Nanoscale Electronic/Energy Materials. (Under the direction of Dr. Christopher B. Gorman).
Two dominant themes in the synthesis of molecules for efficient, thin-layer organic devices are the ability to control their HOMO-LUMO gap and to achieve superior charge mobilities. Two synthetic methodologies have been devised to promote these desirable properties. First, a cascade cyclization route to fused aromatic systems has been designed. Molecules
synthesized via this route can exhibit extended conjugation and efficient, intermolecular orbital overlap. These properties result in decreased HOMO-LUMO gaps and enhanced charge mobility within the bulk material and at metal contacts. The second focuses on the discovery and use of terminal alkyne functional groups capable to promote strong electronic coupling between molecules and metals.
The first approach explores efficient chemistry for cascading cyclization to produce fused, polycyclic aromatic structures. A synthetic methodology was developed for the production of poly-cyano precursor molecules capable of undergoing acid-mediated cyclization to form fused aromatic systems. Polymerization of polycyclic aromatic molecules was complicated by the competition between formation of linear and cyclic oligomers. Subsequently, a route to ketene-imine derivatives of poly-cyano precursors was explored to facilitate cascading cyclization under mild conditions.
by
Jenelle Sue Willett
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
Chemistry
Raleigh, North Carolina 2013
APPROVED BY:
_______________________________ ______________________________ Dr. Christopher B. Gorman Dr. David Shultz
Chair of Advisory Committee
________________________________ ________________________________
DEDICATION
BIOGRAPHY
Jenelle Willett grew up in Waterford, PA and graduated from Fort LeBoeuf highschool in 2000. She received a B.S. in Chemistry from Gannon University in 2007 and worked as a chemical intern at LORD Corporation. She continued her education enrolling in the
ACKNOWLEDGMENTS
The following people are acknowledged for their contribution to this work: Dr. Christopher B. Gorman, advisor
Dr. David Shultz, Dr. Christian Melander, Dr. Bruce Novak and Dr. Walter Weare, members of the advisory committee
Dr. Anil Sharma assistance with organic synthesis Dr. James LeBeau and Bahar M. Alipour
Materials Science, NCSU TEM measurements
Dr. Roberto Garcia AIF NCSU TEM measurements
Victoria Brown Chemistry, NCSU
MALDI-MS
Dr. Daniel Dougherty and Sean Stuart Physics, NCSU
TABLE OF CONTENTS
LIST OF TABLES ...x
LIST OF FIGURES ... xi
LIST OF SCHEMES ... xix
Chapter 1: Organic materials for electronic and energy use 1.1 Introduction ...1
1.2 Organic thin-layer film devices ...1
1.3 Organic thin-film transistors ...2
1.4 Bandgap engineering ...5
1.5 Low bandgap materials ...7
1.6 Functional groups as molecular alligator clips ...10
1.7 Research impetus ...13
1.8 References ...14
Chapter 2: Synthesis of fused polycyclic aromatic molecules 2.1 Introduction ...18
2.2 Synthesis of cyano-containing small molecules ...20
2.3 Prior efforts on the cyclization of poly-cyano containing molecules ...21
2.5 Cyclization of N-alkyl ketene-imines ...31
2.6 Synthesis of N-acyl ketene-imines ...32
2.7 Conclusion ...33
2.8 Experimental ...34
2.9 References ...39
Chapter 3: Progress toward the synthesis of fused polycyclic aromatic polymers 3.1 Introduction ...41
3.2 Synthesis of fused poly-cyclic A-B monomers ...42
3.3 Polymerization of fused poly-cyclic A-B monomers ...43
3.4 Effect of alkyl substituent on polymerization products ...47
3.5 Optimization of polymerization conditions ...49
3.6 Effect of pendant phenyl on polymerization products ...51
3.7 Polymerization under dilute conditions ...54
3.8 Conclusion ...59
3.9 Experimental ...60
3.10 Crystallographic data for cyclic trimer ...69
Chapter 4: Adventitious replacement of ferrocene terminated thiol and alkyne self-assembled monolayers on gold
4.1 Introduction ...73
4.2 Adventitious replacement monitored by scanning tunneling microscopy ...75
4.3 Adventitious replacement monitored by cyclic voltammetry...77
4.4 Synthesis of ferrocene terminated thiol and alkyne molecules ...80
4.5 FcC11S- SAMs on gold on glass substrates ...83
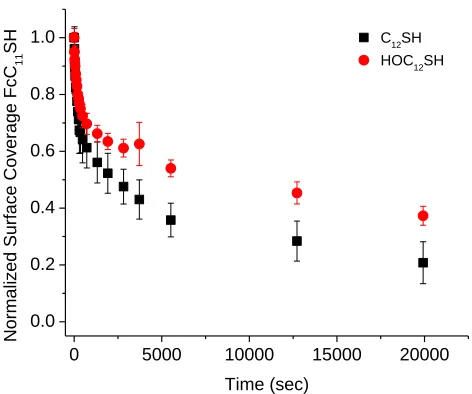
4.5.1 Replacement with C12SH molecules ...85
4.5.2 Replacement with HOC12SH molecules ...90
4.6 FcC11S- SAMs on gold bead substrates ...94
4.6.1 Replacement with HOC12SH molecules ...95
4.6.2 Gold bead cleaning techniques ...99
4.6.3 Replacement with C11≡ molecules ...102
4.7 FcC11≡ SAMs on gold bead substrates ...104
4.7.1 Replacement with HOC12SH molecules ...104
4.7.2 Replacement with C11≡ molecules ...106
4.8 Control experiments with FcC11S- and FcC11≡ SAMs on gold bead substrates ...107
4.9 C12S- SAMs on gold bead substrates ...109
4.9.2 Replacement of C12S- SAMs ...110
4.10 C11≡ SAMs on gold bead substrates...111
4.10.1 Electrochemical blocking by C11≡ SAMs ...112
4.10.2 Replacement of C11≡ SAMs ...112
4.11 Conclusion ...113
4.12 Experimental ...115
4.13 References ...123
Chapter 5: Terminal alklynes as stabilizing ligands for gold nanoparticles 5.1 Introduction ...127
5.2 Synthesis of nanoparticles ...128
5.3 Stabilization of nanoparticles ...130
5.4 Localized surface plasmon band ...130
5.5 Previous investigations of alkyne stabilized gold nanoparticles ...133
5.6 Synthesis of 1-dodecyne stabilized gold nanoparticles ...134
5.7 Characterization of 1-dodecyne stabilized gold nanoparticles ...142
5.7.1 Spectroscopic characterization ...142
5.7.2 Size determination of 1-dodecyne stabilized gold nanoparticles ...147
5.8.1 Spectroscopic characterization of alkyne stabilized gold nanoparticles ...150
5.8.2 Size comparison of alkyne stabilized gold nanoparticles ...151
5.8.3 Thermal characterization of alkyne stabilized gold nanoparticles ...153
5.9 Relative stability of alkyne stabilized gold nanoparticles ...155
5.9.1 Chemical stability ...156
5.9.2 Thermal stability ...165
5.10 Bonding mode ...168
5.10.1 Conversion of bonding modes monitored by ATR-IR ...169
5.10.2 Conversion of bonding modes monitored by 1H NMR ...170
5.11 Gold nanoparticles stabilized by bulky terminal alkyne ligands ...172
5.11.1 Synthesis of 3-phenyl-1-propyne stabilized gold nanoparticles ...173
5.11.2 MALDI-MS of 3-phenyl-1-propyne stabilized gold nanoparticles ...175
5.12 Conclusion ...178
5.13 Experimental ...180
LIST OF TABLES
Chapter 3: Progress toward the synthesis of fused polycyclic aromatic polymers
Table 3.1. Variation in polymerization conditions ...49 Table 3.2. Results of thermally heated reactions (monomer 15c, R =H) ...51 Table 3.3. Results of polymerization under dilute conditions ...55
Chapter 5: Terminal alkynes as stabilizing ligands for gold nanoparticles
Table 5.1. Summary of fitting of 1s carbon peak of XPS data...147 Table 5.2. Summary of TGA data and calculated physical properties of terminal alkyne stabilized gold nanoparticles with ligands; 1-dodecyne, 1-decyne and 1-octyne ...155 Table 5.3. Fit data for NaCN digestion of gold nanoparticles stabilized by ligands:
dodecyne, dodecanethiol, and triphenylphosphine ...161 Table 5.4. Fit data for NaCN digestion of gold nanoparticles stabilized by terminal alkyne ligands of differing chain lengths; dodecyne, decyne and octyne ...164
LIST OF FIGURES Chapter 1: Organic materials for electronic and energy use
Figure 1.1. Schematic diagram of an organic thin-film transistor (OTFT). S = source and
D=drain. ...3
Figure 1.2. Schematic representation of the band diagrams of the following types of materials; insulator (A), semiconductor (B), and metal (C). ...4
Figure 1.3. Mesomeric forms of polythiophene; aromatic (A) and quinoid (B). ...6
Figure 1.4. Structures of regioregular polythiophene (A) and polyisothionapthelene (B)...8
Figure 1.5. Structure of polyacene ...9
Figure 1.6. Proposed binding modes of terminal alkynes complexed with gold. η2 -Acetylene (A), η1 -Acetylide (B), and η1-Alkylidene (C) ...11
Figure 1.7. Configuration of surface-bound molecule ethynyl benzene: (a) final state (b) vinylidene intermediate, and (c) flat intermediate. Reprinted with permission from J. Phys. Chem. B 2005, 109, 20387-20392. Copyright 2005 American Chemical Society...12
Chapter 2: Synthesis of fused polycyclic aromatic molecules Figure 2.1. Molecules containing 4- and 5-cyano groups ...21
Chapter 3: Progress toward the synthesis of fused polycyclic aromatic polymers Figure 3.1. ESI-MS of product obtained from polymerization of monomer 15a. ...44
Figure 3.2. Proposed products obtained from polymerization of monomer 15a ...45
Figure 3.3. ESI MS of polymerization of molecule 15c ...48
Figure 3.4. ESI-MS of product obtained from NAS reaction conditions (monomer 15c, R=H) ...50
Figure 3.6. ESI-MS results from polymerization of monomer 15d (R=H no pendant
phenyl) ...52 Figure 3.7. Polymerization of monomer 15c at 0.023M concentration ...55 Figure 3.8. Polymerization of monomer 15c at 0.1 M concentration ...56 Figure 3.9. ORTEP drawing of trimer of monomer 15c showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were omitted for clarity. ...57 Figure 3.10. Polymerization of monomer 15c with original work-up (MeOH/H2O) ...58 Figure 3.11. Polymerization of monomer 15c with alternative work-up (MeOH/Acetic acid) ...58 Chapter 4: Adventitous replacement of ferrocene terminated thiol and alkyne
self-assembled monolayers on gold
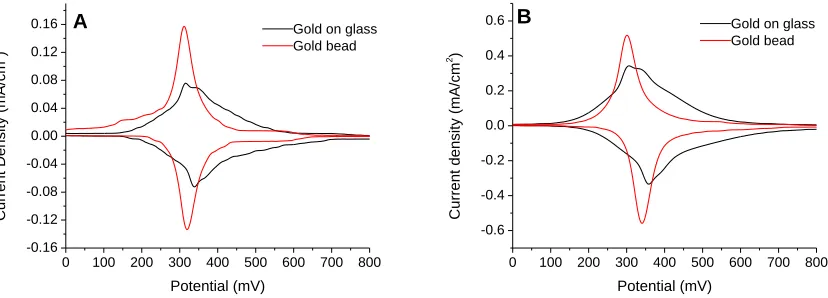
Figure 4.1. Cyclic voltammograms at 100 mV/s of Fe(CN)63-/4- at bare gold bead and SAM modified gold bead electrodes with terminal alkynes. Reprinted with permission from J. AM. CHEM. SOC. 2007, 129, 4876-4877. Copyright 2007 American Chemical Society. .74 Figure 4.2. Comparison of adventitious replacement with 10 µM of
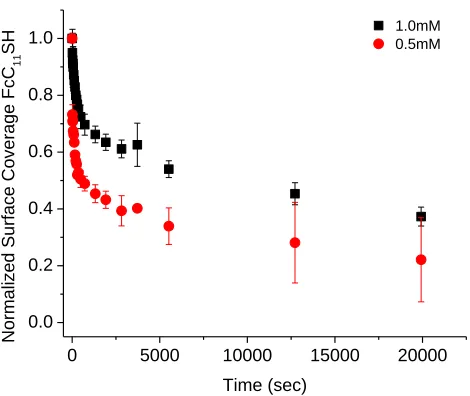
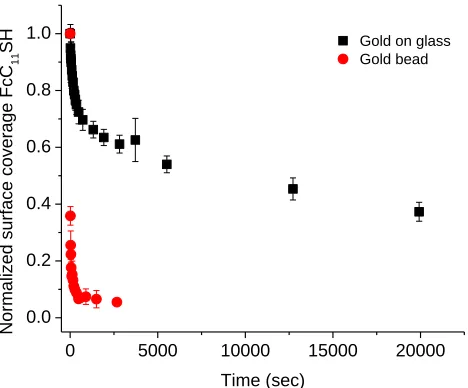
Figure 4.5. Fractional surface coverage of FcC11S- SAMs (molecules/cm2) replaced by C12SH at varying solution concentrations. Surface coverage was normalized for
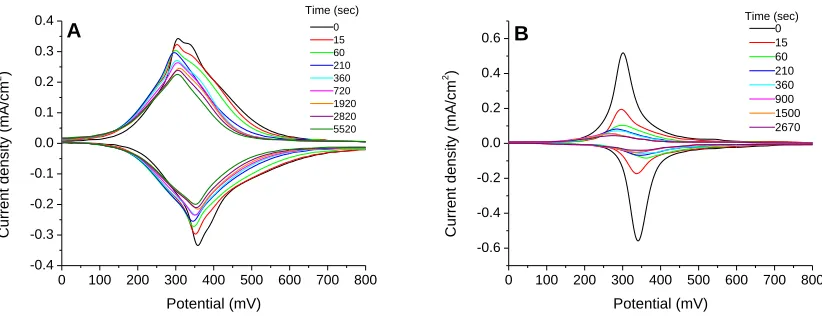
comparison. Error bars represent one standard deviation from multiple trials ...87 Figure 4.6. Cyclic voltammograms of FcC11S- SAMs after replacement by 1mM C12SH (A) or HOC12SH (B) ethanol solutions. Scan rate 500 mV/s; electrolyte 1M HClO4 ...89 Figure 4.7. Fractional surface coverage of FcC11S- SAMs (molecules/cm2) replaced by 1mM C12SH or HOC12SH. Surface coverages were normalized for comparison. Error bars represent standard deviation from multiple trials ...90 Figure 4.8. Fractional surface coverage of FcC11S- SAMs (molecules/cm2) replaced by HOC12SH of different concentrations. Surface coverage was normalized for comparison. Error bars represent standard deviation from multiple trials ...91 Figure 4.9. Cyclic voltammograms of initial FcC11S- SAMs on different types of gold substrates at 100 mV/s (A) and 500 mV/s (B) scan rates. Electrolyte: 1M HClO4 ...95 Figure 4.10. Cyclic voltammograms of FcC11S- SAMs replaced by 1mM HOC12SH. Gold on glass sample (A) and gold bead sample (B). Scan rate 500 mV/s; 1M HClO4 ...96 Figure 4.11. Comparison of replacement of FcC11S- SAMs by 1mM HOC12SH on different gold substrates. Surface coverage was normalized for comparison. Error bars represent standard deviation of multiple trials ...97 Figure 4.12. FcC11S-SAMs replaced by HOC12SH of varying concentrations. Surface coverage was normalized for comparison. Error bars represent standard deviation of multiple trials ...98 Figure 4.13. Cyclic voltammetry of electrochemical cleaning process (0.1 M H2SO4/0.01 M KCl) ...100 Figure 4.14. Comparison of cleaning methods. FcC11S-SAMs replaced by 1mM HOC12SH after cleaning by hydrogen flame annealing (HFA) only or HFA followed by
Figure 4.16. FcC11S-SAMs replaced by 1mM of HOC12SH or C11≡. Surface coverage was normalized for comparison. Error bars represent standard deviation of multiple trials ....104 Figure 4.17. FcC11≡SAMs replaced by 0.1mM HOC12SH. Surface coverage was
normalized for comparison ...105 Figure 4.18. FcC11≡SAMs replaced by C11≡ of varying concentrations. Surface coverage was normalized for comparison. Error bars represent standard deviation of multiple trials ...106 Figure 4.19. Comparison of FcC11≡ SAMs replaced by 0.1mM HOC12SH or C11≡. Surface coverage was normalized for comparison. Error bars represent standard deviation of multiple trials ...107 Figure 4.20. Change in surface coverage upon exposure to ethanol or electrolyte of
FcC11S- SAMs (A) or FcC11≡ SAMs (B). Surface coverage was normalized for comparison108 Figure 4.21. Cyclic voltammograms of K3[Fe(CN)6] blocking experiments by C12S- SAMs when exposed to ethanol (A) or aqueous electrolyte 1M HClO4 (B). Scan rate 100mV/s. Scan acquired with bare gold bead electrode for comparison of K3[Fe(CN)6] signal ...110 Figure 4.22. C12S- SAMs replaced by varying concentrations of FcC11SH. Surface
coverage was normalized for comparison ...111 Figure 4.23. Cyclic voltammograms of K3[Fe(CN)6] blocking experiments by C10≡ SAMs when exposed to ethanol (A) or aqueous electrolyte 1M HClO4 (B). Scan rate 100mV/s. Scan acquired with bare gold bead electrode for comparison of K3[Fe(CN)6] signal ...112 Figure 4.24. C11≡ SAMs replaced by varying concentrations of FcC11SH (A) and FcC11≡
(B). Surface coverage was normalized for comparison ...113 Figure 4.25. Schematic illustration of the electrochemical cell used in this research ...121
Chapter 5: Terminal alkynes as stabilizing ligands for gold nanoparticles
Figure 5.2. Depiction of localized surface plasmons (LSPs). Regions of greater electron density are represented by lighter shading. Electric fields are represented by arrows. Adapted with permission from J. Chem. Edu. 2007, 84(1), 91-96. Copyright 2007
American Chemical Society ...131 Figure 5.3. Extinction spectra of 10 nm, 50 nm, and 100 nm Au particles. The spectra are scaled to the same maximum absorbance. Note the red shift of the plasmons as particle size increases. The colloids were obtained from British Biocell International, Cardiff, Britian. Reprinted with permission from J. Chem. Edu. 2007, 84(1), 91-96. Copyright 2007
American Chemical Society ...132 Figure 5.4. 1H NMR of 1-dodecyne-gold nanoparticles synthesized with TOAB ...134 Figure 5.5. Adapted Brust-Schiffrin synthetic method for the production of 1-dodecyne stabilized gold nanoparticles ...135 Figure 5.6. UV/vis absorption spectra of 1-dodecyne stabilized gold nanoparticles
synthesized with reducing agent, NaBH4, identified as new or old. Absorption values were normalized for comparison ...136 Figure 5.7. UV/vis absorption spectra of 1-dodecyne stabilized gold nanoparticles
synthesized with varying concentrations of reducing agent, NaBH4. Absorption values were normalized for comparison ...137 Figure 5.8. UV/vis absorption spectra of 1-dodecyne stabilized gold nanoparticles
synthesized with different reducing agents, NaBH4 or TBAB. Absorption values were normalized for comparison ...138 Figure 5.9. UV/vis absorption spectra of 1-dodecyne stabilized gold nanoparticles
synthesized at different reduction temperatures. Absorption values were normalized for comparison ...139 Figure 5.10. UV/vis absorption spectra of 1-dodecyne stabilized gold nanoparticles
synthesized with different gold to ligand ratios. Absorption values were normalized for comparison ...140 Figure 5.11. UV/vis absorption spectra of 1-dodecyne stabilized gold nanoparticles
synthesized with different gold(III) salts. Absorption values were normalized for
comparison ...141 Figure 5.12. 1H NMR spectra of 1-dodecyne (top) and 1-dodecyne stabilized gold
Figure 5.13. ATR-IR spectra of free ligand 1-dodecyne (A) and1-dodecyne stabilized gold nanoparticles (B) ...144 Figure 5.14. ATR-IR spectra of 1-dodecyne nanoparticles of C≡C region initially and after 14 days in toluene solution. Absorbance values were normalized and offset for
comparison ...145 Figure 5.15. XPS scan of 1-dodecyne stabilized gold nanoparticles on silicon substrate with native oxide (A) and deconvolution of carbon 1s peak (B) ...146 Figure 5.16. TEM image of 1-dodecyne stabilized gold nanoparticles with corresponding diameter histograms with average diameter of 1.68 + 0.61 nm ...148 Figure 5.17. Dynamic light scattering (DLS) measurements of dodecyne-Au
nanoparticles… ...149 Figure 5.18. Uv/vis absorption spectra of terminal alkyne stabilized gold nanoparticles with ligands; 1-dodecyne, 1-decyne, and 1-octyne for comparison ...150 Figure 5.19. ATR-IR comparison of terminal alkyne stabilized gold nanoparticles using ligands 1-octyne (A), 1-decyne (B), and 1-dodecyne (C) ...151 Figure 5.20. TEM images of gold nanoparticles stabilized by different terminal alkyne ligands with corresponding diameter histograms; dodecyne 1.68 + 0.61 nm diameter(A), decyne 1.84 + 0.84 nm diameter (B), and octyne 1.72 + 0.66 nm diameter (C) ...152 Figure 5.21. TGA of terminal alkyne stabilized gold nanoparticles with ligands;
1-dodecyne, 1-decyne, and 1-octyne ...154 Figure 5.22. Stability of gold nanoparticles stabilized by ( dodecyne, dodecanethiol, and triphenyl phosphine) ligands in toluene (A) or tetrahydrofuran (B) at room
Figure 5.24. Stability of gold nanoparticles stabilized by (dodecyne, decyne, and octyne) ligands in toluene (A) and tetrahydrofuran (B) at room temperature. Changes in absorbance and position of surface plasmon band were monitored over time. Absorbance was normalized for comparison ...159 Figure 5.25. Stability of gold nanoparticles stabilized by (dodecyne, decyne, and octyne) ligands in excess acetic acid (A) and excess pyridine (B). Changes in absorbance and position of surface plasmon band were monitored over time. Absorbance was
normalized for comparison ...160 Figure 5.26. NaCN etching of gold nanoparticles stabilized by (dodecyne,
dodecanethiol, and triphenyl phosphine) ligands. The decrease in absorbance of the localized surface plasmon band was monitored over time. Absorbance was normalized for comparison ...161 Figure 5.27. NaCN etching of gold nanoparticles stabilized by (dodecyne, decyne , and
octyne) ligands. The decrease in absorbance of the localized surface plasmon band was monitored over time. Absorbance was normalized for comparison ...163 Figure 5.28. Stability of gold nanoparticles stabilized by (dodecyne, dodecanethiol, and triphenyl phosphine) ligands heated to 50°C (A) and 80°C (B) in toluene. Change in absorbance and position of surface plasmon band was monitored over time. Absorbance was normalized for comparison ...166 Figure 5.29. Stability of gold nanoparticles stabilized by (dodecyne, decyne, and octyne) ligands heated to 50ºC (A) 80°C (B) in toluene. Change in absorbance and position of surface plasmon band was monitored over time. Absorbance was normalized for
comparison ...167 Figure 5.30. TEM images and corresponding histograms of dodecyne stabilized gold nanoparticle before (top) and after heating in toluene at 80°C for 65 hours (bottom) ...168 Figure 5.31. Proposed bonding modes of terminal alkynes complexed with gold. η2
-Acetylene (A), η1
LIST OF SCHEMES Chapter 1: Organic materials for electronic and energy use
Scheme 1.1. Synthesis BBL polymer ...9 Chapter 2: Synthesis of fused polycyclic aromatic molecules
Scheme 2.1. Proposed synthetic route to fused aromatic small molecules ...18 Scheme 2.2. Proposed route to novel graphite type, aromatic polymer ...19 Scheme 2.3. Synthesis of cyano containing compounds ...20 Scheme 2.4. Two routes to amino-iso-quinoline derivatives from o-cyano-α-tolunitrile….21 Scheme 2.5. Cyclization utilizing excess HBr/HOAc ...22 Scheme 2.6. Cyclization of poly-cyano molecules with 10 equivalents of nBuLi ...24 Scheme 2.7. Proposed formation of tert-methyl silyl ketene-imine intermediate...25 Scheme 2.8. Cyclization utilizing n-BuLi and TBDMSCl ...26 Scheme 2.9. Conversion of nitrilium salt to amide or amidine ...27 Scheme 2.10. Conversion of diphenylacetonitrile to N-tert-butyl ketene-imine ...28 Scheme 2.11. Conversion of poly-cyano compound 1 to N-tert-butyl ketene-imine
derivative.. ...29 Scheme 2.12. Formation of terminal acetamide product during purification ...30 Scheme 2.13. Conversion of poly-cyano compound 2 to N-tert-butyl
Chapter 3: Progress toward the synthesis of fused polycyclic aromatic polymers
Scheme 3.1. Proposed polymerizations of fused poly-cyclic aromatic A-B monomers ...42 Scheme 3.2. Synthetic route to fused poly-cyclic aromatic A-B monomers ...43 Scheme 3.3. Polymerization of fused poly-cyclic aromatic A-B monomers ...44 Scheme 3.4. Results of polymerization of fused-ring A-B monomers: formation of cyclic oligomers ...46 Scheme 3.5. Synthesis of fused poly-cyclic aromatic A-B monomer 15c ...47 Scheme 3.6. Synthesis of fused poly-cyclic aromatic A-B monomer 15d...52 Scheme 3.7. Oligomerization of monomer 15d ...53 Scheme 3.8. Coupling results of a similar A-B monomer under dilute conditions by
Yamamoto ...54 Chapter 4: Adventitious replacement of ferrocene terminated thiol and alkyne
self-assembled monolayers on gold
Chapter 1: Organic materials for electronic and energy use
1.1Introduction
Interest into organic materials for electronic and energy applications started over 30 years ago. It began with the realization of the metallic conducting properties of doped
polyacetylene in 1977.1, 2 In recent years the neutral, semiconducting (undoped) forms of polymers has constituted the main area of research. Both oligomers and polymers containing heteroaromatic units (i.e. pyrrole, aniline and thiophene) are of interest as charge transport materials as they demonstrate increased stability as compared to the original carbon based molecules.3-5
These charge transport materials are currently being used in organic thin-layer film devices (OTFDs). Organic materials with superior charge transport throughout the bulk material are necessary in order for these devices to compete with current technologies. Another challenge is to develop metal-organic interfaces that are also efficient at charge transport. This requires that the work function of the metal matches well with the energy levels of the organic material. Development of molecular ‘alligator clips’ at the metal-organic interface are crucial to device performance.
1.2 Organic thin-layer film devices
The most popular OTFDs include organic thin-film transistors (OTFTs), organic light emitting diodes (OLEDs) and organic photovoltaic cells (OPVs). OLEDs have recently received commercial success when used in high resolution flat-screen displays. The
compatibility with flexible substrates.6, 7 The efficiency and lifetime of these devices still do not compete with current technologies, however.
The research presented here focuses on the development of novel methodologies for the synthesis of organic materials with superior charge mobilities. Although the fabrication of devices utilizing these materials is not proposed at this time, it is still important to
understand the way in which these devices function. As mentioned previously, organic materials designed for charge transport have been utilized in multiple thin-layer devices. The specific electronic and photonic properties of these materials will differ with each device. A thorough discussion of the properties for each device is beyond the scope of this document. However, a basic overview of the materials required for OTFTs is discussed.
1.3 Organic thin-film transistors
OTFTs are electronic switches and are the basic building blocks for integrated
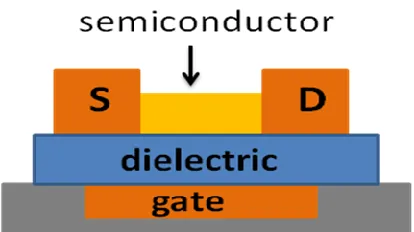
circuits. A schematic diagram of the structure is shown in Figure 1.1. The current that flows between the source and drain electrodes is controlled by the magnitude of the gate bias. The type of charge flow is dominated by the type of semiconductor used. Semiconductors can be of either the p-type which transfers mainly holes or the n-type which transfers mainly
Figure 1.1. Schematic diagram of an organic thin-film transistor (OTFT). S = source and D=drain.
OTFTs are composed of several different types of materials. The drain, source, and gain electrodes require conducting materials while semiconductors are used for the active transport channel. The dielectric layer is composed of an insulator. Figure 1.2 depicts the relationships between band structure diagrams and the predicted electronic properties of a material.8 The valence band or highest occupied molecular orbital (HOMO) is separated from the conduction band or lowest unoccupied molecular orbital (LUMO) by an energy gap often referred to as the band gap (Eg). A material that does not transport charge has a large energy gap between the HOMO and LUMO and is characterized as an insulator (A). A
Figure 1.2. Schematic representation of the band diagrams of the following types of materials; insulator (A), semiconductor (B), and metal (C).
The transistor performance is based on two important factors. The first is charge carrier mobility (µ) which is a measure of how fast the electrons or holes pass within the semiconductor material when an electric field is applied. The second is the on-off ratio (Ion:Ioff) which is a measure of how efficiently the current is modulated by the source-gate bias. Two voltages (typically 0V and 20V) are used to determine this ratio.9
1.4 Bandgap engineering
Minimizing the bandgap control of π-conjugated oligomers and polymers has been the major focus when developing organic materials capable of charge transport. However, the use of π-conjugated materials in thin-layer devices such as OLEDs, OFETs, or OPVs as discussed previously has introduced major changes into bandgap engineering. Focus has shifted to control both the HOMO and LUMO energy levels as well as the bandgap energy. These energy levels affect the charge-transport properties of the materials but also the absorption and/or emission properties that are essential for electronic and photonic
applications. The bandgap of a linear π-conjugated system can be altered based on the five factors as shown in Equation 1.112
E
g= E
BLA+ E
Res+ E
Sub+ E
Θ+ E
Int (Equation 1.1)The second factor is the resonance effect and is applicable to aromatic systems. It is dependent upon the resonance stabilization energy of the aromatic unit. Aromatic systems, such as polythiophene (PT), have a non-degenerate ground state. The two mesomeric forms are obtained by converting the aromatic structure to the quinoid structure as seen in Figure 1.3. These two forms are not energetically equivalent as the aromatic form is generally more stable with a lower energy. The quinoidal form, however, is higher in energy and therefore has a lower energy gap.14 The energy required to convert between the two forms is directly related to the aromatic stabilization resonance energy of the aromatic unit. This resonance effect confines the π-electrons within the aromatic unit preventing delocalization along the chain.
S S
S
A
S
S
S
B
Figure 1.3. Mesomeric forms of polythiophene; aromatic (A) and quinoid (B).
The third factor involves the rotational disorder between the aromatic units of the backbone. Rotation of the aromatic units limits the amount of delocalization of the
while electron withdrawing groups tend to lower these energy levels. The final factor is related to intermolecular interactions between chains which, in some cases, may have a large impact on the bandgap. This equation relating the different energy factors to the bandgap energy is purely qualitative and provides a direction for the design of an appropriate molecular structure. Examples of tailoring molecular structure of materials to achieve desired electronic and photonic properties are prevalent in the literature. The research presented here is mainly focused on two of the factors in the equation; EBLA and EΘ. A brief review of synthetic methodologies affecting these two energy factors follows.
1.5 Low bandgap materials
form is responsible for reducing the bandgap as it increase the double bond character of the thiophene-thiophene linkages.17
Figure 1.4. Structures of regioregular polythiophene (A) and polyisothionapthelene (B).
The second approach is to decrease the rotational disorder between the aromatic units by fusing the aromatic subunits. Rigidification of these conjugated systems will increase the overall planarity of the molecule and therefore increase p-orbital overlap allowing for
increased π-electron delocalization. Increased planarity will also enhance the interchain interactions between the oligomers or polymer chains.
* * R R R R R R Figure 1.5 Structure of polyacene.
A synthetic methodology has been developed to avoid the need to process insoluble materials. A soluble precursor oligomer or polymer is synthesized and then can be converted to the insoluble fused polycyclic material once it has been processed. Several examples of this exist in the literature. Ruan and Litt20-23 prepared the fused aromatic polymer Poly(1-methylcyclohexa-1,3-diene-2,3-diyl-5,6-diylidene-5-methylidyne-6-nitrilo) (BBL) shown in Scheme 1.1. The soluble precursor polymer was synthesized by the reaction of
diaminotoluene with aldehydes in acidic aqueous solutions. This precursor polymer was then cyclized with poly phosphoric acid (PPA) at high temperatures to form the final fused ring aromatic polymer. However, characterization data indicate incomplete cyclization and low molecular weight precursor polymers due to a lack of solubility.
Scheme 1.1. Synthesis BBL polymer
H2N
CH3
NH2 H2N
CH3 NH2 * * R N CH3 R * * n
+ R CHO H2O, HCl PPA
heat
1.6 Functional groups as molecular alligator clips
Modification of the electrode surface with an organic thin-film is often used to enhance the charge carrier mobility of thin-film devices and to alter the morphology at the metal-organic interface.24 Self-assembled monolayers (SAMs) are a common method of creating thin films on metals.25 SAMs are formed by organic molecules that spontaneously absorb onto a surface. This self-assembly is driven by the attractive interaction between the organic binding group and the substrate as well as the collective intermolecular Van der Waals interactions between the alkyl chains. When the surface is exposed to a solution containing SAM forming molecules, the initial absorption occurs within seconds. This is followed by slow reorganization of the initial monolayer to form a more ordered and stable SAM.
In order to develop efficient devices it is necessary to bridge the molecular scale with the macroscale without sacrificing the electronic properties. In order to enhance charge carrier mobility at the metal-organic interface, the Fermi level of the metal electrode should align with the molecular LUMO energy level. Metals with high Fermi levels such as gold or palladium are often used to achieve this alignment. The Fermi level of gold aligns most closely to selenium followed by sulfur and tellurium.26 Due to electronic overlap and strength of the gold sulfur bond this combination is often used for the synthesis of SAMs. However these binding groups are not optimal for charge transfer.27 Isonitriles have also been explored as binding groups however, there are disagreements as to whether they offer enhanced
dipole moment (ca. 4.1 D for n-propyl isonitrile)29 which may have a negative effect on the electronic coupling between the metal and organic material.30
The use of alkynes presents an opportunity for enhanced electronic communication at the metal-organic interface. The bonding of an alkyne-derived functional group should occur with a low interfacial dipole moment and with both dative (σ-type) and π-type components. Three options for the bonding of a terminal alkyne to gold have been proposed as shown in Figure 1.6. The first proposed bonding mode is a side-on η2- acetylene (1.6A). The second is an end-on η1- acetylide (1.6B) that would result from deprotonation of the terminal alkyne. The third is an end-on η1-alkylidene (1.6C) which results from a 1,2-hydrogen shift of the terminal hydrogen to form a carbine type bonding group.
Figure 1.6. Proposed bonding modes of terminal alkynes complexed with gold. η2 -Acetylene (A), η1
-Acetylide (B), and η1-Alkylidene (C)
different alkynes might bond in different modes and undergo different chemical transformations on metal surfaces.
Figure 1.7 Configuration of surface-bound molecule ethynyl benzene: (a) final state (b) vinylidene intermediate, and (c) flat intermediate. Reprinted with permission from J. Phys. Chem. B 2005, 109, 20387-20392. Copyright 2005 American Chemical Society.
1.7 Research impetus
Two dominant themes in the synthesis of molecules for use in thin-layer electronic devices are the ability to control the HOMO-LUMO gap and to achieve superior charge mobilities. Controlling the HOMO and LUMO energy levels will also allow for control of the bandgap and therefore the electronic and photonic properties of the materials. Devising a method to achieve superior charge mobilities within the material and at the metal-organic interface is also important. This work covers two synthetic methodologies devised to promote these desirable properties.
The first synthetic methodology focuses on the development of a route to fused aromatic systems. A single nucleophile initiated cascading cyclization of a series of poly-cyano molecules to afford fused aromatic oligomers was explored. Cyclization of a ketene-imine derivative with milder nucleophiles was also investigated. The synthesis of a partially fused-ring isoquinoloine based polymer was attempted.
The second approach focuses on the discovery and use of terminal alkyne functional groups capable to promote strong electronic coupling between molecules on the nanoscale and the metal contacts on the macroscale. SAMs of terminal alkyne molecules on planar gold substrates as well as stabilizing ligands on gold nanoparticles were explored. The stability of the terminal alkyne ligand was of interest and the specific bonding mode adopted once the terminal alkyne was bound to the gold surface or gold nanoparticle was
1.8 References
(1) Chiang, C.K.; Fincher, C.R.; Park, Y.W.; Heeger, A.J.; Shirakawa, H.; Louis, E.J.; Gau, S.C.; Macdiarmid A.G., Electrical-conductivity in doped polyacetylene. Phys. Rev. Lett.
1977, 39, 1098-1101.
(2) Shirakawa, H.; Louis, E.J.; Macdiarmid, A.G.; Chiang, C.K.; Heeger, A.J., Synthesis of electrically conducting organic polymers-halogen derivatives of polyacetylene, (CH)X. J. Chem. Soc. Chem. Commun. 1977, 16, 578-580.
(3) Diaz, A.F.; Kanazawa, K.K.; Gardini, G.P., Electrochemical polymerization of pyrrole.
J. Chem. Soc. Chem. Commun. 1979, 14, 635-636.
(4) Roncali, J., Conjugated poly(thiophenes) – synthesis, functionalization, and applications.
Chem. Rev.1992, 92, 711-738.
(5) Genies, E.M.; Lapkowski, M.; Tsintavis, C., Preparation, properties and applications of polyaniline. New J. Chem. 1988, 12, 181-196.
(6) Bao, Z.; Locklin, J., Organic field effect transistors. Eds.; Taylor and Francis Group, LLC, 2007.
(7) Ling, M.M.; Bao, Z.N., Thin film deposition, patterning, and printing in organic thin film transistors. Chem. Mater. 2004, 16, 4824-4830.
(8) Anslyn, E.V.; Dougherty, D.A., Modern Physical Organic Chemistry. Eds.; University Science Books, 2006.
(9) http://www.polyera.com/basic-devices/organic-thin-film-transistors
(10) Bao, Z.; Dodabalapur, A.; Lovinger, A.J., Soluble and processable regioregular poly (3-hexylthiophene) for thin film field effect transistor applications with high mobility. Appl. Phys. Lett. 1996, 69, 4108-4110.
(11) Sirringhaus, H.; Tessler, N.; Friend, R.H., Integrated optoelectronic devices based on conjugated polymers. Science 1998, 280, 1741-1744.
(12) Roncali, J., Molecular engineering of the band gap of π-conjugated systems: facing technological applications. Macromol. Rapid Commun. 2007, 28, 1761-1775.
(14) Bredas, J-L., Relationship between band gap and bond length alternation in organic conjugated polymers. J. Chem. Phys. 1985, 82, 3808-3812.
(15) Bredas, J.L.; Chance, R.R.; Baughman, R.H.; Silbey, R., Ab initio effective
Hamiltonian study of the electronic properties of conjugated polymers. J. Chem. Phys. 1982, 76, 3673-3678.
(16) Bredas, J.L.; Silbey, R.; Boudreaux, D.S.; Chance, R.R., Chain-length dependence of electronic and electrochemical properties of conjugated systems: polyacetylene,
polyphenylene, polythiophene, and polypyrrole. J. Am. Chem. Soc., 1983, 105, 6555-6559. (17) Roncali, J., Synthetic principles for bandgap control in linear pi-conjugated systems.
Chem. Rev. 1997, 97, 173-205.
(18) Bredas, J.L.; Themans, B.; Andre, J.M., Valence effective Hamiltonian technique for nitrogen containing polymers- electronic structure of polypyrrole, pyrolized polyacrylonitrile, paracyanogen, polymethineimine, and derivatives. J. Chem. Phys., 1983, 78, 6137-6148. (19) Teyssie, P.; Korngirard, A.C., Synthesis of new monomers and polymers. 4. Synthesis and properties of polydiphenyldiacetylenes. J. of Polymer Science A. 1964, 2, 2849.
(20) Ruan, J.Z.; Litt, M.H., Electronic properties of the linear ladder polymer, poly[4-(3-pyridyl),8-methyl-2,3-6,7-quniolino] (PPMQ). J. Polymer Science A: Polymer Chem. 1987, 25, 285-297.
(21) Ruan, J.Z.; Litt, M.H., Synthesis and characterization of poly[3,5’-(3-pyridyl methyl)-2,6-diamainotoluene] and its condensation product, poly[4-(3- pyridyl),8-methyl-2,3-6,7-quniolino]. J. Polymer Science A: Polymer Chem. 1987, 25, 299-309.
(22) Ruan, J.Z.; Litt, M.H., Synthesis and characterization of poly[3,5’-(3-pyridyl methyl)-2,6-diamainotoluene] and its condensation product, poly(1-methylcyclohexa-1,3-diene-2,3-diyl-5,6-diylidene-5-methylidyne-6-nitrilo), a linear ladder aromatic polymer.
Macromolecules. 1988, 21, 876-882.
(23) Ruan, J.Z.; Litt, M.H., Electronic properties of poly(1-methylcyclohexa-1,3-diene-2,3-diyl-5,6-diylidene-5-methylidyne-6-nitrilo). Macromolecules. 1988, 21, 882-890.
(24) Gundlach, d.J.; Jia, L.L.; Jackson, T.N., IEEE Electr. Dev. 2001, 22, 571-573. (25) Kymissis, I.; Dimitrakopoulos, C.D.; Purushothanman, S., IEEE Trans. Elec. Dev.
(26) Seminaro, J.M.; Zacarias, A.G.; Tour, J.M., Molecular alligator clips for single
molecule electronics. Studies of group 16 and isonitriles interfaced with Au contacts. J. Am. Chem. Soc. 1999, 121, 411-416.
(27) (a) Seminario, J. M.; Zacarias, A. G.; Tour, J. M., Molecular alligator clips for single molecule electronics. Studies of group 16 and isonitriles interfaced with Au contacts. J. Am. Chem. Soc. 1999, 121 p. 6 (b), 411-416; (b) Seminario, J. M.; De la Cruz, C. E.; Derosa, P. A., A theoretical analysis of metal-molecule contacts. J. Am. Chem. Soc. 2001, 123 (23), 5616-5617.
(28) (a) Stapleton, J. J.; Daniel, T. A.; Uppili, S.; Cabarcos, O. M.; Naciri, J.; Shashidhar, R.; Allara, D. L., Self-assembly, characterization, and chemical stability of isocyanide-bound molecular wire monolayers on gold and palladium surfaces. Langmuir 2005, 21 (24), 11061-11070; (b) Kiguchi, M.; Miura, S.; Hara, K.; Sawamura, M.; Murakoshi, K., Conductance of a single molecule anchored by an isocyanide substituent to gold electrodes. Appl. Phys. Lett.
2006, 89 (21); (c) Kiguchi, M.; Miura, S.; Hara, K.; Sawamura, M.; Murakoshi, K.,
Conductance of single 1,4-disubstituted benzene molecules anchored to Pt electrodes. Appl. Phys. Lett. 2007, 91 (5); (d) Venkataraman, L.; Klare, J. E.; Tam, I. W.; Nuckolls, C.; Hybertsen, M. S.; Steigerwald, M. L., Single-molecule circuits with well-defined molecular conductance. Nano Lett. 2006, 6 (3), 458-462; (e) Chu, C.; Ayres, J. A.; Stefanescu, D. M.; Walker, B. R.; Gorman, C. B.; Parsons, G. N., Enhanced conduction through isocyanide terminal groups in alkane and biphenylene molecules measured in
molecule/nanoparticle/molecule junctions. J. Phys. Chem. C 2007, 111 (22), 8080-8085. (29) Fuller, M. J.; Wilson, E. B., Microwave spectrum and rotational isomerism of n-propyl isocyanide. J. Mol. Spectrosc. 1975, 58 (3), 414-426.
(30) (a) Selzer, Y.; Cahen, D., Fine tuning of Au/SiO2/Si diodes by varying interfacial dipoles using molecular monolayers. Adv. Mater. 2001, 13 (7), 508-511; (b) Vilan, A.; Shanzer, A.; Cahen, D., Molecular control over Au/GaAs diodes. Nature 2000, 404 (6774), 166-168; (c) Cahen, D.; Kahn, A., Electron energetics at surfaces and interfaces: Concepts and experiments. Adv. Mater. 2003, 15 (4), 271-277; (d) Salomon, A.; Cahen, D.; Lindsay, S.; Tomfohr, J.; Engelkes, V. B.; Frisbie, C. D., Comparison of electronic transport
measurements on organic molecules. Adv. Mater. 2003, 15 (22), 1881-1890.
(31) Ford, M. J.; Hoft, R. C.; McDonagh, A., Theoretical Study of Ethynylbenzene
Adsorption on Au(111) and Implications for a New Class of Self-Assembled Monolayer. J. Phys. Chem. B 2005, 109 (43), 20387-20392.
(33) (a) Wakatsuki, Y., Mechanistic Aspects Regarding the Formation of Metal Vinylidenes from Alkynes and Related Reactions. J. Organomet. Chem. 2004, 689 (24), 4092-4109; (b) Bullock, R. M., Rearrangement of a Metal (η2-Alkyne) Complex to a Metal Vinylidene and Subsequent Reaction of the Metal Vinylidene to Regenerate the Alkyne. J. Chem. Soc.-Chem. Commun. 1989, (3), 165-167; (c) Quayle, P.; Rahman, S.; Ward, E. L. M.; Herbert, J., Transition-Metal Promoted Acetylene Isomerization-Reactions in Organic-Synthesis.
Tetrahedron Lett. 1994, 35 (22), 3801-3804; (d) Wakatsuki, Y.; Koga, N.; Yamazaki, H.; Morokuma, K., Acetylene π-Coordination, Slippage To σ-Coordination, and 1,2-Hydrogen Migration Taking Place on A Transition-Metal - The Case of a Ru(II) Complex As Studied By Experiment and Ab-Initio Molecular-Orbital Simulations. J. Am. Chem. Soc. 1994, 116
(18), 8105-8111; (e) Johnson, R. P.; Daoust, K. J., Interconversions of Cyclobutyne, Cyclopentyne, Cyclohexyne, and Their Corresponding Cycloalkylidenecarbenes. J. Am. Chem. Soc. 1995, 117 (1), 362-367; (f) Kluwe, C.; Davies, J. A., Lewis-Acid Catalysis of the Rearrangement of a Dipalladium Acetylene Adduct to a Vinylidene-Bridged Complex.
Organometallics 1995, 14 (9), 4257-4262; (g) Stegmann, R.; Frenking, G., Mechanism of the Acetylene-Vinylidene Rearrangement in the Coordination Sphere of a Transition Metal.
Organometallics 1998, 17 (10), 2089-2095; (h) De Angelis, F.; Sgamellotti, A.; Re, N., Acetylene to vinylidene rearrangements on electron rich d(6) metal centers: a density functional study. Dalton Trans. 2004, (20), 3225-3230.
(34) Tulevski, G. S.; Myers, M. B.; Hybertsen, M. S.; Steigerwald, M. L.; Nuckolls, C., Formation of Catalytic Metal-Molecule Contacts. Science 2005, 309 (5734), 591-594. (35) Chen, W.; Davies, J. R.; Ghosh, D.; Tong, M. C.; Konopelski, J. P.; Chen, S., Carbene-Functionalized Ruthenium Nanoparticles. Chem. Mat. 2006, 18 (22), 5253-5259.
(36) (a) Boorman, T. C.; Larrosa, I., Gold-mediated C-H bond functionalisation. Chem. Soc. Rev. 2011, 40 (4), 1910-1925; (b) Liu, B.; Zhang, N.; Chen, W., Synthesis of Metal N-Heterocyclic Carbene Complexes. Progress in Chemistry 2010, 22 (11), 2134-2146; (c) Marion, N.; Nolan, S. P., N-heterocyclic carbenes in gold catalysis. Chem. Soc. Rev. 2008, 37
Chapter 2: Synthesis of fused polycyclic aromatic molecules
2.1 Introduction
Here, the production of new, candidate molecules for molecular electronics is explored via an efficient, cascading cyclization to produce fused poly-cyclic aromatic structure. This approach first involves the development of a synthetic methodology for the production of poly-cyano precursor molecules. This step is followed by a single nucleophile initiated cyclization to form the fused aromatic molecules as proposed in Scheme 2.1. In order to avoid incomplete cyclization and formation of side products, the cascading cyclization must be initiated at one only terminus and continue along the length of the molecule. Ultimately, the mildest possible conditions are desirable.
Scheme 2.1. Proposed synthetic route to fused aromatic small molecules
X +
R1
N N
X = Br/Cl R1 = H or CN
N N
R1
R1 = H or CN
N
N N
Nu NH2
NH2 Nu
Nu:
R1 =
H
N u: R
1 =
CN
the cascading cyclization of the benzylic nitriles and subsequent tautomerization. The precursor polymer can be derivatized by substituting various solubilizing, electron donating or electron withdrawing groups at the para position of the aromatic ring in each repeat unit. It is anticipated that this will allow for increased solubility and the ability to alter the
electronic properties of the final polymer.1
Scheme 2.2. Proposed route to novel graphite type, aromatic polymer
CN CN X
R
CN CN CN CN
CN
R R R
CN
n
Cyclize X = Br, Cl, I
R = various
Solubilizing group Electron donating group Electron withdrawing group
N N N N N N
X
R R R R
CN NH
n
Tautomerize
N HN N HN N HN
X
R R R R
CN NH
2.2 Synthesis of cyano-containing small molecules
A synthetic methodology for the production of molecules containing two or three cyano groups was previously developed as shown in Scheme 2.3. The molecules 1 and 2 were synthesized by nucleophilic aromatic substitution reactions. The two cyano-containing molecule, 1, was recovered in good yields however the synthesis of 2 resulted in a lower yield and required elevated temperatures as reported in the literature.2,3 This paper indicated that the additional electron withdrawing cyano group of the starting material, σ-cyano-α-tolunitrile resulted in decreased reactivity.
Scheme 2.3. Synthesis of cyano containing compounds2,3
Cl
N N N N
+ KOtBu
THF
1: 82%
Cl
N N N N
+ KOtBu
THF 70oC
N N
2: 41%
determination of the efficiency of the cyclization reactions as the fused aromatic molecules should be soluble to the extent that the product can be isolated and characterized.
n-C6H13
N N N N N N N N N N N N
3 4
n-C6H13
n-C6H13
N
5
Figure 2.1. Molecules containing 4- and 5-cyano groups2,4
2.3 Prior efforts on the cyclization of poly-cyano containing molecules
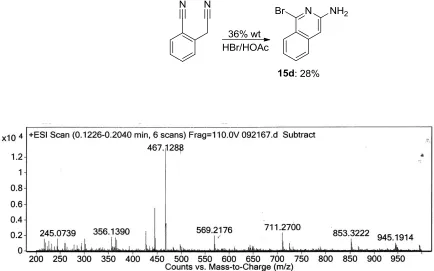
Deady et al.5 and Frohn et al.6 previously reported the synthesis of amino-iso-quinolines from o-cyano-α-tolunitrile through a selective attack of Br- at the aryl cyanide using 36 wt% HBr/HOAc. Tander, et al7 reported the cyclization of the same molecule in the presence of the excess strong base n-butyl lithium. Both cyclization routes are shown in Scheme 2.4.
Scheme 2.4. Two routes to amino-iso-quinoline derivatives from o-cyano-α-tolunitrile. N
R NH2
n-BuLi
HBr/HOAc
R = Br R = nBu
CN CN
Previous group member, Dongchaun Wang, experimented with acid-mediated cyclization utilizing excess hydrogen bromide in acetic acid3. In this reaction the acid presumably catalyzes the ring-closing reaction, but note that the bromide anion acts as a nucleophile, and ultimately this nucleophile initiates the reaction. This method proved reasonably efficient in cyclizing compounds 1 and 2. These results were not translated to longer model compounds, however (Scheme 2.5). Cyclization of 3 resulted in a mixture of cyclized and partially-cyclized molecules. Different acids and halide salts were explored but did not improve the regioselectivity of the reaction. Therefore, these conditions are not applicable for the formation of longer fused polycyclic aromatic molecules.
Scheme 2.5. Cyclization utilizing excess HBr/HOAc3
N N
N N N
1
2
N N N
n-C6H13
N
HBr/HOAc 72%
HBr/HOAc 59%
HBr/HOAc
N
Br NH2
N
Br N NH2
N N N N
Br NH2 Br NH2
N
Br NH2
n-C6H13
n-C6H13
+ H
Scheme 2.6. Cyclization of poly-cyano molecules with 10 equivalents of nBuLi2,4
N N
N N N
1
2
N N N N
N N N N
n-C6H13 n-C6H13
N
-78oC 0oC THF
-78oC 0oC THF
-78oC 0oC THF
-78oC 0oC THF 4 5 96% 97% 77% 25%
N NH2
N N NH2
N N N NH2
N N N N
n-C6H13 n-C6H13
NH2 H H nBuLi nBuLi nBuLi nBuLi
a weaker nucleophile to induce the cascade cyclization. The proposed formation of the silyl ketene-imine intermediate of molecule 1 is shown in Scheme 2.7. However, the silyl ketene-imine intermediates were not stable enough to be isolated and therefore could not be fully characterized.
Scheme 2.7. Proposed formation of tert-methyl silyl ketene-imine intermediate
H
N N
-HB
B
N N N N N N
-Cl -Si
Cl Si
Scheme 2.8. Cyclization utilizing n-BuLi and TBDMSCl4
N N
N N N
1
2
N N N
n-C6H13
N
TBDMSCl
n-BuLi 88%
N NH2
N N NH2
TBDMSCl
n-BuLi 86%
TBDMSCl
n-BuLi 66%
N N
n-C6H13
H2N
3
2.4 Synthesis of N-alkyl ketene-imines
The goal of the research described here is to develop a cascading cyclization via an alkyl ketene-imine derivative that is stable enough to be isolated and fully characterized yet reactive enough to undergo cyclization using a mild reagent. Formation of N-alkyl
ketene-imines from alkyl and aryl substituted nitriles are known.8, 10, 13 Fuks, et al.10 have synthesized N-alkyl ketene-imines by converting benzylic nitriles to nitrilium salts. These nitrilium salts are strong electrophiles and are easily converted to amides with water14 and amidines with amines15 as shown in Scheme 2.9. The nitritlium salt is converted to N-methyl acetamide upon treatment with water or to N-methyl-N-phenyl acetamide upon treatment with aniline.
Scheme 2.9. Conversion of nitrilium salt to amide or amidine
CH3C N CH3 X
X = BF4- or SbCl6
-H2O
CH3C O
H N CH3
C
6H
5NH
2 CH3C N
NH CH3
C6H5
addition of triethylamine to abstract the methine proton and form the corresponding ketene-imine. The product was successfully isolated in a 71% yield after purification by flash chromatography on activated basic alumina.16, 17
Scheme 2.10. Conversion of diphenylacetonitrile to N-tert-butyl ketene-imine
chloride were added bringing the total equivalents to 2.2 and 2.0 respectively. This improved the yield of the ketene-imine product 7 to 40% as shown in Scheme 2.11.
Scheme 2.11. Conversion of poly-cyano compound 1 to N-tert-butyl ketene-imine derivative
N N
AlCl3 tBuCl Et3N NaOH
N N
7: 40%
1
Scheme 2.12. Formation of terminal acetamide product during purification
N N
7
O H
H N O N HN OH
N
H
H HN O
N
The poly-cyano molecule 2 was then converted to the corresponding ketene imine, 8, in 20% yield (Scheme 2.13). An additional equivalent of AlCl3 based on the number of nitrile groups in the molecule was added again (3.2 equiv total). The use of excess AlCl3 and
tert-butyl chloride (5 equivalents) was explored for the conversion, however, no increase in yield was observed. Additional steps were taken to avoid the conversion of the ketene-imine to the acetamide by forgoing the basic workup and purifying the compound immediately, however, these attempts were unsuccessful in increasing the yield.
Scheme 2.13. Conversion of poly-cyano compound 2 to corresponding N-tert-butyl ketene-imine
acid per cyano moiety of the compound and successive decrease in yield made this route unattractive for the synthesis of longer ketene-imine derivatives or as a possible application to the precursor polymer. The formation of acetamide side products and the instability of these ketene-imine compounds may also complicate the proposed cascading cyclization reactions. However, cyclization of the ketene-imine compounds 7 and 8 were explored and are discussed below.
2.5 Cyclization of N-alkyl ketene-imines
The cyclization of the N-tert-butyl ketene-imine 7 was attempted first with lithium piperdide as the nucleophile. Three equivalents of lithium piperidide were required to form the cyclized product 9 in 40% yield as shown in Scheme 2.14. The use of additional lithium piperdide (10 equivalents) was not successful in increasing the yield. Products formed from nucleophilic attack at the ketene-imine carbon followed by cyclization could not be identified in the reaction mixture, rather, only residual starting material was recovered.
Scheme 2.14. Cyclization of N-tert-butyl ketene-imine 7
N N N
Li
N
N NH
9: 40%
7
The cyclization of N-tert-butyl ketene-imine 8 was also attempted using lithium piperdide as nucleophile, however, only starting material was recovered. Several attempts were made at synthesizing the ketene-imine derivative 8 followed by in-situ cyclization with lithium piperdide to produce the desired fused poly-cyclic aromatic molecule 10 as shown in Scheme 2.15. The reaction produced a multitude of products as evidenced by thin-layer chromatography (TLC) of the crude mixture. The desired product was present in the 1H NMR spectrum of the crude material however pure product was not recovered after purification.
Scheme 2.15. Cyclization of N-tert-butyl ketene-imine 8
2.6 Synthesis of N-acyl ketene-imines
the production of the N-acyl ketene-imine as only starting material was recovered. Therefore, the conversions of poly-cyano compounds 1 and 2 to the N-acyl ketene-imines were not attempted.
Scheme 2.16. Proposed conversion of diphenylacetonitrile to N-acyl ketene-imine
2.7 Conclusion
N-tert-butyl ketene-imine compounds 7 and 8 were produced in 40% and 20% yields respectively. The cyclization of compound 7 with lithium piperdide was successful albeit in poor yields. This nucleophile, however, was not sufficient in the cascading cyclization of the ketene-imine compound 8. The reactive nature of the N-alkyl ketene-imines provided an attractive route to the formation of fused polycyclic aromatic structures. However, this reactive nature also made purification of these molecules difficult. Ultimately the
2.8 Experimental
All NMR spectra were obtained on a Varian 300 MHz spectrometer with trimethylsilane as
an internal standard. All IR spectra were taken on a FT-IR Perkin Elmer Spectrum RX I
Spectrometer. Tetrahydrofuran (THF) was distilled prior to use with sodium/benzophenone
ketyl under nitrogen. Dimethylformamide (DMF) was dried over sodium and distilled under
reduced pressure. All commercially available materials were used as received, except for
σ-cyanobenzonitrile which was purified by flash chromatography on silica with 0-2% ethyl
acetate/hexanes mixture. Reagents used to prepare ketene imines, 6, 7 and 8 were purified
prior to use. Dichloromethane (DCM) was purified by shaking with portions of concentrated
sulfuric acid until the acid layer remained colorless. The DCM was then washed with H2O
followed by 5% NaOH and again with H2O. It was then dried with CaCl2 and distilled over
CaH2. Triethylamine (TEA) was dried and distilled over CaH2 under nitrogen. Tert-butyl
chloride was washed with H2O then NaHCO3 then dried and distilled over CaCl2 under
nitrogen.
Synthesis of 2-(Cyano-phenyl-methyl)-benzonitrile (1) This compound was prepared using
a previously reported procedure.2 To a solution of potassium tert-butoxide (0.818g,
7.3mmol) and 2-phenylacetonitrile (0.940g, 8.03mmol) in 4.5 mL dimethyl formamide
(DMF) immersed in an ice bath, a solution of 2-chloro-benzonitrile (0.500g, 3.65mmol) in
5mL of DMF was added dropwise. The mixture was stirred at room temperature overnight,
extracted with ethyl acetate, organic layers were combined and washed with brine. Organic
layer was dried over Na2SO4 and concentrated under reduced pressure. Purified by flash
chromatography 0-15% EtOAc and Hexanes to give 0.654g (82%) of 1; All spectral data
matched reported values.2
Synthesis of 2-(Cyano-phenyl-methyl)-dibenzonitrile (2) This compound was prepared
using a procedure previously reported.3 To a solution of potassium tert-butoxide (1.344g,
12.0mmol) and 2-σ-cyanobenzonitrile (0.852g, 6.0mmol) in 8.5 mL tetrahydrofuran (THF)
was added 2-chloro-benzonitrile (0.548g, 4.0mmol). The mixture was refluxed for 18 hours.
Reaction was allowed to cool and 15mL of saturated aqueous ammonium chloride was
added. Solution was extracted with ethyl acetate, organic layers were combined and washed
with brine. Organic layer was dried over Na2SO4 and concentrated under reduced pressure.
Purified by flash chromatography on basic alumina 20-100% DCM and Hexanes to give
0.396g (41%) of 2; All spectral data matched reported values.3
Synthesis of tert-Butyl-diphenylvinylidene-amine (6) This compound was prepared using
a previously reported procedure.10 To a schlenk flask containing FeCl3 (0.535g 3.3mmol) in
2mL of dichloromethane (DCM) at -30°C, was added a solution of diphenyl acetonitrile
(0.579g 3.0mmol) in 1mL of DCM dropwise. Solution allowed to warm to room temperature
and again cooled to -30°C and tert-butyl chloride (0.276g 3.0mmol) was added dropwise.
Solution then cooled again to -78°C and triethylamine (0.455g 4.5mmol) was added all at
once. Reaction allowed to stir for 1 hour then warmed to -40°C and 10mL 7M NaOH was
added. Reaction extracted with DCM and washed with brine. Organic layers combined and
dried over Na2SO4 and concentrated under reduced pressure. Purified by flash
chromatography on basic alumina with 0-80% DCM and Hexanes to give 0.529g (71%) of 6;
1H NMR ((CD
3)2CO): δ = 1.347 (s, 9H), 7.136 (t, J=7.2Hz, 2H), 7.212 (d, J=1.0Hz, 4H),
7.295 (t, J=8.4Hz, 4H); 13C NMR ((CD3)2CO): δ = 29.9, 60.4, 76.2, 125.9, 127.2, 128.9,
135.4, 181.4; IR (KBr): 3080, 3028, 3057, 2970, 2899, 2005, 1598, 1493, 760, 694 cm1-;
LRMS (ESI) for C18H19N calcd 250.358, found 250.161.
Synthesis of 2-(2-tert-Butylimino-1-phenyl-vinyl)-benzonitrile (7) This compound was
prepared using a previously reported procedure.10 To a schlenk flask containing AlCl3
(0.293g 2.2mmol) in 2mL of dichloromethane (DCM) at -30°C, was added a solution of 1
(0.218g 1.0mmol) in 1mL of DCM dropwise. Solution allowed to warm to room temperature
and again cooled to -30°C and tert-butyl chloride (0.184g 2.0mmol) was added dropwise.
Solution stirred for 30 minutes at -30°C and again allowed to warm to room temperature.
Solution then cooled again to -78°C and triethylamine (0.152g 1.5mmol) was added all at
once. Reaction allowed to stir for 1 hour then warmed to -40°C and 10mL 7M NaOH was
added. Reaction extracted with DCM and washed with brine. Organic layers combined and
dried over Na2SO4 and concentrated under reduced pressure. Purified by flash
1H NMR ((CD
3)2CO): δ = 1.436 (s, 9H), 7.105 (t, J=7.2Hz, 2H), 7.320 (t, J=7.5Hz, 2H),
7.523 (m, 2H), 7.729 (td, J1=1.2Hz, J2=7.8Hz, 1H), 7.849 (dd, J1=1.2Hz, J2=8.1Hz, 1H); 13C
NMR ((CD3)2CO): δ = 29.9, 61.5, 73.6, 113.3, 117.9, 125.4, 125.5, 127.8, 129.1, 131.5,
133.4, 134.1, 135.7, 138.5, 176.5; IR (KBr): 3061, 2972, 2868, 2224, 2014, 1667, 1594,
1493, 1180, 759 cm-1; LRMS (ESI) for C19H18N2 calcd 274.1, found [M+Na+] 297.100.
Synthesis of 2-(2-tert-Butylimino-1-phenyl-vinyl)-dibenzonitrile (8) This compound was
prepared using a previously reported procedure.10 To a schlenk flask containing AlCl3
(0.596g 4.5mmol) in 2mL of dichloromethane (DCM) at -30°C, was added a solution of 2
(0.340g 1.4mmol) in 2mL of DCM dropwise. Solution allowed to warm to room temperature
and again cooled to -30°C and tert-butyl chloride (0.414g 4.5mmol) was added dropwise.
Solution stirred for 30 minutes at -30°C and again allowed to warm to room temperature.
Solution then cooled again to -78°C and triethylamine (0.212g 2.1mmol) was added all at
once. Reaction allowed to stir for 1 hour then warmed to -40°C and 10mL 7M NaOH was
added. Reaction extracted with DCM and washed with brine. Organic layers combined and
dried over Na2SO4 and concentrated under reduced pressure. Purified by flash
chromatography on basic alumina with 10-100% DCM and Hexanes to give 0.080g (20%) of
8; 1H NMR ((CD3)2CO): δ = 1.469 (s, 9H), 7.351 (d, J=8.1Hz, 2H), 7.434 (td, J=1.2Hz,
J=7.5Hz, 2H), 7.666 (td, J1=1.2Hz, J2=7.5Hz, 2H), 7.788 (dd, J1=1.0Hz, J2=7.1Hz, 2H); 13C
NMR ((CD3)2CO): δ = 30.4, 62.4, 112.3, 118.4, 127.9, 130.6, 134.3, 135.3, 139.7; IR (KBr):
Synthesis of tert-Butyl-(4-phenyl-1-piperidin-1-yl-isoquinolin-3-yl)-amine (9)
To a schlenk flask containing 7 (0.17g, 0.62 mmol) was added THF (1.5 mL). The solution
was cooled to 0 °C and then a solution of lithium piperdide (0.17g, 1.86 mmol) in THF (4
mL) was added. The reaction stirred for 2 hours at 0 °C and then allowed to slowly warm to
room temperature. The reaction was cooled again to 0 °C and quenched with 10 mL of 10%
HCl. The pH of the solution was adjusted to 7 with NH4OH. The reaction was extracted
with ethyl acetate and washed with H2O and brine. Organic layers were combined and dried
over Na2SO4 and concentrated under reduced pressure. Purified by flash chromatography on
silic gela with 0-50% ethyl acetate/hexanes. The reaction produced 0.90g (40%) 9; 1H NMR
(CD3Cl): δ = 1.351 (s, 9H), 1.683 (m, 2H), 1.821 (m, 4H), 3.328 (m, 4H), 3.891 (s, 1H),
7.063 (m, 2H), 7.284 (m, 4H), 7.461 (m, 2H), 7.909 (d, 1H); 13C NMR (CD3Cl): δ = 25.1,
26.5, 30.4, 51.7, 53.3, 107.2, 115.1, 120.6, 123.4, 125.8, 127.4, 129.1, 129.5, 131.7, 137.2,
139.4, 151.1, 161.0; IR (KBr): 3425, 3060, 2975, 2932, 2820, 2852, 1612, 1578, 1425, 1232,
1203, 767, 705 cm-1.
Compounds 3, 4, and 5, were synthesized by previous group members. Experimental
procedures and characterization can be found in literature2, 3 and Ph.D. thesis of Dr. William
2.9 References
(1) Eric V. Anslyn, D. A. D., Modern Physical Organic Chemistry. University Science Books: Sausalito, Calif, 2006.
(2) Behof, W. J.; Wang, D. C.; Niu, W. J.; Gorman, C. B., Cascade Cyclization To Produce a Series of Fused, Aromatic Molecules. Org. Lett. 12 (9), 2146-2148.
(3) Wan, Y. J.; Niu, W. J.; Behof, W. J.; Wang, Y. F.; Boyle, P.; Gorman, C. B.,
Aminoisoquinolines as colorimetric Hg2+ sensors: the importance of molecular structure and sacrificial base. Tetrahedron 2009, 65 (22), 4293-4297.
(4) Behof, W.J., Progress towards the synthesis of novel graphite derivatives from a solution processable poly-cyano precursor polymer. NCSU electronic thesis.
www.lib.ncsu.edu/resolver/1840.16/3977.
(5) Deady, L. W.; Ganakas, A. M.; Ong, B. H., Ethoxycarbonylation of alpha-cyano-omicron-tolunitrile and cyclization to iosquinolines and pyrimido[e,5-C] isoquinolines. Aust. J. Chem. 1989, 42 (7), 1029-1034.
(6) Frohn, M.; Burli, R. W.; Riahi, B.; Hungate, R. W., An efficient synthesis of 1,6- and 1,7-dibromo-3-aminoisoquinolines: versatile templates for the preparation of functionalized isoquinolines. Tetrahedron Lett. 2007, 48 (3), 487-489.
(7) Tandel, S.; Biehl, E. R., Convenient synthesis of 1-substituted derivatives of 4-([E]-1-propenyl)- and 4-allyl-3-aminoisoquinolines. Heterocycles 2000, 53 (5), 1183.
(8) Clarke, L. F.; Hegarty, A. F., Regiospecific reactions of the ambident anion of bis(pentamethylphenyl)acetonitrile. J. Org. Chem. 1992, 57 (6), 1940-1942.
(9) Cunico, R. F.; Kuan, C. P., On the metalation silylation of σ-trimethylsilyl aldehyde cyanohydrins. J. Org. Chem. 1992, 57 (4), 1202-1205.
(10) Fuks, R.; Baudoux, D.; Piccinni-Leopardi, C.; Declercq, J. P.; Van Meerssche, M., A new and facile synthesis of ketene imines and their 2-iminoazetidine dimer from nitriles via their nitrilium salts. J. Org. Chem. 1988, 53 (1), 18-22.
(12) Poisson, T.; Gembus, V.; Oudeyer, S.; Marsais, F.; Levacher, V., Product-Catalyzed Addition of Alkyl Nitriles to Unactivated Imines Promoted by Sodium
Aryloxide/Ethyl(trimethylsilyl)acetate (ETSA) Combination. J. Org. Chem. 2009, 74 (9), 3516-3519.
(13) Fuks, R., N-Alkylation of nitriles--I: A general synthesis of substituted amidines.
Tetrahedron 1973, 29 (14), 2147-2151.
(14) Meerwein, H.; Bodenbenner, K.; Borner, P.; Kunert, F.; Müller, K. W.; Sasse, H. J.; Schrodt, H.; Spille, J., Organische Ionenreaktionen. Angew. Chem. 1955, 67 (14-15), 374-380.
(15) Gordon, J.; Turrell, G., Notes- Observations on Methylacetonitrilium and N-Phenylbenzonitrilium Hexachloroantimonates. J. Org. Chem. 1959, 24 (2), 269-271. (16) Green, J. A.; Singer, L. A., Ketenimines via photolysis of diphenyldiazomethane in presence of isonitriles. Tetrahedron Lett. 1969, (58), 5093.
(17) Lee, K. W.; Singer, L. A., Thermally labile ketenimines from triphenylphosphinalkylimines. J. Org. Chem. 1974, 39 (25), 3780-3781.