Ames Laboratory Publications
Ames Laboratory
10-2014
Interfacing the Ab Initio Multiple Spawning
Method with Electronic Structure Methods in
GAMESS: Photodecay of trans-Azomethane
Alexander Gaenko
Iowa State UniversityAlbert DeFusco
University of PittsburghSergey Aleksandrovich Varganov
University of NevadaTodd J. Martinez
Stanford UniversityMark S. Gordon
Iowa State University, [email protected]
Follow this and additional works at:
http://lib.dr.iastate.edu/ameslab_pubs
Part of the
Chemistry Commons
The complete bibliographic information for this item can be found at
http://lib.dr.iastate.edu/
ameslab_pubs/352
. For information on how to cite this item, please visit
http://lib.dr.iastate.edu/
howtocite.html
.
Interfacing the Ab Initio Multiple Spawning Method with Electronic
Structure Methods in GAMESS: Photodecay of trans-Azomethane
Abstract
This work presents a nonadiabatic molecular dynamics study of the nonradiative decay of photoexcitedtrans -azomethane, using the ab initio multiple spawning (AIMS) program that has been interfaced with the General Atomic and Molecular Electronic Structure System (GAMESS) quantum chemistry package for on-the-fly electronic structure evaluation. The interface strategy is discussed, and the capabilities of the combined programs are demonstrated with a nonadiabatic molecular dynamics study of the nonradiative decay of photoexcitedtrans-azomethane. Energies, gradients, and nonadiabatic coupling matrix elements were obtained with the state-averaged complete active space self-consistent field method, as implemented in GAMESS. The influence of initial vibrational excitation on the outcome of the photoinduced isomerization is explored. Increased vibrational excitation in the CNNC torsional mode shortens the excited state lifetime. Depending on the degree of vibrational excitation, the excited state lifetime varies from ∼60–200 fs. These short lifetimes are in agreement with time-resolved photoionization mass spectroscopy experiments.
Disciplines Chemistry
Comments
Reprinted (adapted) with permission fromJournal of Physical Chemistry A118 (2014): 10902, doi:10.1021/ jp508242j. Copyright 2014 American Chemical Society.
Interfacing the Ab Initio Multiple Spawning Method with Electronic
Structure Methods in GAMESS: Photodecay of
trans-
Azomethane
Alexander Gaenko,
†,‡Albert DeFusco,
§Sergey A. Varganov,
∥Todd J. Mart
í
nez,
⊥and Mark S. Gordon
*
,†,‡†Ames Laboratory,‡Department of Chemistry, Iowa State University, Ames, Iowa 50010, United States §Center for Simulation and Modeling, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, United States ∥Department of Chemistry, University of Nevada, Reno, Reno Nevada 89557-0216, United States
⊥Department of Chemistry, Stanford University, Stanford, California 94305-5080, United States
*
S Supporting InformationABSTRACT: This work presents a nonadiabatic molecular dynamics study of the nonradiative decay of photoexcited trans-azomethane, using the ab initio multiple spawning (AIMS) program that has been interfaced with the General Atomic and Molecular Electronic Structure System (GAMESS) quantum chemistry package for on-the-fly electronic structure evaluation. The interface strategy is discussed, and the capabilities of the combined programs are demonstrated with a nonadiabatic molecular dynamics study of the nonradiative decay of photoexcited trans-azomethane. Energies, gradients, and nonadiabatic coupling matrix elements were obtained with the state-averaged complete active space self-consistentfield method, as implemented in GAMESS. The influence of initial vibrational excitation on the outcome of the photoinduced isomerization is explored. Increased vibrational excitation in the CNNC torsional mode shortens the excited state lifetime. Depending on the degree of vibrational excitation, the
excited state lifetime varies from∼60−200 fs. These short lifetimes are in agreement with time-resolved photoionization mass spectroscopy experiments.
1. INTRODUCTION
Solar energy is inherently intermittent,1,2 and an efficient method to store the energy is required for later use; chemical storage is a viable approach for accumulating solar energy at large scales. Hence, understanding and mimicking the naturally occurring solar-to-chemical energy conversion process (photo-synthesis) is a key to the development of renewable energy sources.3For example, the photosynthetic apparatus in plants, algae, and bacteria harvests light through electronic excitation of a multichromophoric antenna array, and then the excitation energy is directed to a reaction center, where it is stored as chemical energy for future use by the organism.4 Computa-tional studies have been extremely valuable in elucidating the relationship between the structure and efficiency of photo-chemical systems. Ab initio molecular dynamics (AIMD) methods provide the capability to simulate the time-dependent conformational and electronic degrees of freedom of a molecular system. With AIMD methods, the time evolution of a photochemical system can be modeled, while the energies of relevant electronic states are computed “on the fly” using high accuracy ab initio methods.5 As the potential energy surfaces of neighboring electronic states of photochemical systems become more closely spaced, the behavior of the electronic and nuclear degrees of freedom become increasingly nonadiabatic. Hence, nonadiabatic dynamics methods that go
beyond the Born−Oppenheimer approximation are necessary to follow the evolution of molecules upon photoexcitation.
Several trajectory-based nonadiabatic dynamics methods exist that have been found to be extremely useful in understanding excited state phenomena. The Ehrenfest approximation6 introduces a quantum-classical coupling via a mean-field back-reaction force acting on classically moving nuclei.7 The trajectory-based Ab initio Multiconfigurational Ehrenfest (AI-MCE) method, developed by Saita and Shalashilin,8 uses adiabatic electronic states and the non-adiabatic coupling matrix element to form the Ehrenfest force. The surface hopping (SH) method developed by Tully9models nonadiabatic processes as stochastic hops between adiabatic potential energy surfaces. The SH method has been extensively utilized10 with both on-the-fly ab initio electronic structure evaluation and fitted potentials. A number of methods go beyond the adiabatic (Born−Oppenheimer) approximation by computing electronic and nuclear wave functions. For example, the nuclear-electronic orbital (NEO) method, implemented in GAMESS, represents the mixed electronic-nuclear wave function as a linear combination of Gaussian basis func-tions;11,12 together with the dynamic reaction path method,13
Received: August 14, 2014 Revised: October 18, 2014 Published: October 20, 2014
Article
pubs.acs.org/JPCA
the NEO method is especially suitable for studying non-adiabatic phenomena involving hydrogen transfer without introducing explicit time-dependence.
Wavepacket propagation schemes provide a more complete description of nonadiabatic dynamics than is possible with trajectory-based methods such as Ehrenfest and SH. The multiconfigurational time dependent Hartree method (MCTDH)14,15 has been used successfully for this purpose. Unfortunately, fully flexible wavepacket schemes scale poorly with dimensionality and generally require analytic functional forms for the potential energy surfaces and their couplings.
Ab initio multiple spawning (AIMS)16is an adaptive basis set nuclear wave function method that has been used to simulate excited state population decay through quantum coherence between electronic states17−19 without empirical parameters. AIMS has been employed to discern competing pathways in observable spectra involving more than one electronic state19 and to simulate time-resolved photoelectron spectra (TRPES).20−26 In some cases, the SH and AIMS methods have been shown to provide comparable results for population decay,27but the AIMS method includes short time coherence effects that cannot be treated in single trajectory methods like SH.16,28 AIMS has been used with a number of different electronic structure packages for on-the-fly ab initio molecular dynamics calculations,18,29,30 including Molpro31for on-the-fly ab initio calculations.30Related methods include those based on Gaussian wavepackets such as the coupled coherent states method32and the frozen Gaussian variant of MCTDH known as vMCG33 and the direct dynamics DD-vMGC variation.34 Recently, the ab initio multiple cloning (AIMC) method35has been developed in which each trajectory basis function is propagated by the Ehrenfest equations of motion.
To understand photochemical processes in complex media, the ability to study photochemical processes in extended molecular systems is essential.36It has been demonstrated that fragmentation methods, such as the fragment molecular orbital (FMO) and effective fragment potential (EFP) methods, available in the GAMESS (General Atomic and Molecular Electronic structure System) package,37,38 are able to treat extended systems, such as solvated molecules, proteins and polysaccharides.39The EFP method has been interfaced to ab initio excited state methods to model solvent effects on absorption spectra40 and photochemical processes. Local multireference methods,41available in the GAMESS package,42 are also able to treat molecular systems in the ground and excited states efficiently. Thus, interfacing the GAMESS package with AIMS facilitates the use of fragmentation methods for AIMD simulations on extended systems. Such applications will be explored in future work.
In the present work, the photodecay of trans-azomethane, CH3NNCH3, from then−π*excited state is used as a
benchmark and demonstration of the new AIMS-GAMESS interface. In 1929, Ramsperger studied the photodissociation of azomethane, to produce methyl radicals.43 The experiments performed by Ramsperger could not determine if the methyl radical products were obtained by a concerted or stepwise mechanism. Since then, many more experimental and theoretical studies of the photodissociation of azomethane have been performed.44−49 Gas phase surface hopping simulations have been carried out, using both fitted ab initio potential energy surfaces50−53and on-the-fly quantum chemical methods.54 While the competition between concerted and stepwise dissociation may not be completely understood, both
time-resolved spectroscopy46 and nonadiabatic dynamics simulations54 have shown that the photodecay of
trans-azomethane from then−π*excited state to the ground state occurs within 500 fs.
This paper is organized as follows: In Section 2, the GAMESS-AIMS interface used in this work is briefly discussed. In Section 3, the computational details and the results of the GAMESS-AIMS study of the photodecay oftrans-azomethane from then−π*excited state are presented. Concluding remarks are presented in Section 4.
2. GAMESS-AIMS INTERFACE
In the present work, an AIMD simulation is performed within a client-server paradigm, in which the dynamics code AIMS acts as a client, providing a molecular geometry and requesting an evaluation of the electronic properties from the server electronic structure code GAMESS. At each time step of an AIMS simulation, the molecular geometry and guess molecular orbital (MO) vectors are provided to GAMESS. After the electronic structure evaluation using the input geometry and MOs, AIMS expects the following quantities from GAMESS: (i) energy and gradient vector for each electronic state of interest, (ii) converged configuration interaction (CI) vectors, MO vectors, and the overlap matrix between the guess and the final MO vectors, (iii) oscillator strengths and transition dipole moments for each electronic state, and (iv) nonadiabatic coupling matrix elements between the electronic states. GAMESS must preserve the phase of the converged CI and MO vectors so that AIMS can correctly integrate the equations of motion. An interface code that facilitates the exchange of the required quantities between GAMESS and AIMS was implemented in Fortran 90.55 Additional details are provided in the Supporting Information (SI).
3. NONADIABATIC DECAY OF THEn−π*EXCITED STATE OFTRANS-AZOMETHANE
3.1. Computational Details.AIMS-GAMESS simulations of the nonadiabatic decay of the n−π* excited state were carried out using the state averaged (SA)-complete active space self-consistentfield (CASSCF) method with an active space of six electrons in four orbitals, denoted CASSCF(6,4), and the 6-31G(d) basis set. The active space contains the CCπandπ* orbitals and the two lone-pair orbitals on the nitrogen atoms. Sellner et al.54used the SA-CASSCF(6,4)/6-31G(d), where the ground state and the n−π* excited state were given equal weight, and multireference configuration interaction (MRCI) methods to study the photodecay oftrans-azomethane from the
n−π* excited state with the Tully surface hopping method. They found good agreement between the CASSCF method and high-level MRCI methods for the photodecay from the
n−π*excited state.
In this work, the ground state structures of thecisandtrans
isomers have been optimized with the CASSCF(6,4)/6-31G(d) method. The conical intersection geometry has been optimized with SA-CASSCF(6,4)/6-31G(d), where the ground state and then−π*excited state have been given equal weights. Single-point energies for the ground state and for the n−π*excited state were computed with SA-CASSCF(6,4)/6-31G(d) at the ground state optimized geometries of thecisandtransisomers. The energies are reported relative to the trans-azomethane ground state energy.
The Journal of Physical Chemistry A Article
3.2. Excitation Energy and Conical Intersection Region.Good agreement is found between the SA-CASSCF
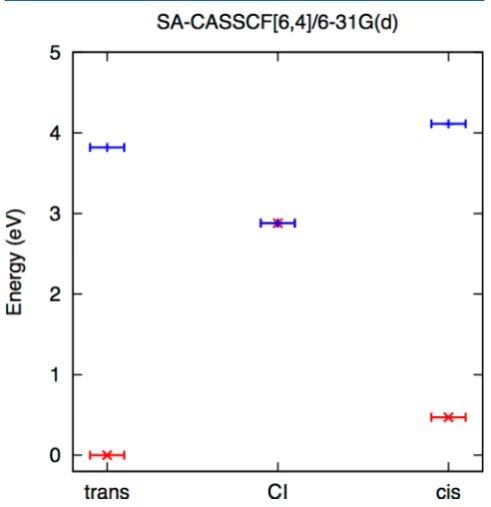
n−π*excitation energy, reported in this work to be 3.82 eV, and the experimental value of 3.6 eV.56The excitation energy is also in agreement with the excitation energies reported by Sellner et al.54 obtained using several high-level methods. Figure 1 shows the ground and excited state energies for
azomethane at the optimizedcis,trans, and conical intersection geometries. The conical intersection geometry was optimized at the SA-CASSCF(6,4)/6-31G(d) level, where the ground state and then−π*excited state were given equal weights, using the NACME gradient projection method.57 The CNNC dihedral angle was found to be 94.2° at the optimized conical intersection geometry shown in Figure 2.
3.3. Conical Intersection Topography.The topography of the potential energy surface near the CASSCF optimized conical intersection is shown in Figure 3 along the tuning
vector g⃗IJ and the coupling vectorh⃗IJ.58The tuning vector is defined in eq 1 to be the nuclear gradient of the difference between the energy of then−π*excited stateIand the energy of the ground stateJ. The coupling vector, defined in eq 2, is proportional to the nonadiabatic coupling matrix element (NACME) between the ground state andn−π*excited state.
⃗ = ∂ ∂ ⃗ − g
R(E E)
IJ I J (1)
ψ ψ
⃗ = − | ∂
∂ ⃗| ⟩
h E E
R
( )
IJ I J I J (2)
In eqs 1 and 2EIandEJare the energies of the ground and excited electronic states,ψIandψJare the wavefucntions of the
[image:5.625.323.565.107.250.2]two states, andRdenotes the nuclear coordinates. The origin of Figure 3 is the CASSCF optimized conical intersection geometry. Theg⃗IJand h⃗IJvectors, which have been calculated with SA-CASSCF(6,4)/6-31G(d) at the optimized conical intersection geometry, are displayed in Figure 4.
Using the topographical indicators introduced by Yark-ony,59,60conical intersections can be classified as either peaked or sloped. A peaked intersection allows for very fast population transfer from the excited state to the ground state in a nonadiabatic process. A sloped intersection may exhibit multiple recrossing events near the conical intersection and slow down the photodecay process. The dimensionless quantity
|s|59,60 in eqs 3−6 is used to classify a conical intersection as either peaked or sloped and is derived using both the seam coordinatesIJ⃗, defined in eq 3, and the gIJ⃗ andh⃗IJ vectors:
⃗ = ∂ ∂ ⃗
+ ⎛ ⎝
⎜ ⎞⎠⎟
s R
E E
2
IJ
I J
(3)
[image:5.625.58.304.151.405.2] [image:5.625.64.296.552.732.2]= ⃗ · ⃗
| ⃗ | s
s g
g
x IJ IJ IJ
2
(4)
Figure 1.Ground (S0, red font) andn−π*(S1blue) energies of thecis,
trans, andS0/S1conical intersection optimized geometries. All energies are relative to thetrans ground state energy. The geometries were optimized with SA-CASSCF(6,4)/6-31G(d).
Figure 2. SA-CASSCF(6,4)/6-31G(d) optimized S0/S1 conical intersection geometry. The CNNC dihedral angle is 94.2°.
Figure 3.Energies of the ground state and then−π*excited state near the S0/S1 conical intersection along displacements in the branching plane along theg⃗IJandh⃗IJvectors. The energies are shown relative to the total energy of the degenerate ground and excited states at the optimized conical intersection geometry. ThegIJ⃗ andh⃗IJvectors were calculated with SA-CASSCF(6,4)/6-31G(d) at the optimized conical intersection geometry and are shown in Figure 4.
The Journal of Physical Chemistry A Article
= ⃗ · ⃗ | ⃗ |
s s h
h
y IJ IJ
IJ2 (5)
| | =s s sx x +s sy y (6) Conical intersections with values of |s| less than unity can be considered to be peaked. The value of |s| for azomethane calculated with SA-CASSCF(6,4)/6-31G(d) is 0.14; therefore, azomethane is considered to have a peaked conical intersection, leading to the prediction that the photodecay of the n−π*
excited state to the ground state should be very fast once a nonadiabatic dynamics simulation approaches the conical intersection geometry.
3.4. Nonadiabatic Dynamics: AIMS Initial Conditions. One-photon excitation to the n−π* state is symmetry forbidden in trans-azomethane. As shown by Szalay et al.,61 the 1Bgn−π*state can borrow intensity from a 1Bu Rydberg
state as low as 6.4 eV through the AuCNNC dihedral angle vibrational mode. The intensity borrowing spectrum calculated by Szalay et al. is in accordance with the Herzberg−Teller effect62since, for theC2hpoint group, the inner product of the Au and the Bg irreducible representations is the Bu
representation. Following the work of Sellner et al.,54 a torsional bias in the AIMS initial conditions has been introduced in this work to mimic intensity sharing. Three sets of initial conditions for the AIMS simulations were prepared: (i) the Wigner distribution63in thev= 0 vibrational state, (ii) a quasi-classical distribution64with 5 quanta of bias in the CNNC torsion angle, and (iii) a quasi-classical distribution with 12 quanta of bias in the CNNC torsion angle. Ten sets of position and momenta were taken from each of the three initial condition ensembles and used as starting points for the AIMS simulations. A total of 30 AIMS simulations were run, each of which spawned an average of 18 trajectories to simulate population transfer from then−π*excited state to the ground state.
3.5. Ab initio Multiple Spawning Simulation. The AIMS method approximates the nuclear Schrödinger equation by expansion in a basis of frozen Gaussian functions called trajectory basis functions (TBFs).16In the present work, each TBF was propagated classically along a potential energy surface
computed with the SA-CASSCF(6,4)/6-31G(d) method for either the ground state or then−π*excited state, starting from the torsionally biased trans-azomethane minimum energy structure, as described in Section 3.4. The AIMS simulations each started with a single trajectory basis function propagated along the n−π* excited state potential energy surface. The nonadiabatic coupling matrix elements (NACME) were computed at every time step during the propagation of the trajectory. More basis functions were spawned onto the ground state potential energy surface in regions of high nonadiabatic coupling. The time-dependent population transfer between the
n−π* excited state and the ground state was computed by solving the nuclear Schrödinger equation within the TBF basis set at each time-step. As the primary interest here is in the excited state lifetime, TBFs that are spawned to the ground electronic state are terminated when their population reaches 0.99. The electronic wave function ansatz employed here is insufficiently flexible to describe dissociation on the ground state (a larger active space would be required), as evidenced by discontinuities in the PES when the molecule attempts to extrude a CH3 group. Each simulation was ended when the population of then−π* excited state decreased to 0.01. The velocity Verlet algorithm with a time step of 0.5 fs was used for the classical propagation of the TBF phase space centers.
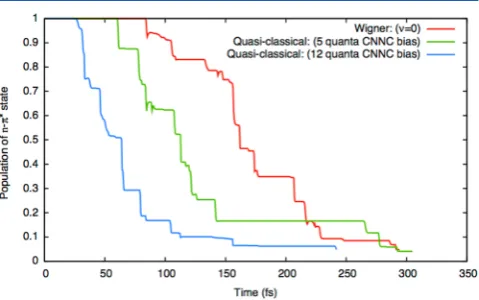
3.6. Population Decay ofn−π*Excited State oftrans -Azomethane.The photodecay from then−π*excited state to the ground state as a function of time is plotted for each of the three sets of initial conditions in Figure 5. In each case, the
plotted population corresponds to an average over 10 distinct initial conditions. Figure 5 shows that vibrational excitation of the mode corresponding to the CNNC torsion decreases the lifetime of then−π*excited state by more than 100 fs. The decrease in the lifetime of then−π*excited state is attributed to increased energy localized in the mode leading from the Franck−Condon structure to the conical intersection geometry shown in Figure 2. With 12 quanta of energy in the CNNC torsion angle, the population of the ground state reaches 90% within 150 fs, which is in agreement with previous surface-hopping simulations.54
At each spawning event, four quantities were recorded: (i) The CNNC dihedral angle, (ii) the energy gap between the
Figure 4. Branching plane vectors. The tuning vector g⃗IJ is the difference between the energy gradients of the excited state and the ground state at the conical intersection geometry. The tuning vector corresponds roughly to displacement of the CNNC dihedral angle. Theh⃗IJvector is the NACME vector, which is also called the coupling vector. The coupling vector corresponds to displacements of the NN bond and the CNN angle. See eqs 1 and 2 in text for the definitions of theg⃗IJandh⃗IJvectors.
Figure 5.Time evolution of the population of then−π*excited state for the three initial conditions. Red: The Wigner distribution in thev= 0 vibrational state. Green: The quasi-classical distribution in thev= 0 vibrational state with an extra 5 quanta in the CNNC torsion mode. Blue: The quasi-classical distribution in thev= 0 vibrational state with an extra 12 quanta in the CNNC torsion mode.
The Journal of Physical Chemistry A Article
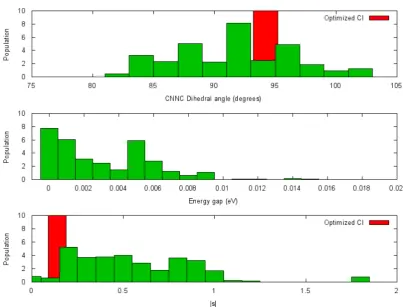
excited state and the ground state, (iii) the value of|s|and (iv) the amount of population that was transferred from the excited state to the ground state. In Figure 6 quantities (i)−(iii) collected from all 30 AIMS simulations have been binned into a histogram weighted by the amount of population that was transferred from the excited state to the ground state. It is found that the population transfer is most efficient for CNNC dihedral angles near the optimized value of 94.2°and for energy gaps less than 0.002 eV. The values of|s|with large population transfers are also found to be very near the optimized conical intersection value of 0.14. These three quantities confirm the prediction presented in section 3.3 that very fast photodecay of azomethane occurs once the simulation approaches the conical intersection region.
4. CONCLUSIONS
To enable the study of nonadiabatic dynamics of large photosystems (such as solar light harvesting systems), an interface between the GAMESS quantum chemistry software package and the AIMS nonadiabatic dynamics program has been developed. The photoinduced dynamics of the n−π*
excited state of trans-azomethane has been studied with the new AIMS-GAMESS interface. Three sets of initial conditions for position and momentum were employed with increasing excitation of the CNNC torsional mode. The simulated lifetime of then−π*excited state starting from the vibrational ground state was found to be∼200 fs. As the CNNC torsional mode is more vibrationally excited, the lifetime of the n−π* excited state decreases. The fastest decay time was found to be 150 fs
when 12 quanta of energy are added to the CNNC dihedral angle, which is in agreement with previous experimental and theoretical results.
No evidence was found for dissociation of CH3 on the
excited state surface; only on the ground state surface was significant elongation of the corresponding CN bond observed. It can therefore be concluded that CH3dissociation occurs on
the ground electronic state. In order to establish whether the loss of both CH3 groups occurs in a concerted or stepwise
fashion, a moreflexible electronic wave functionansatzthat can describe simultaneous dissociation of the CH3 groups is
needed.
■
ASSOCIATED CONTENT*
S Supporting InformationGAMESS-FMS implementation details; (Table S1) list of generalized data exchange API functions with brief descriptions; and (Figure S1) schematic of the client and server interaction via the GDE interface. This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: [email protected].
Notes
[image:7.625.109.513.61.368.2]The authors declare no competingfinancial interest.
Figure 6.Histograms of the spawning quantities from all 30 simulations. In each pane, the heights of the histograms have been weighted by the amount of population transfer. The upper pane plots the binned value of the CNNC dihedral angle in green. The CNNC dihedral angle at the optimized conical intersection geometry is shown in red. The middle pane plots the binned energy gap between the ground state and then−π*
excited state. The lower pane is the binned quantity|s|defined in eqs 3−6. The value of|s|at the optimized conical intersection geometry is shown in red.
The Journal of Physical Chemistry A Article
■
ACKNOWLEDGMENTSThis work was supported by a SciDAC-E grant to MSG from the US Department of Energy, Office of Advanced Scientific Computing Research through the Ames Laboratory and by an NSF grant (OCI-1047577) to TJM. The Ames Laboratory is operated for the US Department of Energy by Iowa State University under contract No. DE-AC02-07CH11358.
■
REFERENCES(1) Song, W.; Chen, Z.; Brennaman, M. K.; Concepcion, J. J.; Patrocinio, A. O. T.; Murakami Iha, N. Y.; Meyer, T. J. Making Solar Fuels by Artificial Photosynthesis.Pure Appl. Chem. 2011,83, 749− 768.
(2) Roy, S. C.; Varghese, O. K.; Paulose, M.; Grimes, C. A. Toward Solar Fuels: Photocatalytic Conversion of Carbon Dioxide to Hydrocarbons.ACS Nano2010,4, 1259−1278.
(3) Reece, S. Y.; Hamel, J. A.; Sung, K.; Jarvi, T. D.; Esswein, A. J.; Pijpers, J. J. H.; Nocera, D. G. Wireless Solar Water Splitting Using Silicon-Based Semiconductors and Earth-Abundant Catalysts.Science 2011,334, 645−648.
(4) Barber, J. Biological Solar Energy.Philos. Trans. A. Math. Phys. Eng. Sci.2007,365, 1007−1023.
(5) Gordon, M. S.; Chaban, G.; Taketsugu, T. Interfacing Electronic Structure Theory with Dynamics.J. Phys. Chem.1996,100, 11512− 11525.
(6) Li, X.; Tully, J. C.; Schlegel, H. B.; Frisch, M. J. Ab Initio Ehrenfest Dynamics.J. Chem. Phys.2005, 123.
(7) Prezhdo, O. V.; Duncan, W. R.; Prezhdo, V. V. Photoinduced Electron Dynamics at the Chromophore-Semiconductor Interface: A Time-Domain Ab Initio Perspective.Prog. Surf. Sci.2009,84, 30−68. (8) Saita, K.; Shalashilin, D. V. On-the-Fly Ab Initio Molecular Dynamics with Multiconfigurational Ehrenfest Method.J. Chem. Phys. 2012,137, 22A506.
(9) Tully, J. C. Molecular Dynamics with Electronic Transitions.J. Chem. Phys.1990,93, 1061−1071.
(10) Barbatti, M. Nonadiabatic Dynamics with Trajectory Surface Hopping Method. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 620−633.
(11) Webb, S. P.; Iordanov, T.; Hammes-Schiffer, S. Multiconfigura-tional Nuclear-Electronic Orbital Approach: Incorporation of Nuclear Quantum Effects in Electronic Structure Calculations.J. Chem. Phys. 2002,117, 4106−4118.
(12) Sirjoosingh, A.; Pak, M. V.; Swalina, C.; Hammes-Schiffer, S. Reduced Explicitly Correlated Hartree-Fock Approach within the Nuclear-Electronic Orbital Framework: Applications to Positronic Molecular Systems.J. Chem. Phys.2013,139.
(13) Maluendes, S. A.; Dupuis, M. A Dynamic Reaction Coordinate Approach to Abinitio Reaction Pathways: Application to the 1,5 Hexadiene Cope Rearrangement.J. Chem. Phys.1990, 93.
(14) Worth, G. A.; Robb, M. A.; Lasorne, B. Solving the Time-Dependent Schrödinger Equation for Nuclear Motion in One Step: Direct Dynamics of Non-Adiabatic Systems. Mol. Phys. 2008, 106, 2077−2091.
(15) Chen, X.; Batista, V. S. Matching-Pursuit/split-Operator-Fourier-Transform Simulations of Excited-State Nonadiabatic Quan-tum Dynamics in Pyrazine.J. Chem. Phys.2006,125, 124313.
(16) Ben-Nun, M.; Martínez, T. J. Ab Initio Quantum Molecular Dynamics.Adv. Chem. Phys.2002,121, 439.
(17) Ben-Nun, M.; Martínez, T. J. Nonadiabatic Molecular Dynamics: Validation of the Multiple Spawning Method for a Multidimensional Problem.J. Chem. Phys.1998,108, 7244−7257.
(18) Ben-Nun, M.; Quenneville, J.; Martínez, T. J. Ab Initio Multiple Spawning: Photochemistry from First Principles Quantum Molecular Dynamics.J. Phys. Chem. A2000,104, 5161−5175.
(19) Martínez, T. J.; Ben-Nun, M.; Levine, R. D. Multi-Electronic-State Molecular Dynamics: A Wave Function Approach with Applications.J. Phys. Chem. A1996,100, 7884−7895.
(20) Thompson, A. L.; Martínez, T. J. Time-Resolved Photoelectron Spectroscopy from First Principles: Excited State Dynamics of Benzene.Faraday Discuss.2011,150, 293−311.
(21) Hudock, H. R.; Levine, B. G.; Thompson, A. L.; Satzger, H.; Townsend, D.; Gador, N.; Ullrich, S.; Stolow, A.; Martínez, T. J. Ab Initio Molecular Dynamics and Time-Resolved Photoelectron Spec-troscopy of Electronically Excited Uracil and Thymine.J. Phys. Chem. A2007,111, 8500−8508.
(22) Hudock, H. R.; Martínez, T. J. Excited-State Dynamics of Cytosine Reveal Multiple Intrinsic Subpicosecond Pathways. Chem. Phys. Phys. Chem.2008,9, 2486−2490.
(23) Tao, H.; Allison, T. K.; Wright, T. W.; Stooke, A. M.; Khurmi, C.; Van Tilborg, J.; Liu, Y.; Falcone, R. W.; Belkacem, A.; Martinez, T. J. Ultrafast Internal Conversion in Ethylene. I. the Excited State Lifetime.J. Chem. Phys.2011,134.
(24) Mori, T.; Glover, W. J.; Schuurman, M. S.; Martinez, T. J. Role of Rydberg States in the Photochemical Dynamics of Ethylene.J. Phys. Chem. A2012,116, 2808−2818.
(25) Kuhlman, T. S.; Glover, W. J.; Mori, T.; M?ller, K. B.; Martinez, T. J. Between Ethylene and PolyenesThe Non-Adiabatic Dynamics ofcis-Dienes.Faraday Discuss.2012,157, 193−212.
(26) Wang, K.; McKoy, V.; Hockett, P.; Schuurman, M. S. Time-Resolved Photoelectron Spectra of CS2: Dynamics at Conical Intersections.Phys. Rev. Lett.2014,112, 113007.
(27) Toniolo, A.; Ciminelli, C.; Persico, M.; Martínez, T. J. Simulation of the Photodynamics of Azobenzene on Its First Excited State: Comparison of Full Multiple Spawning and Surface Hopping Treatments.J. Chem. Phys.2005,123, 234308.
(28) Ben-Nun, M.; Martínez, T. J. A Continuous Spawning Method for Nonadiabatic Dynamics and Validation for the Zero-Temperature Spin-Boson Problem.Isr. J. Chem.2007,47, 75−88.
(29) Martinez, T. J.; Levine, R. D. Dynamics of the Collisional Electron Transfer and Femtosecond Photodissociation of NaI on Ab Initio Electronic Energy Curves.Chem. Phys. Lett. 1996,259, 252− 260.
(30) Levine, B. G.; Coe, J. D.; Virshup, A. M.; Martínez, T. J. Implementation of Ab Initio Multiple Spawning in the Molpro Quantum Chemistry Package.Chem. Phys.2008,347, 3−16.
(31) Werner, H.-J.; Knowles, P. J.; Knizia, G.; Manby, F. R.; Schütz, M.; et al. MOLPRO, Version 2010.1, a Package of Ab Initio Programs, 2010.
(32) Shalashilin, D. V.; Child, M. S. Real Time Quantum Propagation on a Monte Carlo Trajectory Guided Grids of Coupled Coherent States: 26D Simulation of Pyrazine Absorption Spectrum. J. Chem. Phys.2004,121, 3563−3568.
(33) Worth, G. A.; Robb, M. A.; Burghardt, I. A Novel Algorithm for Non-Adiabatic Direct Dynamics Using Variational Gaussian Wave-packets.Faraday Discuss.2004,127, 307−323.
(34) Lasorne, B.; Robb, M. A.; Worth, G. A. Direct Quantum Dynamics Using Variational Multi-Configuration Gaussian Wave-packets. Implementation Details and Test Case. Phys. Chem. Chem. Phys.2007,9, 3210−3227.
(35) Makhov, D. V.; Glover, W. J.; Martinez, T. J.; Shalashilin, D. V. Ab Initio Multiple Cloning Algorithm for Quantum Nonadiabatic Molecular Dynamics.J. Chem. Phys.2014,141, 054110.
(36) Toniolo, A.; Olsen, S.; Manohar, L.; Martínez, T. J. Conical Intersection Dynamics in Solution: The Chromophore of Green Fluorescent Protein.Faraday Discuss.2004,127, 149−163.
(37) Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System.J. Comput. Chem.1993,14, 1347−1363.
(38) Gordon, M. S.; Schmidt, M. W. Advances in Electronic Structure Theory: GAMESS a Decade Later. Theory Appl. Comput. Chem.2005, 1167−1189.
(39) Gordon, M. S.; Fedorov, D. G.; Pruitt, S. R.; Slipchenko, L. V. Fragmentation Methods: A Route to Accurate Calculations on Large Systems.Chem. Rev.2012,112, 632−672.
The Journal of Physical Chemistry A Article
(40) DeFusco, A.; Ivanic, J.; Schmidt, M. W.; Gordon, M. S. Solvent-Induced Shifts in Electronic Spectra of Uracil.J. Phys. Chem. A2011,
115, 4574−4582.
(41) Krisiloff, D. B.; Carter, E. A. Approximately Size Extensive Local Multireference Singles and Doubles Configuration Interaction. Phys. Chem. Chem. Phys.2012,14, 7710.
(42) Gaenko, A. V; Krisiloff, D. B.; Gordon, M. S. Manuscript in Preparation.
(43) Ramsperger, H. C. The Thermal Decomposition of Methtyl Isopropyl Di-Imide: A Homogeneous Unimolecular Reaction. The Thermal Decomposition of Hydrazoic Acid and Methyl Azide.J. Am. Chem. Soc.1929,51, 2134−2143.
(44) Burton, K. A.; Weisman, R. B. Stepwise Phtodissociation of Vapor-Phase Azomethane.J. Am. Chem. Soc.1990,112, 1804−1807.
(45) Fairbrother, D. H.; Dickens, K. A.; Stair, P. C.; Weitz, E. Energy Content of Methyl Radicals Prodcued in the UV Photodissociation of Azomethane.Chem. Phys. Lett.1995,246, 513−520.
(46) Diau, E. W. G.; Zewail, A. H. Femtochemistry of trans -Azomethane: A Combined Experimental and Theoretical Study.Chem. Phys. Phys. Chem.2003,4, 445−456.
(47) North, S. W.; Longfellow, C. A.; Lee, Y. T. The near Ultraviolet Photodissociation Dynamics of Azomethane.J. Chem. Phys.1993,99, 4423−4429.
(48) Hu, C.-H.; Schaefer, H. F. I. I. I. The Mechanism of the Thermal Decomposition and the (n−π*) Excited States of Azomethane.J. Phys. Chem.1995,99, 7507−7513.
(49) Liu, R.; Cui, Q.; Dunn, K. M.; Morokuma, K. Ab Initio Molecular Orbital Study of the Mechanism of Photodissociation of
trans-Azomethane.J. Chem. Phys.1996,105, 2333−2345.
(50) Cattaneo, P.; Persico, M. Diabatic and Adiabatic Potential-Energy Surfaces for Azomethane Photochemistry. Theor. Chem. Acc. 2000,103, 390−398.
(51) Cattaneo, P.; Persico, M. Ab Initio Determination of Quasi-Diabatic States for Multiple Reaction Pathways. Chem. Phys. 1997,
214, 49−60.
(52) Cattaneo, P.; Persico, M. Semiclassical Treatment of the Photofragmentation of Azomethane. Chem. Phys. Lett. 1998, 289, 160−166.
(53) Cattaneo, P.; Persico, M. Semiclassical Simulations of Azomethane Photochemistry in the Gas Phase and in Solution. J. Am. Chem. Soc.2001,123, 7638−7645.
(54) Sellner, B.; Ruckenbauer, M.; Stambolić, I.; Barbatti, M.; Aquino, A. J. A.; Lischka, H. Photodynamics of Azomethane: A Nonadiabatic Surface-Hopping Study. J. Phys. Chem. A 2010, 114, 8778−8785.
(55) American National Standard Programming Language FOR-TRAN, ANSI X3198−1992; ISO/IEC 1539: 1991, 1992.
(56) Engel, P. S. Mechanism of the Thermal and Photochemical Decomposition of Azoalkanes.Chem. Rev.1980,80, 99−150.
(57) Bearkpark, M. J.; Robb, M. A.; Schlegel, H. B. A Direct Method for the Location of a Lowest Energy Point on a Potential Surface Crossing.Chem. Phys. Lett.1994,223, 269−274.
(58) Yarkony, D. R. Diabolical Conical Intersections.Rev. Mod. Phys. 1996,68, 985−1013.
(59) Yarkony, D. R. Nuclear Dynamics near Conical Intersections in the Adiabatic Representation: I. The Effects of Local Topography on Interstate Transitions.J. Chem. Phys.2001,114, 2601−2613.
(60) Ben-Nun, M.; Molnar, F.; Schulten, K.; Martínez, T. J. The Role of Intersection Topography in Bond Selectivity of Cis-Trans Photoisomerization. Proc. Natl. Acad. Sci. U. S. A.2002,99, 1769− 1773.
(61) Szalay, Pe. G.; Aquino, A. J. A.; Barbatti, M.; Lischka, H.̀ Theoretical Study of the Excitation Spectrum of Azomethane.Chem. Phys.2011,380, 9−16.
(62) Herzberg, G.; Teller, E. Vibrational Structure of Electronic Transitions in Polyatomic Molecules.Z. Phys. Chem.1933,B21, 410. (63) Hillery, M.; O’Connell, R. F.; Scully, M. O.; Wigner, E. P. Distribution Functions in Physics: Fundamentals.Phys. Rep.1984,106, 121−167.
(64) Ben-Nun, M.; Martínez, T. J.; Levine, R. D. Dynamical Stereochemistry on Several Electronic States: A Computational Study of Na*+ H2.J. Phys. Chem. A1997,101, 7522−7529.
The Journal of Physical Chemistry A Article