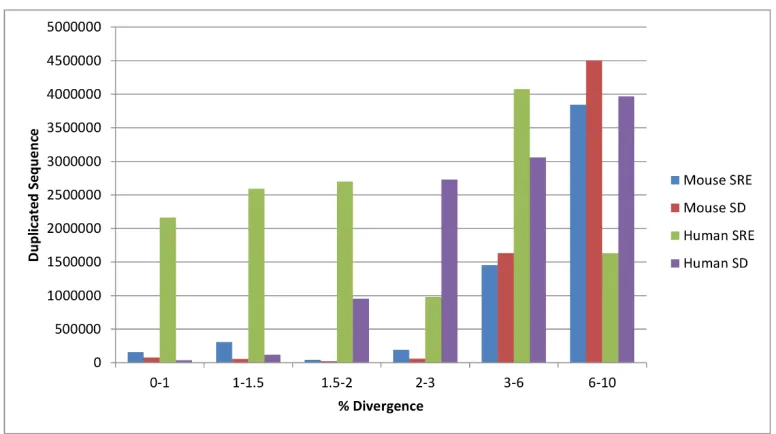
Figure 4.3 − Comparison of duplicated sequence in the mouse and human subtelomere ... 94
Figure 4.4 − Characteristics of interstitial telomere sequences in the mouse (A) and human (B) genome ... 95
Figure 4.5 − Sequence composition of murine subtelomeres ... 96
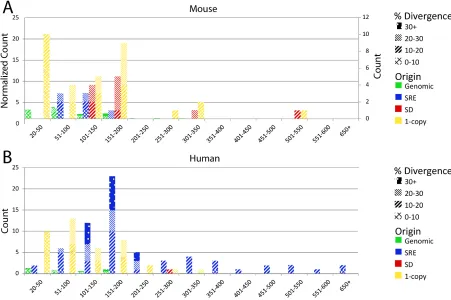
Figure − 4.6 Subtelomere features of mouse 18q relative to TERRA-associated features as described by de Silanes et al. (2014) ... 100
Figure 4.7 − Telomeric BAC Isolation ... 104
Figure 4.8 − Snapshots of Annotated Mouse Subtelomeres ... 104
Figure 4.9 − Mouse 18q Subtelomere Annotated using additional datasets ... 105
Figure 4.10 − Mouse 9q Subtelomere Annotated using additional datasets ... 106
Figure 5.1 − Idealized telomere structure ... 113
Figure 5.2 − Changes in TASER measurement distribution due to telomere shortening ... 115
Figure 5.3 − Box plot of total telomere measurement for normal and cancer samples ... 118
Figure 5.4 − Box plot of boundary measurement for normal and cancer samples ... 119
Figure 5.5 − Box plot of percent perfect measurement for normal and cancer samples ... 120
Figure 5.6 − Box plot of mutation interspersion ratio measurement for normal and cancer samples ... 121
Figure 5.7 − Fitted logistic regression model for tumor classifier ... 123
Figure 5.8 − ROC curve of tumor classifier ... 124
xii
Figure 5.11 − Box plot of percent perfect measurement for normal and cancer samples with
Individual sample points ... 128 Figure 5.12 − Box plot of mutation interspersion ratio measurement for normal and cancer
samples with individual sample points ... 129 Figure 5.13 − Histogram of difference in total telomere measurement between normal and cancer
samples ... 130 Figure 5.14 − Histogram of difference in boundary measurement between normal and cancer
samples ... 131 Figure 5.15 − Histogram of difference in percent perfect measurement between normal and
cancer samples ... 132 Figure 5.16 − Histogram of difference in mutation interspersion ratio measurement between
normal and cancer samples ... 133 Figure 5.17 − Box plot of mutation interspersion ratio measurement for normal and cancer
CHAPTER 1: BACKGROUND
1.1 Telomere Biology
1.1.1 Telomere Structure
In 1938 Herman Muller first hypothesized the presence of a terminal gene with the special function of sealing the end of a linear chromosome, which he termed the telomere. Drawing on years of work trying to understand the mechanisms of chromosome rearrangement after X-ray irradiation in Drosophila, he reasoned that if chromosomal breakage were to occur before rearrangement initiated, then it would be possible for the broken fragments to not find each other before the acentric fragment was lost in mitosis. However having never observed any such fragments he concluded that the telomere was required for chromosome stability[1]. It was also observed in Zea Mays that chromosome breaks created by the divergent pulling of centromeres in a dicentric chromosome results in unstable ends, which go through repeated breakage-fusion-bridge cycles[2]. The chromosome ends resulting from breaks are highly unstable and have a propensity to fuse leading to chromosomal rearrangements. Telomeres found at natural chromosome ends are required for genome stability.
It was several decades before the structure of the telomere began to be unraveled with the advent of DNA sequencing. Elizabeth Blackburn, a former graduate student of Fred Sanger, sequenced the telomeres of rDNA in Tetrahymena thermophilia by use of restriction
endonuclease digestion and found a tandemly repeated nucleotide sequence 5’(CCCCAA)n 3’
2
and is considerably longer in many other verterbrates, including in Mus musculus[9]. A universal feature of telomere sequences is a short single stranded overhang of the 3’ G rich strand[5,11]. In the human genome this overhang is on average 200bp[12] This genomic sequence is responsible for differentiating the ends of linear chromosomes from ends resulting from internal double strand DNA breaks.
Even before the sequence structure of the telomere was known, it was abstracted that the ends of the linear chromosomes could not be replicated completely. The mechanics of the semiconservative DNA replication are unable to fully duplicate template stands of DNA. DNA polymerase requires an RNA primer to begin DNA synthesis, which is later removed by RNAse. This combined with the directionality of DNA polymerase from 5’ to 3’ means the terminal 3’ end of both template strands cannot be duplicated [13–15]. Alexey Olovnikov and Jim Watson both independently described this as the end replication problem. This results in the shortening of the resulting chromosome by at least 12-15bp, if the RNA primer is placed at the last base of the 3’ end of the template strand. Olovnikov went further to argue that marginotomy of DNA (the shortening of replicated DNA in respect to the template), and the total loss of telogenes, leads to the elimination of aged cells, and speculated that it was responsible for the depletion of cell populations in the body and therefore a primary cause in disorders of aging[14]. The end replication problem leads to a 3’ overhang characteristic of telomere ends however only at the shortened end of the chromosome. In addition the shortening due to the end replication problem is lesser than the observed average 200bp overhang in the human genome[12]. This indicates that telomeric single stand 3’ overhangs are maintained through exonucleic degradation[16,17]. Telomere shortening occurs with DNA replication due to both the end replication and post replicative processing. This has been observed after cell divisions in both in vitro and in vivo[18,19].
The telomere is able to form a structure that protects it from being recognized as a double strand break by the DNA damage repair machinery through both the physical
The G rich tandem repeat of the telomere matches the sequence motif necessary to form G quadruplexes, a stable structure in which guanine residues form hydrogen bonds in a helical structure of one strand of DNA[20]. Given the single stand overhang at the telomere this conformation is energetically favorable[21]. There is extensive evidence for these structures occurring in vivo[22,23]. The single strand overhang is also able to invade the double stranded region of the telomere, forming a small single stranded displacement loop (D-loop) where the overhang disrupts the DNA duplex, and a large lasso-like loop (T-loop) at the chromosome end[24]. The formation of this loop depends on an interaction with telomere associated proteins[25].
1.1.2 Telomere DNA Interactions
4
CST complex in human cells is required for DNA replication, restarting stalled replication forks[43,44].
While shelterin protects the telomere from being recognized as a double strand break normal telomere shortening occurs with each cell division. In addition accelerated telomere shortening can occur due to oxidative stress[45] or polymerase pausing at sites of single stranded DNA damage leading to premature termination of DNA replication in the telomere[46]. To
overcome these mechanisms of shortening the cell is able to extend telomeres. Telomerase is a ribonucleoprotein that like the telomere sequence itself was first identified in Tetrahymena. Telomerase has terminal transferase activity that is able to directly add the telomeric repeat on to existing telomeres[47]. Telomerase is able to do this because it has an RNA component which contains the priming sequence for the addition of the whole telomere repeat on to the
chromosome end[48]. Telomerase is also responsible for lengthening telomeres in the human genome[49]. The human RNA component of telomerase, hTR, contains an 11 nucleotide template region, (5’-CUAACCCUAAC), which is complementary to the human telomere
sequence[50]. The protein component of telomerase is catalytically active and drives the reverse transcriptase activity of telomerase which adds the telomeric sequence[51,52]. This human telomerase reverse transcriptase (hTERT) subunit is the limiting factor in telomere lengthening. While hTR is expressed in all human tissues[53], hTERT is not expressed significantly in somatic cells[54,55].
response including increased p53 activity[61]. In mammalian cells this response to DNA damage is mediated through two protein kinases ATM and ATR. ATM recognizes double strand breaks and ATR recognizes a single strand from a resected double strand break[62]. As part of the damage response ATM and ATR phosphorylate histone H2AX (known as gamma-H2AX)[63,64], promoting the accumulation of damage response factors 53BP1, MDC1, and the MRN complex. This accumulation of damage response elements can be found at individual critically short telomeres[65–67]. ATM and ATR also phosphorylate Chk1 and Chk2 respectively which can cause G1 and G2 phase arrest and are involved in the activation of p53 which inhibits cell cycle progression through downstream activation of p21[68]. In certain cell types and conditions this same p53 mediated signaling cascade can also lead to apotosis[65,69].
6
is interacting to form a t-loop, resulting in a truncated telomere and leaving the cleaved t-loop as telomere circle (t-circle). The end of the telomere and internal double stranded telomere can form a Holliday junction which is cleaved. The formation of t-circles is dependent on XRCC3 and NBS1, consistent with known mechanisms of HR[79,80]. HR can also occur between nascent chromosomes termed telomere sister chromatid exchange (T-SCE). This unequal exchange of telomere sequence results in one telomere being elongated at the expense of a shortened telomere. Some cells in crisis recover by activating a telomerase independent lengthening mechanism known as alternate lengthening of telomeres (ALT)[81,82]. ALT cells have increased rates of T-SCE[83]. ALT cells have high levels of t-circles and heterogeneous telomere lengths, including some very long telomeres, consistent with a HR mechanism[80,84].
1.1.3 Telomeres and Disease
deficiencies caused by mutations in TR and TERT. Mutations are found in 8-15% of familial and in 1-3% of sporadic idiopathic pulmonary fibrosis (IPF)[95,96], 3-5% of aplastic anemia[97–99], and some familial cases of liver cirrhosis[100]. These adult onset conditions are also
complications that can occur in patients with DC [91].
These conditions represent a spectrum of disease in which the most severe cases have the shortest telomeres[101]. As expected this results in deterioration or in the manifestation of symptoms over time as telomere length continues to erode. Mutations that occur in TR or TERT are inherited in an autosomal dominant manner which is due to telomerase
haploinsufficieny[102]. As telomerase is unable to sufficiently extend telomeres in germ cells affected families display genetic anticipation in which subsequent generations have more severe disease[89]. This results in the spectrum of disease occurring in one family, with earlier
generations suffering from IPF later in life, and successive generations having complications of bone marrow failure occur at a younger age, and dykeratosis congentina in further
generations[103]. Genetic anticipation is also seen in telomerase knockout mouse models. In founder mice which have long (~50kb) telomeres severe defects in highly proliferative tissues only occur in the 5th and 6th generations[104–107].
8
due to telomere shortening and other factors leading to senescence. In a model system of telomere shortening, the telomerase knockout mouse, continued replicative capacity granted through p53 mutation abates the telomere dysfunction phenotype, restoring reproductive capacity and allowing later generations of telomerase deficient mice[122]. However the loss of
proliferative capacity is a suppressor of carcinogenesis.
Overcoming the effects of telomere shortening allows for limitless replicative potential of a cell, which occurs in cell immortalization and in developing neoplasms. In a cell population that is not dividing as a result of telomere shortening mutation of p53 and RB by viral oncoproteins or other mechanisms allows further cell division resulting in further telomere erosion, genomic instability and likely cell death, a state known as crisis[123,124]. Cells that escape crisis have an activated mechanism to extend telomeres, usually through activation of telomerase. Over 90% of cancers and immortalized cell lines express active telomerase[125,126]. In those immortalized cell lines and cancer cells that do not have activated telomerase telomere lengthening is achieved by the ALT mechanism[81,82,127]. Enabling a mechanism of telomere elongation is necessary for a developing cancer, as it is the only way to acquire limitless replicative potential, one of the six hallmarks of cancer[128]. Telomere lengthening alleviates genomic instability, resulting in near elimination of chromosome end-to-end fusions[129] and FISH signal free ends[130] observed in crisis cells. Immortalized human fibroblasts that escape crisis have a shorter average telomere length than cells in crisis[129]. Telomerase most efficiently extends critically short telomeres[131] which leads to telomere length stabilization, not necessarily an average increase, resolving the telomere dysfunction caused by the an individual critically short
telomere[132]. In cancer cells telomerase elongation is distributed across telomeres adding 50-100 bp to maintain telomere length[133].
Carcinogenesis is a multistep process in which cells sequentially acquire mutations which contribute to the ability of resulting cell population to continue its uncontrolled
the context of mutational deficiencies in DNA damage response elements, cells experiencing telomere dysfunction are able to avoid replicative senescence or apoptosis[122]. A model of this situation exists in telomerase knockout mice compounded with a homozygous or heterozygous p53 knockout. In late generation mice with telomere dysfunction this continued cellular division creates bridge-fusion-breakage cycles leading to abnormal karyotypes. These mice have accelerated rates of tumor formation, in different types of tissues. In the heterozygous p53 mutant mice carcinomas, mainly skin, breast, and gastrointestinal tract, are the main tumor type[137]. Analysis of these tumors shows reciprocal translocations between
non-homologous chromosomes, which can lead to copy number changes[138], and are a major cause of oncogene duplication and tumor suppressor deletion[139,140]. There is also evidence of non-reciprocal translocation in human fibroblasts in response to telomere damage due to TRF2 depletion leading to NHEJ[141].
There is extensive evidence of telomere shortening in human cancers. Early
measurements such as telomere restriction fragment length, which uses southern blot to find an average bulk telomere length of a sample on a gel, showed breast and colon tumor samples had shorter telomere lengths than adjacent normal tissue[142–144]. More accurate measurement techniques have been developed through the use fluorescent in situ hybridization. Using this technique PNA probe fluorescence is measured from individual telomere ends in an individual cell. Short telomeres can be measured in invasive breast, prostate, and pancreatic cancers[145– 148]. Short telomeres can also be found in a majority of pre-invasive cancerous growths in bladder, cervix, colon, esophagus, and oral cavity, indicating that telomere shortening occurs in the early steps of carcinoma development[149]. Telomere length can also be measured using less laborious PCR based methods. Average telomere length can be assayed by a quantitative PCR method which compares the amount of telomere sequence signal as measured by qPCR to the amount of signal generated from a single copy locus in the genome[150]. Individual
10
however this is limited to telomeres on chromosomes with a unique subtelomeric priming site[151].
1.1.4 Subtelomere Structure
Despite the crucial role telomeres play in genome stability and their contribution to cancer and disease, telomere length varies widely in the human population. qFISH measurements of individual telomeres have shown that telomere length is heterogeneous[152,153], even between homologous chromosomes[154]. However the length profile is largely consistent in discrete tissues of an individual, and the profile is predominantly heritable[155]. Telomere length profiles are set in the zygote and maintained throughout life[156]. This telomere length regulation is controlled in part by cis-acting factors in the telomere adjacent sequence[157–159]. This directly adjacent sequence, the subtelomere, contains the telomere associated repeat (TAR) repeats which were described early on as being telomere adjacent in the human genome[160,161]. Parts of the TAR1 repeat are found within 2kb of nearly all telomeres, and similar sequences are also found in the pericentromere[162]. While the TAR repeats are found directly adjacent to the telomere, a larger proximal region, the first 500kb adjacent to each telomere makes up the subtelomere sequence. In the human genome the subtelomere is a hotspot of recombination, with sister chromatid exchange enriched 160 fold in the terminal 100kb [163,164]. As such the subtelomere contains a high degree of segmental duplication. These duplicated sequences are termed subtelomere duplicon blocks. Patterns of duplicated blocks in the first 25kb of the
subtelomere are shared between related subtelomeres, defining six subterminal duplicon families (A-F). There are seven distinguishable single copy subtelomeres (7q, 8q, 11q, 12q, 14q, 18q, XpYp), and the rest are identifiable to their pertaining family[162].
The subtelomere repeat elements (SRE) are segmental duplications of sequence from other subtelomeres found on separate chromosome ends. SRE sequence makes up 25% of the subtelomere and 80% of the most distal 100kb[165]. There is also extensive segmental
pericentromic regions, and the ancestral chromosome fusion on chromosome two[166]. The segmental duplications in the subtelomere are larger and more abundant than elsewhere in the genome[167]. The subtelomere is also enriched in internal telomere sequence (ITS) sites. These are (TTAGGG)n-like sequences that are found through the genome, however they are enriched 25 fold in the subtelomere and tend to be longer and more similar to the perfect canonical telomere repeat[165]. In the yeast genome these ITS sites play a role in subtelomere transcriptional regulation and recombination, especially in the context of ALT telomere
maintenance[168,169]. A similar role for these sequences has been hypothesized in the human genome[170,171]. The subtelomere also varies in the human population, it was one of the first genomic regions to be identified to contain copy number variants[172,173]. Variant combinations of subtelomere duplicons define diverse subtelomere alleles which can have an effect on
subtelomere regulation and transcription[174].
1.1.5 Subtelomere Transcripts
The telomere and subtelomere were once thought to be maintained in a repressive heterochromatic state, as is the case in drosophila[175] and budding yeast[176] due to what is known as the telomere position effect; however studies exploring this effect in mammalian genomes have been inconclusive. The subtelomere contains many transcripts including throughout the SRE. While some appear to be noncoding or pseudogene copies, some contain open reading frames that can encode proteins[162]. An important subtelomere transcript in the human genome is a subclass of the Wiscott-Aldrich Syndrome Protein family, Wiscott-Aldrich Syndrome Protein and Scar homolog (WASH). The WASH gene family is made up of different duplicated isoforms throughout the subtelomere, most of which end within 5kb of the telomere tract. The WASH gene is an ancient conserved protein whose function is as an actin
12
hot spot of recombination; it may be a mechanism by which new genes are generated[167,178]. The subtelomere has also been shown to contain the transcription start sites of TERRA.
Telomeric repeat containing RNA (TERRA) is a long non-coding RNA that is transcribed from the subtelomere through the telomeric sequence. It is highly conserved and has been detected in Homo Sapiens, Mus musculus, Danio rerio, plants, and yeasts[179,180]. TERRA transcripts contain both subtelomeric sequences and the canonical telomere repeat, but are heterogeneous in length, ranging from 100bp to 9kb[181,182]. Inhibition of RNA Polymerase II (RNAP2) abolishes TERRA transcription in human U2OS cells[182]. TERRA transcripts are 5’ capped by 7-methylguanosine[183], however only 7% of transcripts are polyadenylated[181,182]. Subtelomere fragments that were previously isolated from 10q and XqYq[161] were used to show a CpG island (61-29-37 repeats) within the SRE sequence acted as a promoter sequence and was capable of transcribing a reporter gene placed between the 61-29-37 repeat and the telomere[184]. TERRA localizes to the nucleus where it forms discrete foci at
telomeres[181,182]. TERRA associates with shelterin components TRF1 and TRF2 and
heterochromatin found at telomeres[185,186]. In yeast TERRA overexpression has been shown to lead to shortening of the transcribed telomere through exonucleic activity[187], or DNA replication dependent loss[188]. TERRA has been shown to act in coordination with hnRNP A1 to help loading of POT1 on single stranded telomere overhangs[189]. Knockdown of both TERRA and hnRNP A1 lead to significant increases in telomere dysfunction induced foci (TIF)[185,190,191]. TERRA expression is deregulated in human cancers, with some studies showing cancer cell lines and certain cancer types have decreased TERRA expression[192,193], and others showing increased TERRA expression overall, with loss of expression from certain subtelomeres[194]. TERRA transcription is tightly regulated in healthy cells.
1.2 Cohesin and CTCF
holds distant or disjointed regions of the genome within the ring[195,196]. Cohesin also
associated with accessory proteins SA 1, 2, and 3[197]. There is evidence that cohesin has roles beyond sister chromatid cohesion, and is involved in transcriptional regulation. In yeast SMC mutants have disrupted insulator function[198] and in Xenopus and human cells cohesin is associated with chromatin at points in the cell cycle with no sister chromatid cohesion[199–201], and cohesin subunits are expressed in post-mitotic murine neurons where there will be no more sister chromatid cohesion[202,203]. Cohesin has been found to frequently colocalize with the zinc finger domain protein CCCTC binding factor (CTCF) in the mouse and human genomes, and these sites of colocalization are enriched in regions within 2kb of genes[202,204–206].
CTCF was originally found as a protein bound to the promoter region of the MYC
oncogene[207]. CTCF has been identified as a transcriptional activator and repressor, as it has been found at sites between transcriptionally active and inactive chromatin[208–210]. CTCF binding is inhibited by CpG island methylation[211], and conversely CTCF binding inhibits denovo CpG methylation[212]. It has been postulated that CTCF is the main factor controlling spatial positioning of genomic segments, grouping disparate segments in condensing or decondensing nuclear territories[213]. Chromsome confirmation capture (3C) experiments have shown long range interactions play an important role in the expression of other repetitive sequences such as olfactory genes[214,215] and CTCF was shown to be involved in regulating gene expression through paternal imprinting at the IGF2/H19 locus by controlling long range interactions[216–218].
1.3 High Throughput Sequencing
1.3.1 Technology
14
genome either physically through sonication or enzymatically through restriction digests. In order to generate a large enough signal suitable for detection identical copies of individual DNA
fragments are generated and fixed at a single physical site that can be imaged. To accomplish the amplification of these individual fragments universal priming sites are first ligated to the fragments before they are PCR amplified simultaneously. The details of the PCR reaction conditions and how the fragments are fixed differentiate the existing technologies.
The first HTS method that was developed was based on pyrosequencing, which was commercialized by 454. This technology employs emulsion PCR (emPCR) to amplify individual fragments. The fragments are captured by beads containing the complement of the annealed universal primer under conditions such that you would expect at most one fragment per bead. The PCR reaction can then amplify each fragment in an individual microreactor, coating the bead with copies[219]. Pyrosequencing takes advantage of the release of inorganic phosphate during DNA synthesis. When nucleotides are added to a nascent strand of DNA they release an inorganic phosphate which can be measured by its conversion to visible light through enzymatic reactions [220,221]. Beads are sequestered in wells on a chip and washed with consecutive washes of dNTPs. Fluorescent signals are captured, with homopolymers generating an
increased signal[222]. As pyrsoquencing does not rely on recurrently terminating DNA synthesis it is able to generate the longest reads currently widely available.
Life technologies platform, SOLiD, also employs the use emPCR, however the sequencing reaction does not rely on the measurement of bases incorporated by DNA
The most popular HTS platform is technology was developed by Solexa which was later acquired by Illumina. Instead of binding fragments to a bead as in ePCR, fragments are bound to primers that are covalently attached to a glass slide. These fragments are then are then
amplified directly on the slide by bridge amplification, generating clusters of amplified DNA distributed on the chip. Illumina sequencing relies on reversible terminator chemistry, permitting the addition of one fluorescently labeled nucleotide in each cycle. After unincorporated
nucleotides are washed away, fluorescence is measured through imaging, and the terminating group is chemically cleaved, allowing for the next cycle of nucleotide addition[225].
Further advances in HTS are allowing for direct sequencing of single molecules to eliminate the noise introduced by amplification (PacBio, Nanopore), and allowing for nonoptical measurements of base interrogation to allow for higher throughput (IonTorrent, Nanopore). The second generation HTS technologies generate reads between 36 and 250 bases in length. These reads are too short and biased to find stretches of overlapping fragments for de-novo assembly of complex genomes, instead these reads are aligned to the existing reference sequence.
1.3.2 Alignment
16
Another strategy to allow both mismatches and gaps within the seed is to use q-gram filtering, which is implemented in SHRiMP[233]. Building a hash table of an entire reference genome takes a significant amount of space and holding it in memory for searching can be taxing on computational resources. A different strategy for aligning sequences relies on suffix trees. These have the advantage of being able to be represented by more efficient data structures, a prefix array[234], and further compressed in structures that are based on the Burrows-Wheeler Transform[235], a FM-index[236]. Using an FM-index the human genome reference can be built to take up 2-8Gb of memory. The most popular short read alignment algorithms, bowtie, SOAP2, and BWA use an FM-index strategy[237–239]. All these mapping strategies allow for
mismatches and gaps in the final alignment to account for sequencing errors, and variation and mutation found in sample sequences compared to the reference. However a major issue that arises in aligning these sequences is the presence of repetitive sequence in the reference genomes, for example nearly half of the human genome is repetitive sequence[240,241]. Reads generated from these parts of the genome that are completely identical at the length of the read generated cannot be mapped unambiguously. The standard analysis pipelines do not consider these multimapping reads and instead focus only those reads which map uniquely. This limits the application of the data sets in analyzing the telomere and subtelomere.
1.3.3 Applications
Whole genome sequencing (WGS) has allowed for in depth observations of the variants and mutations that exist in the human population. Many software tools have been developed to call single nucleotide polymorphisms (SNP)[242], small indels[243], and copy number variants (CNV)[244]. This has allowed us to find rare variants and denovo mutations, and study rare Mendelian disorders with small numbers of affected individuals. It has also allowed for the study of cancer genomics and acquired mutations that are common to certain cancers, and the
RNA-seq allows analysis of whole transcriptomes by sequencing of cDNA made from reverse transcription of poly(A) selected, or ribosome repeat depleted RNA[246]. The analysis of these datasets is similar to WGS, except that reads must either be mapped to the known
transcriptome, or allow for large gaps within the genome mapping to account for transcript splicing. Gene expression levels are then estimated by calculating the number of reads per kilobase of transcript per million reads. The problem of multi-mapping reads is exacerbated in RNA-seq data as there are both gene families made up of similar psuedogenes, and a large degree of transcript heterogeneity in the form of alternatively transcribed and spliced isoforms of genes. This problem has been addressed in most implementations of RNA-seq analysis, and a common strategy is to assign partial reads to all of its possible mapping positions. ERANGE assigns location weighting based on the expression level of adjacent unique regions[246]. Methods that quantify individual isoform levels, such as RSEM, go beyond this to assign partial reads to different isoforms representing the same mapping position[247,248].
ChIP-seq is a method to measure protein-DNA interaction and epigenetic marks genome wide. In Chromatin immunoprecipitation (ChIP) experiments DNA binding proteins are cross linked to DNA in vivo by treating cells with formaldehyde before the genome is fragmented. Antibodies targeted to the selected protein of interest, or directly targeting chromatin marks, immunoprecipitate the DNA-protein complex. Once the crosslinks are reversed the resulting DNA fragments are used to build a HTS library that is enriched for the targeted protein or histone mark. The resulting sequenced reads are aligned to the reference genome in the same way as WGS datasets. An important caveat of ChIP-seq dataset is that they are only enriched for the chosen protein or mark. As such they are compared to an input or ChIP (IGG) control. Regions
18
1.4 Outline of Dissertation
In chapter 2 I present the initial mapping and analysis of ChIP-‐seq datasets to the 15kb
of sequence proximal to the telomere in the human genome, which was my key contribution to
the Deng et al 2012 paper I co-‐authored. CTCF and cohesin colocalization within 2kb of the
telomere was found on the majority of human subtelomeres. The results of follow-‐up functional
studies carried out by Zhong Deng in the Lieberman lab for this paper are summarized briefly,
and demonstrate the importance of CTCF and cohesin binding for TERRA transcription and
telomere stability. In chapter 3 I show the results of extending this analysis to the entire 500kb
human subtelomere. A complete multi-‐mapping ChIP-‐seq analysis pipeline was developed.
ChIP-‐seq results for a number of datasets were generated and are all available on a Wistar
mirror of the UCSC genome browser focused on the subtelomere.
In chapter 4 I extended the ChIP-‐seq pipeline for use on murine samples. I describe the
sequence characteristics of the recently completed mouse subtelomere sequence, and the
differences between the human and mouse subtelomere repeat structure. A number of ChIP-‐
seq datasets were analyzed to provide an online resource similar to the human subtelomere
browser. Additionally RNA-‐seq datasets were analyzed to look for evidence of TERRA
transcription. Differences in human and mouse TERRA regulation and transcription are
explored.
In chapter 5 I present Telomere Analysis from SEquencing Reads (TASER), a pipeline to
capture telomere sequence information from HTS data sets. This pipeline was used to analyze
53 paired tumor normal samples from the prostate cancer genome sequencing project. The
results of the analysis were used to show the feasibility of the strategy and show a trend in the
samples as tumor or normal. In chapter 6 I conclude with an overall discussion of the
20
CHAPTER 2: PROXIMAL 15KB ANALYSIS
2.1 Introduction
The ends of eukaryotic chromosomes form specialized chromatin structures that are essential for chromosome stability and genome maintenance[1]. The terminal TTAGGG repeats of mammalian telomeres bind to a set of proteins that are nucleated by the DNA-binding proteins TRF1, TRF2, and Pot1, and are collectively referred to as shelterin[2,3] or telosome[4,5]. These terminal repeat binding factors regulate telomere length homoeostasis and DNA damage repair processing at the chromosome termini[6]. Loss or damage of the terminal repeats can initiate a DNA damage response and trigger cellular replicative senescence[7,8]. DNA damage and senescence can also be elicited by mutation or depletion of telomere repeat binding proteins[9]. Dynamic remodelling of telomere repeat factors and telomere DNA conformation is also required for normal telomere length regulation and telomerase accessibility[10–13].
In addition to shelterin and telomerase, telomere maintenance depends on the proper assembly and regulation of telomeric chromatin[5,14,15]. Traditionally, telomeres have been thought of as highly heterochromatic structures associated with condensed chromatin and transcriptional silencing[14,16–18]. More recent studies have revealed that many eukaryotic telomeres, including human and yeast, can be transcribed, indicating that telomeric silencing is incomplete and telomere chromatin is dynamic[19–23]. The chromatin structure of telomeres is further complicated by the variations in the subtelomeric DNA structures, suggesting that telomeric heterochromatin structure and regulation may vary among different chromosomes[24– 26]. In budding yeast, telomeric silencing is mediated by Sir proteins that interact with telomere repeat binding factor Rap1[27]. In mammalian telomeres, nucleosomal arrays commonly
previously shown that TRF2 can bind directly to telomeric repeat-containing RNA (TERRA) to recruit heterochromatin proteins including ORC and HP1 and maintain histone H3K9me3 enrichment at telomeres[33]. TERRA expression is itself dependent on histone H3K4
methyltransferase MLL[36], as well as DNA methylation status and CpG-island promoter found in many subtelomeric regions [37–39]. In fission yeast, the expression of TERRA and other
subtelomeric transcripts are subject to diverse regulation by chromatin regulatory factors[40,41]. The dynamic interplay between shelterin, telomere chromatin structure, TERRA expression, and telomere biology appears to be an essential and universal component of chromosome stability.
The chromatin organizing factor CTCF has been implicated in numerous aspects of chromosome biology, including chromatin insulation, enhancer blocking, transcriptional activation and repression, DNA methylation-sensitive parental imprinting, and DNA-loop formation between transcriptional control elements[42–44]. CTCF has been implicated in the transcriptional
repression of the D4Z4 macrosatellite repeat transcript found ∼30 kb from the telomere repeats of chromosome 4q[45]. At D4Z4, CTCF interacts with lamin A and tethers the chromosome 4q telomere to the nuclear periphery [46,47]. A more general role for CTCF has been found in its ability to colocalize with cohesin subunits at many chromosomal positions[48–51]. Cohesin is a multiprotein complex consisting of core subunits SMC1, SMC3, Rad21, and SCC3 (referred to as SA1 or SA2 in humans), which can form a ring-like structure capable of encircling or embracing two DNA molecules[52,53]. Cohesin was originally identified as a regulator of sister-chromatid cohesion, but subsequent studies in higher eukaryotes indicate that they have functions in mediating long-distance interactions between DNA elements required for transcription
22
telomeres or subtelomeres in addition to binding the D4Z4 gene repeat, nor if it can interact with cohesin at these locations.
The chromosome region immediately adjacent to the terminal repeats has been referred to as the subtelomere. In humans, the distal subtelomeres consist of a variety of degenerate repeat elements with a few discrete gene transcripts interspersed at various distances from the terminal TTAGGG repeat tracts[24–26,58,59]. TERRA transcription initiates from within the subtelomeres, and a promoter containing a CpG-island and subtelomeric 29- and 61-bp repeat element has been identified in plasmid reconstitution assays[38]. DNA methylation and DNA methyltransferases have been shown to inhibit TERRA expression since TERRA levels are highly elevated in cells where DNMTs have been genetically disrupted or depleted [38,60], as well as in Immunodeficiency-Centromeric instability-Facial abnormalities (ICF) Syndrome cells that are genetically defective in DNA methyltransferase 3B (DNMT3B) [37,39,61]. CTCF binding is known to be DNA methylation sensitive but it is not yet known whether CTCF associates with
transcriptional regulatory elements important for TERRA regulation or telomere maintenance. Herein, we investigate the role of CTCF and cohesin at human subtelomeres and their role in regulating TERRA expression, telomere chromatin organization, and telomere DNA end protection.
2.2 Methods
2.2.1 ChIP-Seq data
ChIP-Seq was performed using 1 × 107 BCBL1 cells per assay with either rabbit anti-cellular SMC1 (Bethyl A300-055A-3) or CTCF antibody (Millipore 07–729), or control rabbit IgG (Santa Cruz Biotechnology), using Illumina-based sequencing as described[249].
2.2.2 Mapping ChIP-Seq data to human subtelomeres
The human subtelomere reference assemblies used for the mapping studies represent the most distal 15 kb of DNA sequence adjacent to the (TTAGGG)n terminal repeat tract for the indicated telomeres. Each assembly is oriented with the telomere end on the left with nucleotide position 1 corresponding to the first (CCCTAA) of the tract, which continues to the left of this position but was truncated for mapping consistency purposes. Some of these sequences were available in HG19 [165] whereas others were assembled by merging new fosmid sequence data with HG19 to bridge remaining gaps. In several instances, structural variants corresponding to alternative subtelomere alleles were also included in the set of subtelomere assemblies used here because they differed substantially from the original reference telomere. The full set of subtelomere assemblies are described in detail in Chapter 3. All of the sequences in the described orientation are available in FASTA format in Supplementary File S1.
Reads were mapped to the 15kb subtelomere reference using bowtie[238]. Many subtelomeres are duplicon rich with duplicon-specific nucleotide sequence similarities ranging from 90 to 99% between individual members of duplicon families that occur on separate
subtelomeres[162,164,252,253]. To deal with this issue, we required a perfect match to retain a read, and all perfect matches of a given read to positions within the reference assemblies were recorded. Multiply mapping reads were dealt with as described previously[254], by assigning weights to reads such that multiple mapping positions sum to one read. Mapping likelihood was added to the reads as the inverse of the number of mapping positions. Picard
24
used in Supplementary File S2. All figures were generated on the subtelomere reference genome hosted at the Wistar mirror of the UCSC genome browser, http://vader.wistar.upenn.edu.
2.3 Results
2.3.1 CTCF, cohesin, and RNAPII binding to the CpG-island promoters in
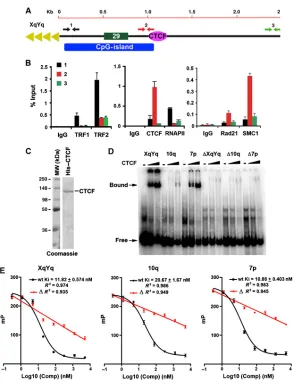
human subtelomeres
Genome-wide analyses of CTCF, cohesin, and RNA polymerase II (RNAPII) have been performed in several different cell lines from various laboratories, including those generating the human ENCODE database [1–5]. In these published studies, the complete human subtelomeric DNA was not available for ChIP-Seq data mapping, with gaps immediately adjacent to the start of terminal repeat tracts for many telomeres[6]. We have generated complete assemblies of human subtelomeres for most of these chromosome ends and here we use these reference assemblies to map the read sequences from data sets, including our own, for CTCF, Rad21, SMC1, and RNAPII (Figure 2.1;Supplementary Figures 2.3 and 2.4). We found that most but not all human chromosome ends have a major CTCF-binding site within 1–2 kb from the TTAGGG repeat tracts. These CTCF sites consistently mapped to a region just upstream (centromeric) to the CpG-islands and 29 bp repeats, often overlapping 61 bp repeat element (Supplementary Figures 2.3 and 2.4). In the few exceptions to this pattern, CTCF sites were observed at positions ∼10 kb from the TTAGGG repeats (7p, XYp) or several CTCF-binding sites with relatively low peak scores (3p, 7q, 8q, and 12q) (Supplementary Figure 2.4). We refer to these two different subtelomeres as type I (with major CTCF peaks at ∼1–2 kb) or type II (lacking obvious CTCF peaks proximal to the telomere repeat tracts). In almost all cases, including those of type II, we observed an overlap of CTCF-binding sites with cohesin subunit Rad21 (Figure 2.1;
26
Due to the complex duplications in subtelomeric sequence, we permitted multimapping signals weighted according to the number of perfect subtelomeric mapping sites to contribute, along with uniquely mapping reads, to subtelomeric ChIP-seq signals. We found that the
remaining unique signals recapitulated the ChIP-Seq peak positions in most cases when multiple mappings were eliminated (Supplementary Figure 2.3E), suggesting that most of the binding sites can be uniquely assigned to specific subtelomeres. Some unique signals are lost, as expected for perfect duplications. This was sometimes the case with the 29-mer repeats over which RNAPII signal is centred and which a portion of the CTCF and cohesin read peaks was formed at many subtelomeres. Supplementary Figure 2.3D illustrates this effect for the example subtelomeres shown in the Supplementary Figure 2.3E. At the same time, Supplementary Figure 2.3D also shows the clear enrichment of RNAPII ChIP-seq reads mapping to the 29-mer variable number tandem repeat (VNTR) over the IgG controls, a true binding peak that would have been missed if multimapping signal contributions were disallowed.
2.3.2 CTCF binds directly upstream of the CpG-island and 29 repeat
element found in subtelomeres
To verify the ChIP-Seq data for CTCF, cohesin, and RNAPII, we performed conventional ChIP-qPCR with primers spanning the first 3 kb of the XYq (Figure 2.2A and B) and 10q
28
ChIP-seq peaks in subtelomere XYq, 10q, and 7p, as well as control oligonucleotides containing substitution mutations in the putative CTCF consensus sites, ΔXYq, Δ10q, and Δ7p, were synthesized as 46 mers for EMSA probes (Supplementary Table S3). Purified CTCF protein bound efficiently to the XqYq and 7p probes, less efficiently to 10q probe, but not to the mutated
ΔXYq, Δ10q, and Δ7p probes (Figure 2.2D), indicating that these subtelomere ChIP-Seq peaks contain bonafide CTCF recognition sites. The relative binding affinities of these subtelomeric CTCF-binding sites was further quantified by a fluorescence polarization based competitor assay (Figure 2.2E). The wild-type CTCF-binding sites from XYq, 10q, and 7p showed robust
competition against a FAM6-labelled probe containing a CTCF-binding site with high similarity to the consensus motif as defined previously[255]. Inhibitory constants (Ki) for each binding sites were equal to 11.82, 20.67, and 10.88 nM, respectively. On the other hand, the mutant ΔXYq,
Figure 2.2− Identification of CTCF-binding site elements in the 61-bp element of human
subtelomeres. (A) Schematic of the type I subtelomere showing the relative positions of the 29- and 61-bp repeat element, CpG-island, and TTAGGG terminal repeats. (B) ChIP-qPCR for TRF1, TRF2, CTCF, RNAPII, Rad21, and SMC1 relative to IgG controls using primers for the XYq subtelomere at positions close (∼150 bp) to TTAGGG repeat (black), at CpG-island (red), or ∼3 kb from terminal repeats (green). Bar graph represents the average value of percentage of input for each ChIP from three independent PCR reactions (mean±s.d.). (C) Purified recombinant CTCF protein analysed by Coomassie staining of SDS–PAGE gel. (D) EMSA with CTCF protein binding to DNA oligonucleotide probes containing putative binding sites from subtelomere XYq, 10q, or 7p, as well as with oligonucleotides containing point mutations in CTCF
30
2.3.3 Summary of Results of Dr. Zhong Deng’s functional experiments in
Dr. Paul Lieberman’s lab that were included in the Deng et al., 2012
publication
Using the CTCF and cohesin binding to subtelomeres suggested by initial mapping and analysis experiments as a starting point, Dr. Deng led a series of functional studies establishing the importance of subtelomeric CTCF and cohesin binding for TERRA transcription and genome integrity. These experiments indicated that (1) CTCF recuits RNAPII to subtelomeres; (2) CTCF and cohesin stabilize TRF binding to subtelomere; and (3) CTCF and cohesin are required for protection of telomeres and prevention of telomere DNA damage signaling. A subtelomere construct was used to show that both mutation of the CTCF binding site, and siRNA knock down of CTCF results in a decrease in TERRA transcription. CTCF and RAD21 knock downs were also used to show a subsequent loss of RNAPII binding in the subtelomere. Telomere damage foci were observed as a result of the depleted TERRA transcription. Dr. Deng’s experiments are detailed in Deng et al. 2012 and are not included in this dissertation.
2.4 Discussion
2.4.1 A foundation for a chromatin atlas of the human subtelomeres
several ChIP-Seq data sets to the most distal parts of human subtelomeres (Figure
2.1; Supplementary Figures 2.3 and 2.4). We focused on CTCF and cohesin subunits because of their general importance in chromosome organization throughout vertebrate evolution. We found that CTCF and cohesin colocalized at a position immediately adjacent to the CpG-islands implicated in TERRA promoter regulation[184] (Figures 2.1 and 2.2). We confirmed this binding by generating a new experimental data set for CTCF and SMC1 ChIP-Seq in a B-lymphoma cell lines. In addition, we mapped RNAPII binding and found that it localized more broadly across the subtelomeres, but had an average enrichment at the telomeric side of the CpG-island promoter for TERRA expression. CTCF and cohesin bound just centromeric to the CpG-island, and were further investigated for their role in TERRA expression and telomere end protection. The genome browser and methods established for mapping next-generation sequence data to the subtelomere provides a foundation for building a more complete atlas of epigenetic marks and chromatin organization at human subtelomeres.
2.4.2 Supplemental Figures
Large multipage figure available as figure S1 at
http://onlinelibrary.wiley.com/doi/10.1038/emboj.2012.266/suppinfo
Figure 2.3 − Summary of ChIP-Seq analysis of CTCF, cohesin, and RNAPII binding to type I human subtelomeres. Fragment density profiles were generated for samples and matched IgG controls as described in Methods. The fold enrichment of sample over IgG is shown. The Y axis for each track is auto-scaled to highest peak in each chromosome region shown. Subtelomere identity is indicated in the top left of each panel. (A-D) CTCF_W and SMC1_W were newly generated ChIP-Seq data using human pleural effusion lymphoma cell line BCBL1. CTCF, RNAPII, and Rad21 were derived from human encode data sets using B-lymphoblastoid cell lines. (E) Example enrichment profiles for ChIP-Seq analysis, comparing the standard multi-mapping method used vs allowing only unique mappings. The top track of each dataset pair permitted multimapping in the indicated ChIP and control IgG dataset, and the bottom (designated by _U) permitted only unique mappings in both ChIP and IgG control dataset. Binding in the first 15 kb
subtelomeres of chromosome arms 10q, 13q, 15q, and XYq are shown.
Large multipage figure available as figure S2 at
http://onlinelibrary.wiley.com/doi/10.1038/emboj.2012.266/suppinfo
Figure 2.4 − Summary of ChIP-Seq analysis on type II human subtelomeres. Same as in Figure S1,
32
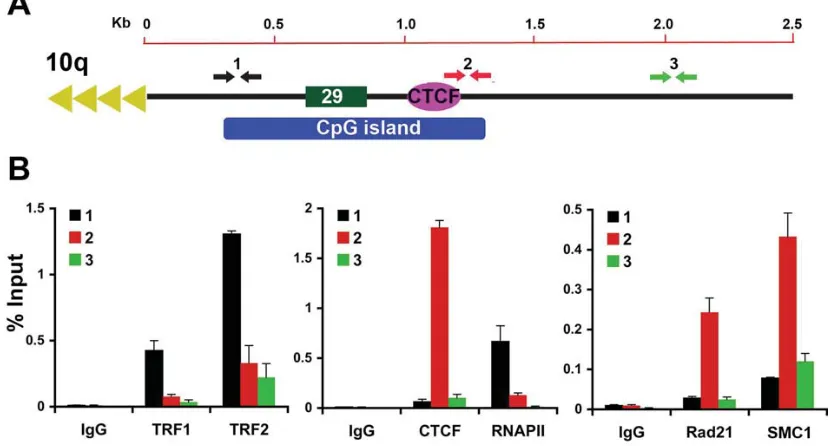
Figure 2.5 − Validation of CTCF binding site at 10q human subtelomeres in BCBL1 cells. (A)
Schematic of the 10q subtelomere showing the relative positions of the 29 bp repeat element, CpG island, and TTAGGG terminal repeats. (B) ChIP-qPCR for TRF1, TRF2, CTCF, RNAPII, Rad21, and SMC1 relative to IgG controls using primers for the 10q subtelomere at positions close (~400 bp) to TTAGGG repeat (black), at CpG island (red), or ~2 kb from terminal repeats (green). Bar graph represents the average value of percentage of input for each ChIP from three independent PCR (Mean + SD).
CHAPTER 3: HUMAN SUBTELOMERE ANALYSIS
3.1 ABSTRACT
34
3.2 INTRODUCTION
Subtelomeric DNA is crucial for telomere (TTAGGG)n tract length regulation and telomeric chromatin integrity. A telomeric repeat-containing family of RNAs (TERRA) is transcribed from subtelomeres into the (TTAGGG)n tracts [181,183,182] and forms an integral component of a functional telomere; perturbation of its abundance and/or localization causes telomere dysfunction and genome instability [181,185]. Telomere dysfunction caused by critically short telomere DNA sequence or by disruption of telomeric chromatin integrity induces DNA Damage Response pathways that cause cellular senescence or apoptosis (depending on the cellular context) in the presence of a functional p53 tumor suppressor pathway [256]. Only one or a few critically short telomeres in a cell are sufficient to induce DDR-mediated senescence or apoptosis [66,257]. Senescence or apoptosis of somatic cells can disrupt tissue
microenvironments, and senescence or apoptosis of stem cell populations can prevent proper replenishment of rapidly dividing cellular lineages, both impacting aging phenotypes and age-related diseases including cancer [258–261].
Subtelomeric DNA elements regulate both TERRA levels and haplotype-specific
(TTAGGG)n tract length and stability [157–159,184,185], with accumulating evidence for specific epigenetic modulation of these effects [184,185,262–264]. Heterogeneously-sized TERRA transcripts with as yet ill-defined transcription start sites and potential splice patterns originate in many, perhaps all human subtelomere regions [181,183,265], with the sizes of the larger
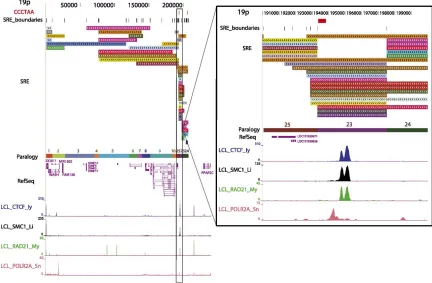
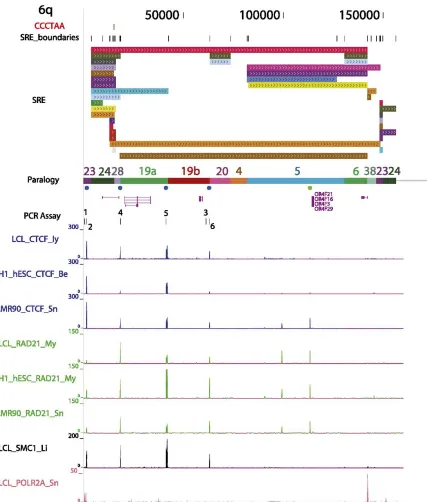
transcripts (greater than 15 kb) suggesting structural overlap with some transcribed subtelomeric gene families [167,266]. While many details of the dynamic interplay between shelterin, telomere chromatin structure, TERRA expression, and telomere biology remain unclear, recent work from our group indicates that CTCF and cohesin are integral components of most human