warwick.ac.uk/lib-publications
A Thesis Submitted for the Degree of PhD at the University of Warwick
Permanent WRAP URL:
http://wrap.warwick.ac.uk/132613
Copyright and reuse:
This thesis is made available online and is protected by original copyright.
Please scroll down to view the document itself.
Please refer to the repository record for this item for information to help you to cite it.
Our policy information is available from the repository home page.
and associate with multiple organelles
throughout the replicative lifespan of the yeast
cell
by
Oliver Paul Sinfield
Thesis
for the degree of
Doctor of Philosophy
To
MRC Doctoral Training Partnership, Warwick Medical
School, University of Warwick
Supervisors: Prof. John E G McCarthy and Prof. Robert Cross
2
Table of Contents
Table of Contents ... 2
Table of Figures ... 6
Acknowledgements ... 9
Declaration... 10
Abbreviations ... 11
Abstract... 12
1 Introduction ... 13
1.1 Foreword ... 14
1.2 The mRNA life cycle in yeast ... 16
1.2.1 Transcription and nuclear processing of mRNA ... 16
1.2.2 Localisation of mRNA ... 17
1.2.3 Translation of mRNA ... 20
1.2.4 mRNA Decay in Yeast ... 22
1.3 Processing bodies ... 30
1.3.1 Discovery ... 30
1.3.2 Structure and Composition ... 32
1.3.3 Assembly and regulation ... 34
1.3.4 Function ... 36
1.3.5 Transport within the cell ... 37
1.4 Replicative ageing in budding yeast ... 39
1.4.1 Conserved mechanisms of extended lifespan ... 39
1.4.2 Sirtuins and ERCs ... 42
1.4.3 Asymmetric inheritance of protein aggregates ... 43
1.4.4 Dietary restriction and downstream signalling pathways ... 45
1.5 Objectives ... 46
2 Materials and Methods ... 49
2.1 Molecular Cloning ... 50
2.1.1 DNA Manipulation ... 50
2.1.2 Yeast Strain construction ... 57
2.1.3 GFP Library replication ... 60
2.1.4 PCR Clone screening ... 61
3
2.1.6 Fluorescence screening ... 63
2.1.7 Yeast strain stocking ... 64
2.2 Microfluidic system fabrication ... 64
2.2.1 Microfluidic mask design ... 64
2.2.2 Su-8 Mould fabrication ... 65
2.2.3 PDMS Chip fabrication... 66
2.2.4 Epoxy/Polyurethane replica moulding ... 67
2.2.5 Fluid reservoir fabrication ... 67
2.2.6 Fluidic connection fabrication... 69
2.3 Microscopy ... 70
2.3.1 CellASIC microfluidic platform set-up ... 70
2.3.2 Microfluidic chip platform set-up ... 70
2.3.3 Microscope Set-up ... 73
2.4 Image analysis ... 74
2.4.1 Segmentation of Cells ... 74
2.4.2 Counting p-bodies ... 75
2.4.3 Measuring p-body size and intensity ... 77
2.4.4 Manual Curation ... 77
2.4.5 Replicative age quantification ... 77
2.4.6 P-body inheritance ... 78
2.4.7 Cell cycle phase determination ... 78
2.4.8 Co-localization of p-bodies and organelles ... 79
2.4.9 P-body composition analysis ... 80
2.4.10 P-body tracking and velocity measurement ... 80
3 Development of a robust protocol for analysis of p-bodies in Saccharomyces cerevisiae during replicative ageing ... 81
3.1 Existing imaging methodologies ... 82
3.2 Microfluidic dissection devices available for evaluation ... 84
3.3 Testing and evaluation of microfluidic dissection devices ... 88
3.3.1 CliC retains cells longer than HYAA ... 89
3.3.2 P-body numbers and morphology ... 91
3.4 Improved designs for microfluidic dissection devices to observe p-bodies ... 96
4 Processing body localisation during growth and ageing ... 98
4
4.2 Colocalisation of processing bodies and cellular compartments ... 100
4.2.1 Sac6 / Actin patches... 103
4.2.2 Cop1, Anp1, Chc1 / Early, Mid, Late Golgi ... 105
4.2.3 Mitotag / Mitochondria ... 110
4.2.4 Further organelle markers ... 112
4.3 Age-induced changes in processing body localisation ... 114
4.3.1 Sac6 / Actin Patches ... 116
4.3.2 Cop1, Anp1, Chc1 / Early, Mid, Late Golgi ... 117
4.3.3 Pex3 / Peroxisomes... 120
4.3.4 Mitotag/Mitochondria ... 121
5 Inheritance and motility of Processing Bodies in Saccharomyces cerevisiae ... 124
5.1 Movement and inheritance of processing bodies in Saccharomyces cerevisiae 125 5.2 P bodies are mobile in S. cerevisiae and move in a direction manner. ... 126
5.2.1 Procesing bodies undergo rapid fusion events ... 131
5.3 P bodies are inherited through multiple generations during caloric restriction 132 5.4 P body inheritance depends on mRNA transport systems and Dcp1 ... 132
5.5 P body inheritance is maintained in replicatively aged cells. ... 135
6 Discussion ... 138
6.1 CliC2 microfluidic chips are suitable for analysis of processing bodies during replicative ageing ... 139
6.2 Subcellular Localisation and associations of processing bodies ... 140
6.2.1 Processing bodies localise to ER and Golgi membranes during log phase growth, implications for function ... 140
6.2.2 Replicative age causes a shift in processing body membrane association .... 141
6.3 Processing body transport on actin filaments ... 143
7 Conclusions and future work ... 146
8 References ... 148
Appendix ... 161
List of Yeast Strains used in this study ... 161
List of Bacterial strains used in this study ... 165
List of Primers for yeast integrations ... 165
ImageJ Analysis Macros ... 168
6
1
Table of Figures
Figure 1: The central dogma of molecular biology ... 16
Figure 2: The main processes involved in transcription ... 18
Figure 3: mRNA is transported through the cell by the "locasome" complex ... 20
Figure 4: The initiation of Eukaryotic translation ... 21
Figure 5: Elongation (top) and termination (bottom) of eukaryotic translation ... 23
Figure 6: Simplified progression of mRNA decay in budding yeast and eukaryotes ... 25
Figure 7: Simplified representation of a processing body ... 31
Figure 8: Conditions inducing processing body formation... 35
Figure 9: Cell cycle progression (upper) and measures of ageing (lower) in budding yeast ... 41
Figure 10: Regulatory interactions affecting cellular ageing in budding yeast ... 47
Figure 11: An overview of the Gibson assembly cloning process ... 52
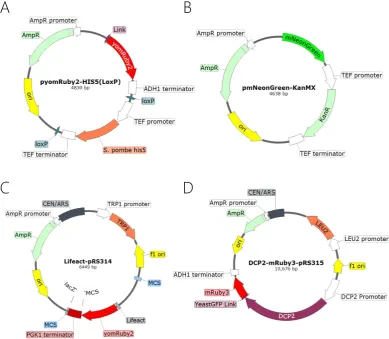
Figure 12: The main plasmids used in this study ... 57
Figure 13: Plasmids used in the CRISPR-Cas9 Genome modification process ... 59
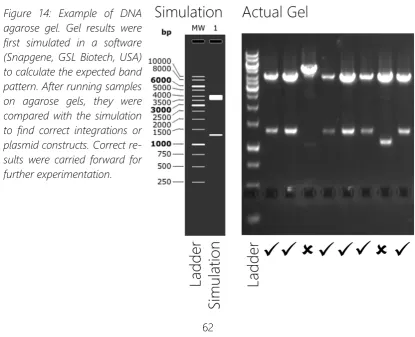
Figure 14: Example of DNA agarose gel. ... 62
Figure 15: Example micrographs of transformed yeast strains ... 64
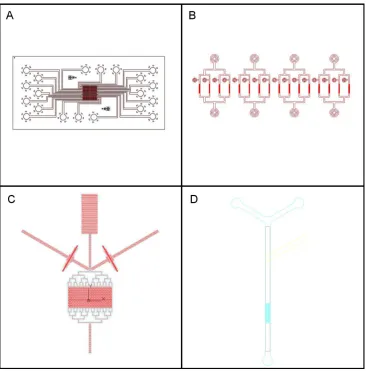
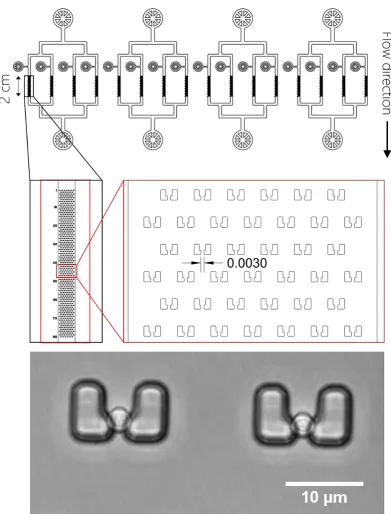
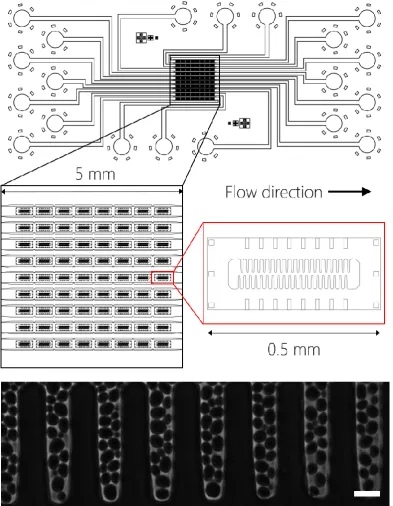
Figure 16: Designs of microfluidic masks fabricated for this study... 66
Figure 17: Diagram of the microfluidic platform as used in this study. ... 71
Figure 18: Demonstration of the cell segmentation and p-body analysis routine ... 76
Figure 19: Cell cycle phase was determined based on shape ... 78
Figure 20: Microfluidic Dissection Device of Lee et al ... 83
Figure 21: The High throughput Yeast Ageing Array device ... 85
Figure 22: The ALCATRAS microfluidic device ... 86
Figure 23: The CliC High throughput ageing device ... 87
Figure 24: Cell retention in the Clic2 and HYAA microfluidic dissection devices. ... 89
Figure 25: Loss of retention in the HYAA (top row, 20-minute intervals) and CliC 2 (bottom row, 10-minute intervals)... 90
7
Figure 27: New Microfluidic dissection device designs ... 96
Figure 28: Overview of structures tagged in this study... 101
Figure 29: Time Lapse Micrographs of Sac6-mNeonGreen (left), marking actin patches, and Edc3-mRuby2, marking processing bodies ... 103
Figure 30: Time Lapse Micrographs of Cop1-mNeonGreen, early Golgi and Edc3-mRuby2, marking processing bodies. ... 105
Figure 31: Time Lapse Micrographs of Anp1-mNeonGreen, marking Golgi, and Edc3-mRuby2, marking processing bodies. ... 106
Figure 32: Time Lapse Micrographs of Chc1-mNeonGreen, marking late Golgi, and Edc3-mRuby2. ... 107
Figure 33: – Time Lapse Micrographs of Sec13-mNeonGreen, marking ER-Golgi Vesicles, and Edc3-mRuby2, marking p-bodies (left). ... 108
Figure 34: Time Lapse Micrographs of Edc3-mNeonGreen, marking processing bodies, and MitoTag-mRuby2, marking mitochondria (left). ... 111
Figure 35: Overview of processing body localisation (Mander’s M2 coefficients) for cellular compartments... 113
Figure 36: Micrograph of aged cell (top of channel) tagged with Sac6-mNeonGreen and Edc3-mRuby2. ... 116
Figure 37: Micrographs of aged cells labelled with Sec13 mNeonGreen and ... 117
Figure 38: Micrographs of aged cells labelled with Cop1-mNeonGreen and Edc3-mRuby2 ... 118
Figure 39: Micrographs of Aged cells tagged with Pex3-mNeonGreen marking peroxisomes and Edc3-mRuby2 marking processing bodies ... 120
Figure 40: Micrographs of aged yeast cells tagged with MitoTag-mRuby2 and Edc3-mNeonGreen ... 121
Figure 41: Summary of processing body localisation in aged cells... 122
Figure 42: Change in localisation of processing bodies during cellular ageing... 123
Figure 43: Anisotropic movement of large processing bodies ... 127
8
Figure 45 Colocalisation of actin filaments and processing bodies. ... 129
Figure 46: Processing bodies undergo fusion events ... 130
Figure 47: Multigenerational inheritance of processing bodies ... 133
Figure 48: mRNA Transport mutants disrupt processing body inheritance ... 134
Figure 49: Dcp1 is required for processing body inheritance ... 135
Figure 50: Processing bodies are inherited in cells of advanced replicative age... 136
9
Acknowledgements
Firstly, I would like to thank Professor John McCarthy for the opportunity to perform this work in his laboratory and his assistance and guidance throughout the work. I would also like to thank Professor Robert Cross for his valuable advice, scientific and otherwise, throughout his study.
Special thanks go to the members of the McCarthy Lab at the University of Warwick. Dr
Helena Firczuk who has trained me since my initial bachelor’s project and taught me all
the yeast techniques I know, Dr John Duncan who taught me peaked my interest in mi-croscopy and Dr Mark Walsh for his assistance with image analysis coding. Dr Estelle Dacheux was a continuous source of support in all areas both scientific and personal. Thanks also go to the Warwick Integrative Synthetic Biology Centre for use of their equip-ment and facilities to conduct this work.
I would like to thank the original designers of the microfluidic chips used in this study. Dr Sandrine Morlot and Gilles Charvin of IGBMC, France, for the use of their unpublished CliC 2 device. Professor Peter Swain and Dr Matthew Crane of Edinburgh University for the designs of the ALCATRAS 2. Dr Weiwei Dang of Baylor College of Medicine and Dr’s Lidong Qin and Myeong Chan Jo of Houston Methodist Hospital for the designs of the HYAA device. Finally, Dr Matthias Heinemann of the University of Groningen who provided the designed from Lee et. Al.
I would also like to thank Dr Marco Polin and Dr Raphael Jeanneret of the University of Warwick Physics department who produced the microfluidic moulds for this study.
10
Declaration
This thesis is submitted to the University of Warwick in support of my application for the degree of Doctor of Philosophy. It has been composed by myself and has not been sub-mitted in any previous application for any degree.
11
Abbreviations
RNA ribonucleic acid
mRNA messenger ribonucleic acid
mRNP messenger ribonucleoprotein
GTP Guanosine triphosphate
poly-A poly-adenosine
DNA Deoxyribonucleic acid
ER endoplasmic reticulum
tRNA Transfer ribonucleic acid
GDP Guanosine diphosphate
LLPS liquid liquid phase separation
NMD nonsense mediated decay
NSD non-stop decay
NGD No-go decay
PB processing body
SG stress granule
RLS replicative lifespan
CLS chronological lifespan
rDNA ribosomal DNA
ERC Extrachromosomal rDNA circles
DR dietary restriction
TOR target of rapamycin
PKA protein kinase A
PDMS polydimethylsiloxane
PCC Pearson's correlation coefficient
12
Abstract
Processing bodies (p-bodies) are cytoplasmic messenger ribonucleoprotein granules con-taining components of the mRNA degradation machinery, that form during stress
condi-tions in the budding yeast Saccharomyces cerevisiae. P-bodies are conserved in eukaryotes
and are related to other mRNP granules such as stress granules and neuronal granules. mRNP granules and their components self-assemble through a process of liquid-liquid phase separation, facilitated by protein-protein interaction by low complexity regions. Er-rors in this assembly process can cause a build-up of aggregated protein and have been implicated in the pathology of human neurodegenerative diseases such as amyotrophic lateral sclerosis. The exact role of bodies in normal cellular function is not known, but p-bodies are induced in conditions of stress, and can store mRNA that later re-enters trans-lation, leading to the theory that they are sites of mRNA storage. A contrasting theory suggests them as sites of mRNA decay, due to the presence of deadenylation, decapping and exonuclease complexes within p-bodies. In this work, time-lapse fluorescence micros-copy, using optimised high brightness, low photobleaching fluorescent protein fusions, was
used to allow long term imaging of p-body localisation throughout the cell cycle of S.
cerevisiae. Imaging was combined with microfluidic dissection of mother and daughter
13
14
2.1
Foreword
Through all the advances in modern medicine over the past century, we now live longer and more prosperous lives that we ever have before. The advent of germ theory, modern hygiene standards and powerful interventions such as antibiotics, have made the infectious diseases of the past largely insignificant to mortality statistics in the developed world. But with this newfound longevity previously unrecognised ailments have arisen, caused in part by our own body’s lack or preparedness for life beyond historical limits. The most widely publicised of these conditions are the cancers, diseased of uncontrolled cellular prolifera-tion triggered by the build-up of genetic mutaprolifera-tions and therefore exacerbated in their prevalence by virtue of our lengthening lifespans. Large amounts of research investment have gone into understanding the mechanisms of cancer development and pathogenesis over the last few decades. This has led to a significant reduction in the morbidity and mortality caused by cancers, due to successful interventions and early screening pro-grammes.
While cancers affect all parts of the body, the recent shift in media and academic attention has been towards the brain, with projects such as the BRAIN initiative in the US and the Human Brain Project in Europe hoping to significantly advance our understanding of the human brain. What, one might ask, is the point of living to our 100s, curing our various physical maladies along the way, only to lose our identities long before that due to neuro-degeneration of the brain? Understanding the pathology of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease, is therefore integral to the improvement of quality of life in an ever ageing population.
15
sequestration of mRNA and proteins within these granules leading to changes in gene expression and cellular function that ultimately lead to the death of the cell. mRNP granules have also been implicated in a range of cancers, highlighting their potential as agents of disease.
16
2.2
The mRNA life cycle in yeast
The creation and utilisation of mRNA plays a fundamental role in the central dogma of molecular biology, in which DNA is transcribed to mRNA which is subsequently translated into proteins.
2.2.1 Transcription and nuclear processing of mRNA
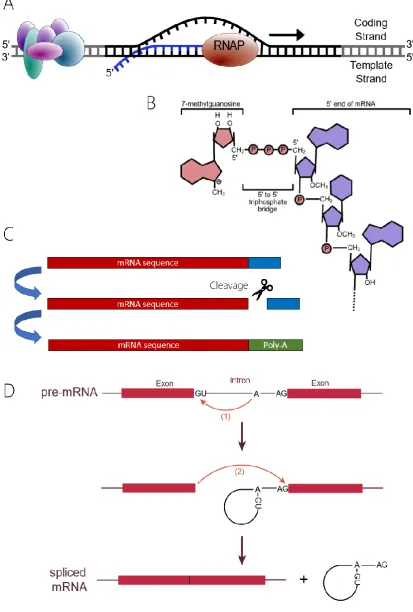
In eukaryotes, mRNA is created in the nucleus in a process called transcription, in which RNA polymerase II catalyses the polymerisation of ribonucleotides complementary to the sequence of DNA being transcribed to create a single stranded RNA molecule. This process is facilitated by proteins known as transcription factors. Once sequence transcription is complete, the RNA undergoes a series nuclear processing events (T. I. Lee & Young, 2000).
A 5’ cap consisting of a 7’methylguanosine is added at the 5’ end of the mRNA molecule, which stabilises the mRNA and prevents exonuclease degradation. The capping process occurs while transcriptional elongation is still taking place and involves 3 steps. The ex-posed triphosphate of the first nucleotide in the RNA sequence is hydrolysed by RNA tri-phosphatase and a guanosine monophosphate nucleotide derived form GTP is fused to the exposed phosphate through a 5’-5’ triphosphate bond by mRNA guanylyl transferase. Finally, the N7 position nitrogen of the guanosine nucleotide is methylated by mRNA (gua-nine-N7-)-methyltransferase (Fong, 2001). Non-coding regions of the RNA, called introns, are removed in a process called splicing carried out by a ribonucleoprotein complex known as the spliceosome. The spliceosome contains catalytically active RNA that recognises
[image:17.595.221.419.556.753.2]in-tronic sequences within the mRNA and catalyses their removal. As with 5’ capping this
17
process happens co-transcriptionally, while the mRNA is still attached to RNA polymerase 2 (Görnemann et al., 2011).
Once transcription of the sequence is complete, a poly-A tail, consisting solely of adeno-sine nucleotides is attached completing the maturation of the mRNA. Almost all coding mRNAs in yeast contain a poly-A tail comprising on average 70 poly-adenosines at the 3’ end of the sequence. Poly-A polymerases add these adenosine monophosphate nucleo-tides after mRNA has been cleaved at the 3’ end in a processes that is instigated by the cis regulatory elements in the DNA sequence (Ares & Proudfoot, 2005; Bentley, 1999).
mRNA is exported from the nucleus where it can be recognised by components of the mRNA translation machinery and translated to produce proteins. The export process is controlled by the nuclear pore complex (NPC) made up of nucleoporin proteins that form a channel in the nuclear membrane (C. Smith et al., 2015a). To facilitate mRNA export, mRNAs combine with export factors to form ribonucleoproteins that then translocate across the NPC into the cytoplasm. These mRNPs can be remodelled to regulate direc-tionality and nuclear export rate (C. Smith et al., 2015b).
2.2.2 Localisation of mRNA
Translation can occur freely in the cytoplasm or can be localised to specific areas of the cytoplasm or compartmentalised in specific organelles. The advantage of limiting transla-tion to localised positransla-tions within the cell are numerous; for example, localised translatransla-tion can be used to introduce polarity and facilitate polarised cell growth. It could also be used to facilitate asymmetric cell division as in budding yeast. Other examples of localised trans-lation include production of protein involved in neuronal processes and daughter cell spe-cific transcription inhibition (Niessing et al., 2018).
18
[image:19.595.108.522.119.732.2]of this process is that of the Ash1 mRNA which encodes a transcription inhibitor that
Figure 2: The main processes involved in transcription. A - The elongation of the mRNA
transcript by RNA polymerase II. B – Addition of the 5’ methyl guanosine cap. C – Splicing
19
prevents mating type switching when present. Ash1 is trafficked to the tip of the emerging bud during cell division and prevents mating type switching in the daughter cell by local-ised translation, while the mother cell retains the ability to switch mating types (Bobola et al., 1996). ASH1 mRNA has several localisation elements contained in both the coding re-gion and 3’ untranslated rere-gion (Bohl, 2000). These rere-gions are recognised by the mRNA binding protein She2 in the nucleus, with a relatively low affinity that is enhanced by the addition of a second protein, She3, upon nuclear export. She3 binds both She2 and the mRNA to stabilise the interaction and increase its specificity (Edelmann et al., 2017; Marisa Müller et al., 2011). She3 also binds constitutively to the myosin motor protein Myo4. To-gether the 3 proteins form a messenger ribonucleoprotein complex capable of transport along actin filaments (Bohl, 2000; M. Müller et al., 2007).
She2 has also been shown to bind to the membrane of the endoplasmic reticulum, specif-ically the cortical ER which in yeast is transported to the bud tip. The observation that cortical ER transport is dependent on the Myo4/She3 complex revealed an associated role in mRNA transport (Genz et al., 2013; Schmid et al., 2006). Multiple mRNAs have been shown to be co-transported with the cortical ER into the budding daughter cell with all these mRNAs capable of being bound by She2 (Fundakowski et al., 2012). Although the specific mechanism of ER binding by She2 is not known the association with the ER has been shown to be specific to the ER and no other organelle membranes (Genz et al., 2013).
In order to prevent translation of mRNA during transportation, the She2/She3/Myo4 mRNP is complexed with translation inhibitors such as the p-body component Dhh1 (Zhang et al., 2017). Release on inhibition by the local environment at the destination then allows trans-lation initiation to take place.
20
2.2.3 Translation of mRNA
Translation of mRNA to produce proteins is a multistep process. The major conserved proteins in eukaryotic translation are outlined below. Translation begins with initiation, the formation of 80S ribosomes capable of elongating amino acid sequences, via the joining of a 48S initiation complex to a 60S ribosomal subunit (Jackson et al., 2010). The 5’ cap of the mRNA is initially bound by the cytoplasmic cap binding complex, composed of initia-tion factors 4E, 4G and 4A. The cap binding complex works to unwind secondary structures in the 5’ region of the mRNA and create a circular mRNA structure through binding to the poly-A binding protein associated with the 3’ poly-A region. The cap-binding complex associated mRNA is then bound by the 43S pre-initiation complex of the 40S ribosome
and ternary complex of eIF2-GTP Met-tRNAmet to form a 48S initiation complex before
scanning the mRNA to identify a start codon.
21
Upon recognition of the start codon at the P-site of the 40S ribosome, the GTP bound to
Figure 4: The initiation of Eukaryotic translation. The mRNA 5’ cap is initially bound by the
22
eIF2 is hydrolysed to GDP, releasing it, along with the other initiation factors (Dever et al., 2016; Wilson et al., 2000).
To begin elongation of the peptide, the A site of the ribosome is populated with a eEF1A-GTP-aminoacyl-tRNA complementary to that of the mRNA sequence, and the eEF1A-GTP is hydrolysed to eEF1A-GDP. A peptide bond is formed between the two amino acids and movement of the ribosome shifts acceptor ends of these tRNAs into the E and P sites respectively. Hydrolysis of eEF2-GTP provides the energy for movement of the anticodon ends into the same sites. The E site tRNA is released and a new eIF1A-GTP-aminoacyl-tRNA populates the A site to begin the process again. Eventually a stop (or nonsense) codon is reached and translation termination is initiated (Dever et al., 2016).
Once the A site of the ribosome is populated with a stop codon, eRF1, a functional tRNA mimic binds the ribosome along with eRF3-GTP. Hydrolysis of the GTP of eRF3-GTP causes its dissociation and accelerates peptide release from the ribosome (Eyler et al., 2013). The ATPase Rli1 then binds eRF1 and catalyses the hydrolysis of the peptide-tRNA bond to release the nascent polypeptide. Further hydrolysis by Rli1 releases the 60S subunit from the mRNA and further ribosome associated proteins promote the dissociated of the 40S unit. This release process is known as recycling and these ribosomal subunits can then re-enter translation using the same or new mRNAs.
2.2.4 mRNA Decay in Yeast
A key stage in the control of gene expression in Saccharomyces cerevisiae, as in other
23
undergo either 3’ to 5’ exonuclease degradation by the exosome (J. S. J. Anderson & Parker, 1998) or decapping by the decapping complex consisting of Dcp1/Dcp2 followed by 5’ – 3’ exonuclease degradation by Xrn1 (Travis Dunckley & Parker, 1999; Hsu & Stevens, 1993; Kenna et al., 1993; D Muhlrad et al., 1995; Denise Muhlrad & Parker, 1994; E. Van Dijk et al., 2002). The exosome is also capable of endonuclease activity, although the mapping
of endonuclease sites in S. cerevisiae to mRNA sequences showed that very few are
Figure 5: Elongation (top) and termina-tion (bottom) of eukaryotic translatermina-tion. Elongation - A polypeptide is produced by a recurring process of aminoacyl-tRNA binding to the ribosome A site, fol-lowed by peptide bond formation to the existing peptide. The mRNA is translo-cated through the ribosome to move the tRNA between sites, allowing release of the tRNA from the E site and ingress of
a new aminoacyl-tRNA. Termination –
24 degraded in this way (Parker, 2012).
Decapping followed by degradation by Xrn1 is thought to be the major pathway for mRNA degradation in yeast due to both observations of mutant strains and direct measurement of degradation rates. Mutations in the 5’-3’ exonuclease or decapping factors show signif-icant changed in mRNA decay rates compared with mutants in the 3’-5’ pathway (Beelman et al., 1996; X. He & Moore, 2005) indicating that the decapping pathway may influence the degradation of a greater volume of mRNA. Mutations in the decapping proteins also cause severe growth restrictions and are lethal in some strains of yeast (Beelman et al., 1996; Travis Dunckley & Parker, 1999; Giaever et al., 2002). Computation modelling of the degradation pathways also implied that deadenylation had the largest influence on mRNA levels and changing the rate of deadenylation had a larger effect on levels than changing the activity of the 3’-5’ exonuclease pathway (Cao & Parker, 2001).
For the non-standard degradation of mRNA, for example in cases of aberrant mRNA se-quences, yeast also possess quality control mechanisms capable of acting independently of the usual degradation pathways that allow specific targeting and degradation of these mRNAs. These pathways can act by bypassing the usual restriction of deadenylation through deadenylation-independent decapping, such as in nonsense-mediated decay, or through rapid 3’-5’ exonuclease (non-stop decay) or endonuclease cleavage (no-go de-cay) of the mRNA (Doma & Parker, 2006, 2007; D Muhlrad et al., 1994; Van Hoof et al., 2002).
As deadenylation, decapping and Xrn1 degradation factors have been shown to accumu-late in cytoplasmic p-bodies, this pathway is explored further below.
Deadenylation
Deadenylation is the process of removing adenine nucleobases from the 3’ UTR of an
25
et al., 2004). The Not proteins may function in adapting the complex to different mRNAs, as mutations in these proteins can affect the deadenylation of specific subsets of mRNA (Morgan Tucker et al., 2002). A secondary deadenylase complex consisting of Pan2, the catalytic subunit, and Pan3, thought to bind the poly-A tail region via a zinc finger domain (Schäfer et al., 2014) is also present in yeast. This complex is promoted by the poly-A bind-ing protein, Pab1 (Boeck et al., 1996), which also inhibits Ccr4 activity, and may be respon-sible for the initial shortening of exposed poly-A tails bound to Pab1, as deletion mutants of Pan2 lead to longer poly-A tails (Brown & Sachs, 1998). A partial redundancy exists between these 2 deadenylase complexes, as deletion of either does not lead to fatal phe-notypes, but growth defects in which growth rate is limited. Pan2 can continue past its initial 25 nucleotide region in a Ccr4 deletion mutant but arrests at around 20-25 remaining adenine nucleotides. No further deadenylases are known and none are likely to exist given
Figure 6: Simplified progression of mRNA decay in budding yeast and eukaryotes. The Pan2/Pan3 complex is responsible for initial deadenylation, followed by continued dead-enylation by the Ccr4/Not1 complex. Decapping proteins Dcp1 and Dcp2 catalyse the
re-moval of the 5’ mRNA cap, in a process enhanced by Edc3. Finally, Xrn1 exonuclease activity
[image:26.595.123.535.370.676.2]26
that a double deletion of Ccr4 and Pan2 results in the loss of deadenylation activity (M Tucker et al., 2001).
27
bound by Ccr4/Dhh1 (Dhh1 again being a p-body component) during normal growth are subject to regulation during starvation (J. E. Miller et al., 2017).
To summarise, deadenylation is a two-stage process in which the Pan2/Pan3 complex first shortens the initial nucleotides and then has its activity is promoted by the presence of Pab1, introducing a point of regulation. The Ccr4/Not complex removes the remaining poly-A nucleotides not bound by Pab1 triggering either the decapping process or 3’-5’ degradation by the exosome. Multiple factors affect Ccr4/Not complex binding to the mRNA, and sequence specific features can regulation the specific decay rates of individual mRNAs. This process can be globally regulated by environmental factors with some envi-ronmental factors such as nutrient starvation having specific effects on distinct mRNA pop-ulations.
Decapping
Active eukaryotic mRNAs feature a 5’ structure called the cap, a methylated guanine nu-cleotide that is linked to the 5’ end of the mRNA by its own 5’ carbon. This unique structure stabilises the mRNA, preventing 5’-3’ exonuclease activity. For complete degradation of mRNA via the 5’-3’ pathway this 5’ cap must therefore be removed to allow access to the exonuclease Xrn1. The process of removing this 5’ cap, known as decapping, involves the hydrolysis of the cap structure, leaving an exposed 5’ monophosphate that is targeted by Xrn1. In yeast decapping is performed by the decapping complex, made up of the catalytic pyrophosphatase Dcp2 (E. Van Dijk et al., 2002) and the decapping promotor subunit Dcp1 (Deshmukh et al., 2008; She et al., 2004). The first 300 amino acids of Dcp2 are responsible for the hydrolysis of the phosphate bond (Travis Dunckley & Parker, 1999), and fold into a 2 domain structure which closes into a more active structure when associated with Dcp1 (Deshmukh et al., 2008; Floor et al., 2010; She et al., 2008). Dcp2 also features binding sites for other activators of decapping such as the Edc3 (Harigaya et al., 2010).
Regulatory functions of decapping fall into three categories, the catalytic action of Dcp2, the removal of the cap-binding complex eIF4F to allow access to the cap and the recruit-ment of the decapping complex to the cap.
28
catalysis (Harigaya et al., 2010; Nissan et al., 2010), Edc1 and its paralog Edc2 bind instead
to Dcp1, and have been shown in vitro to enhance Dcp2’s catalytic activity in this complex
(Borja et al., 2011). During log phase, stress free growth, the catalytic activity of Dcp2 is not rate limiting for decapping, as demonstrated by the lack of decapping-deficient pheno-types in Edc1/2/3 deletion mutants (T Dunckley et al., 2001; Kshirsagar & Parker, 2004). Other mutations affecting Dcp2 catalysis give similar phenotypes showing little loss of decapping activity, including Pat1 C-terminal domain deletion (Nissan et al., 2010; Pilkington & Parker, 2008) and mutations in Dcp1 and Dcp2 themselves (Steiger et al., 2003; Tharun & Parker, 1999).
Further regulatory proteins can increase the rate of decapping by promoting the binding of the decapping complex to the 5’ cap or recruiting additional enhances of decapping. Protein interaction studies have identified 2 major groups of proteins that interact to form decapping complexes. One complex binds to the 3’ end of deadenylated mRNAs in vitro and consists of Pat1, the sub complex of Lsm1-7 and the Xrn1 exonuclease (Chowdhury et al., 2007). This complex is important in maintaining the order to degradation, ensuring that decapping enhancement occurs only after deadenylation by binding to oligoadenylated rather than polyadenylated mRNA. It also preferentially activates the decapping and 5’-3’ degradation pathway, as a Pat1 or Lsm1 deletion causes exonuclease activity at the 3’ end of the mRNA indicative of exosome activity (W. He & Parker, 2001)). The Dcp1/Dcp2 com-plex can also recruit or be recruited by additional proteins including Edc3, Scd6 and Dhh1 (Parker, 2012). Several of the proteins involved in these complexes, including Lsm4 and Edc3 contain low complexity domains that can lead to aggregation of these mRNP com-plexes into larger structures such as p-bodies.
29
Ramirez et al., 2002). The proteins regulating this competition are diverse in function and many are also involved in the formation of, or localise to, cytoplasmic p-bodies during cellular stress (Nissan & Parker, 2008). Enhancers of decapping in yeast are summarised in table x. For enhancers that work by inhibiting translation, the exact mechanism that leads to an increase in decapping rate is not clear. It could be a simple matter of increasing the time available for the dissociation of the cap binding complex or may be a more complex remodelling of the mRNP (Parker, 2012). This set of proteins would be classed under the final category of ‘removing the 5’ cap binding complex’. Further evidence that the cap binding complex is the key inhibitor of decapping comes from studies of mutants with aberrant copies of its constituent proteins, in which mRNA stability is significantly reduced (Linz et al., 1997; McCarthy, 1998; Ramirez et al., 2002; Schwartz & Parker, 1999, 2000; C. Vilela et al., 2000; Cristina Vilela et al., 1999).
In the 5’-3’ pathway, the initiation of decapping is triggered by the completion of dead-enylation. As the poly-A tail is reduced it loses the ability to bind to Pab1 but increases its
affinity for the Pat1/Lsm complex as demonstrated by the in vitro studies of Pat1/Lsm and
Pab1 poly-A binding (Chowdhury et al., 2007; Sachs et al., 1987). This reliance on Pab1 is supported by the observation that decapping and deadenylation are uncoupled in Pab1 deletion mutants (Caponigro & Parker, 1995).
30
Once exposed by the decapping process, the 5’ monophosphate can be recognised by the Xrn1 exonuclease, beginning a process of rapid 5’-3’ exonuclease degradation (Kenna et al., 1993; Stevens & Poole, 1995; E. L. Van Dijk et al., 2011).
2.3
Processing bodies
Processing Bodies (p-bodies) are non-membranous cellular compartments, part of a family of similar bodies/granules known as mRNP granules, consisting of an aggregated mixture of mRNA and proteins, some conserved and others variable depending on the state of the cell. Similar mRNP granules are present in a variety of organisms and hold different roles for cellular development and survival, these include Stress Granules (P. Anderson & Kedersha, 2008) Germ Granules (Gallo et al., 2008) and Neuronal Granules (Kiebler & Bassell, 2006). The structure, composition and function of p-bodies has been studied in
both human cell lines as well as multiple model organisms such as the yeast Saccharomyces
cerevisiae.
2.3.1 Discovery
P-bodies were originally observed as cytoplasmic foci of mRNA decay and decapping pro-teins, identified by fluorescent protein fusions and immunofluorescence experiments ini-tially in mammalian cells (Kedersha et al., 2005) and subsequently in yeast and other eu-karyotes (U Sheth & Parker, 2003). Fluorescence microscopy represents an invaluable tool
for the study of p-bodies given the difficulty of purifying or reproducing them in vitro. The
31
[image:32.595.106.546.341.698.2]Around the same time as the discovery of p-bodies, a closely related mRNP known as a stress granule was also identified and has been shown to be conserved in a similar range of model organisms and cell lines (P. Anderson et al., 2015; Nover et al., 1989). Stress gran-ules are not constitutive components of cells and instead are induced by a range of cellular stresses including heat shock, starvation and other translation inhibiting factors (J. R. Buchan et al., 2011). Although initially proposed to be formed from existing p-bodies, fur-ther work has highlighted key distinctions between the composition and function of these two mRNPs. For simplicities sake this section focuses only on p-bodies, but their relation-ship with stress granules should be kept in mind given their various similarities which are highlighted throughout.
32
2.3.2 Structure and Composition
Studies using electron microscopy to investigate the ultrastructure of stress granules and p-bodies found that they are often closely associated with one another in HeLa cells. How-ever, the two mRNPs have distinct, non-overlapping structures, with p-bodies showing a fibrillar ultrastructure compared with a fibrillo-granular stress granule ultrastructure (Souquere et al., 2009). These results point to distinct functions for SGs and p-bodies that are backed up by their differing protein composition.
There are also differences between the p-bodies observed in mammalian cells and in the
budding yeast Saccharomyces cerevisiae, in which a large number of studies have been
conducted. Mammalian p-bodies are constitutive components of most cells but vary in number and composition depending on the cell type. In contrast, yeast p-bodies have been observed to form only under certain conditions, similar to SGs, such as starvation or caloric restriction (Decker & Parker, 2012). Mammalian p-bodies disassemble during mitosis in a similar fashion to some other organelles and reform in G1, whereas in yeast, p-bodies can be present throughout mitosis if the inducing conditions remain in place (Garmendia-Torres et al., 2014; Yang, 2004). The composition of yeast p-bodies has been extensively documented since their initial discovery. With further immunofluorescence experiments and fluorescent protein fusions, a wide range of proteins have been observed to localise to p-bodies, some constitutively while others only during certain stresses.
33
yeast p-bodies. Finally, yeast p-bodies contain a currently unknown group of mRNAs, alt-hough some mRNAs have been individually identified to localise to p-bodies (Aizer et al., 2014). mRNA is a requisite component of p-bodies, and when translation is blocked exper-imentally using cycloheximide) leading to a reduction in mRNA abundance, p-bodies dis-assemble, presumably into smaller mRNP complexes. More recently, a study of human p-bodies, in which a sorting method similar to fluorescence associated cell sorting (FACS) was used to purify p-bodies, the mRNA content of p-bodies in human cells was obtained through mRNA sequencing (Hubstenberger et al., 2017). This study showed that almost 1/3 of coding mRNA transcripts could be found within the constitutive p-bodies of the human cell line, mainly made up of highly abundant but poorly translated sequences. As well as this it was also found that another 1/3 of coding mRNAs, and the majority of non-coding mRNAs were significantly excluded from p-bodies indicating that mRNA entry into p-bod-ies was a specific rather than a global phenomenon. This study also discovered a number of new protein components of human p-bodies via mass-spectrometry based analysis of the purified bodies. Many of these fell into the traditional categories of mRNA repression and decay, but some new components were identified including a number of myosin
pro-teins of which one example was shown to localise to p-bodies in vivo. A full list of the yeast
PB components as derived from the Saccharomyces Genome Database can be found in
in the Appendix.
34
2.3.3 Assembly and regulation
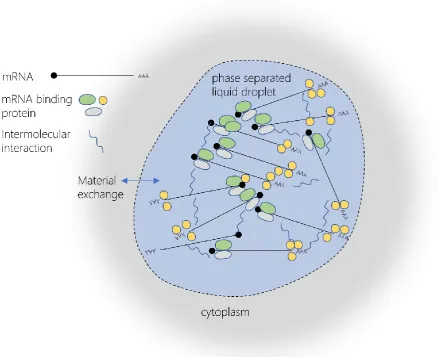
Although PB regulation appears to vary between organisms, with yeast p-bodies being stress-induced and metazoan p-bodies always present, the proteins involved in the regu-lation of size and abundance appear to be conserved across species. Assembly of p-bodies depends on the concentrations of their constituent proteins (Franks & Lykke-Andersen, 2008; Y. Luo et al., 2018), with low complexity region of Edc3 and Lsm4 being particularly important in yeast p-bodies (Decker et al., 2007; Reijns et al., 2008). These regions of low complexity have been shown to be involved in both protein-protein interaction and RNA binding and are thought to contribute to a process of liquid-liquid phase separation (LLPS), in which non-membranous aggregates of proteins become phase separated into a dense inner core and aqueous outer layer(Schutz et al., 2017). A complex of Dcp1/Dcp2, mRNA and Edc3 has been shown to undergo LLPS in vitro.
35
Multiple p body associated proteins have also been shown to be susceptible to aberrant protein aggregation when the normal mRNA surveillance mechanisms are disrupted. In a study of the composition of wild type protein aggregates and those formed in mutants of the mRNA surveillance pathways, multiple p-body components were identified (Jamar et al., 2018). Two major p-body components, Dhh1 and Xrn1, were found to commonly form protein aggregates in wild type cells, and Dhh1, a regulator of p-body assembly and dis-assembly (J. R. Buchan et al., 2008; Mugler et al., 2016) also aggregated in NMD, NSD and NGD mutants. The p-body components Pat1, Ccr4 and Dcp2 were also shown to form protein aggregates in cells deficient in NSD, NMD and MD respectively. This propensity to form aggregates can be seen as both advantageous, in the formation of phase separated p-bodies, and potentially dangerous, as uncontrolled aggregation can lead to cellular damage.
Yeast p-bodies differ from mammalian p-bodies in their regulation during the cell cycle. While mammalian bodies appear to be dissolved during mitosis (Yang, 2004), yeast p-bodies are maintained, and trafficked into the emerging bud in a unidirectional manner that appears to confer a selective advantage on the daughter cells, at least under condi-tions of caloric restriction (Garmendia-Torres et al., 2014). Outside of mitosis regulation of p-bodies appears similar across species, with increases in their numbers under conditions that generally inhibit translation such as oxidative stress and starvation. In yeast, Dcp2 is
36
phosphorylated in response to stress which causes its localisation to p-bodies while also stabilising a set of mRNAs encoding ribosomal proteins (Yoon et al., 2010). Mammalian DCP1a is also phosphorylated in response to stress, affecting its p-body localisation, as is human DCP1a during cellular differentiation (Chiang et al., 2013; Rzeczkowski et al., 2011).
2.3.4 Function
The function of p-bodies has been much debated in the decades since their discovery. While the initial evidence pointed towards a clear role as hubs of mRNA degradation, fur-ther investigation has cast doubt on that role, and a more general function in mRNA stor-age and regulation of translation has taken shape.
The initially observed composition of P-bodies was the main driver of the theory that p-bodies were sites of cytoplasmic mRNA degradation. This was backed up by the observa-tion that the inhibiobserva-tion of 5’ – 3’ mRNA decay by removal of the Xrn1 exonuclease caused a build-up of p-bodies that were also larger than wild type p-bodies and were thought to contain mRNAs stalled in degradation (U Sheth & Parker, 2003) and in human cells were shown to be enriched in polyA mRNA (Cougot et al., 2004). It was also found that mRNA decay intermediates localised to p-bodies in Xrn1-deficient yeast strains (U Sheth & Parker, 2003) supporting the theory of degradation occurring in p-bodies. The inhibition of trans-lation, through the stabilisation of mRNA complexes using cycloheximide, caused a marked decrease in p-bodies. This supported their role in mRNA decay, as a reduction in available mRNA, due to mRNA becoming trapped on in polysomes rather than available for binding, would likely reduce the size of p-bodies reliant on mRNA for their formation. The lack of translation initiation factor in p-bodies also points to a role outside general storage of mRNAs, unlike SGs where translation factors are found during stress conditions and mRNAs are thought to be stored ready to re-enter translation (Brengues et al., 2005).
37
intermediates in p-bodies, while a study using a new imaging technique to image mRNA decay as it occurs showed a lack of mRNA degradation events in p-bodies (Horvathova et al., 2017). It has also been shown that the inhibition of PB formation by removal of core formation regulator DDX6 (human, Ddh1 in yeast) has little effect on RNA stability for p-body localised transcripts (Hubstenberger et al., 2017), and that nonsense-mediated mRNA decay does not require p-bodies to occur (Stalder & Mühlemann, 2009). An alternative approach to mRNA degradation analysis identified both stabilisation and degradation of mRNAs that were localised to p-bodies on glucose starvation in yeast (Wang et al., 2018).
The structure of p-bodies could also be an influencing factor in their function. Recent stud-ies into LLPS showed that enzyme activity of the core PB component Dcp2 is reduced in
an in vitro model of LLPS, as is the activity of RNAse A. This could imply that the dense
liquid droplet core of the PB reduces the activity of decay factors enough for them to function as storage sites and protect their mRNA components from decay, either for stor-age or possible for mRNA transport.
Overall the true function of p-bodies still remains unclear. There is a large body of evidence supporting the fact that they are site of mRNA decay, but more recently work has begun to question this, and a role in mRNA storage has also emerged. Given that the initial work on PB function, as well as some of the later work supporting its role in decay, was per-formed in yeast whereas the bulk of the evidence for storage comes from metazoan stud-ies, it could be that p-bodies have taken on divergent roles, with yeast p-bodies only func-tioning as storage sites during cellular stress and funcfunc-tioning as sites of decay under normal conditions. This is supported by the findings that highly expressed mRNA accumulate in yeast p-bodies during various stresses (Lavut & Raveh, 2012) and that p-bodies are re-quired for long-term survival of stationary phase yeast cells (Ramachandran et al., 2011), while also not contradicting the earlier studies of p-bodies that indicate their role in mRNA decay. Further work is clearly required to elucidate the full function of p-bodies.
2.3.5 Transport within the cell
38
mammals and unicellular organisms, association of p-bodies with the cytoskeleton has been reported.
In mammalian cells, p-bodies have been shown to move along microtubules in a direc-tional manner and change microtubule tracks implying association with motor proteins. It was suggested that this association with microtubules could be facilitated by binding to organelles such as mitochondria, which are known to move on microtubules; although the adaptor proteins or cellular motors involved were not identified (Aizer & Shav-Tal, 2008). Microscopic observation of fluorescently tagged p-bodies showed both spatially confined movement and linear translocation across small regions of the cell. Various p-body com-ponents have also been shown to co-localise and bind to the class V myosin motor Myosin Va in mammalian cells, and siRNA knockdown of Myosin Va inhibits p-body assembly. Overexpression of Myosin Va tails also limits the movement of p-bodies in vivo (Lindsay & McCaffrey, 2011). Recently, Nesprin-1 has been identified as a link between p-bodies and microtubules and expression of the Nesprin-1 microtubule binding region disrupts p-body transport on microtubules (Rajgor et al., 2014).
In plants, p-body motion appears to be actin, rather than microtubule based. P-bodies were observed to translocate along actin filaments, with movement inhibited by the over-expression of myosin XI tails. Myosin XI was shown to bind to the p-body component atDCP1, and the Dcp1 myosin interaction was conserved across species, with Yeast Myo2 binding yeast Dcp1, and Mouse MyoVa binding both yeast and human Dcp1 homologues (Alexandra Steffens et al., 2014).
39
work using live cell observation of the p-body marker Edc3 in a microfluidic device showed that p-bodies are directionally trafficked into emerging buds, lingering at the bud site be-fore budding begins. This deliberate inheritance was again dependent on the locasome machinery and a drop in velocity of p-bodies during budding was also inhibited by this loss of the locasome. The inheritance of the p-body was also shown to confer a selective advantage on the daughter cell, whereby cells inheriting a p-bodies reached a larger size before budding, although this study was carried out in conditions of dietary restriction.
2.4
Replicative ageing in budding yeast
Ageing is the continuing change and development of an organism over time, and on a cellular level takes the form of an eventual breakdown in key maintenance systems leading to the eventual death of the cell, and the organism. In budding yeast, ageing is described in terms of replicative or chronological ageing. Replicative ageing refers to the breakdown in cellular function as a mother cell spawns an increasing number of daughter cells, whereas daughters are “reset” in terms of age and capable of producing daughter cells of their own. Chronological ageing is the process in which yeast cells deteriorate when not dividing, under conditions such as starvation. Replicative ageing in yeast represents an attractive model for eukaryotic ageing research, as the mother and daughter cell relation-ship is easily defined, simplifying the tracking of inherited molecules and damaged pro-teins. The genetic tractability of budding yeast and its ease of growth, short cell cy-cle/lifespan and well-developed toolkit of laboratory techniques mean that it has featured heavily in ageing research over the past 30 years.
2.4.1 Conserved mechanisms of extended lifespan
Using budding yeast as a model for research into eukaryotic ageing processes would make little sense unless these mechanisms were conserved in general amongst eukaryotes. Re-search to date has identified a number of genetic and environmental factors that impact RLS in yeast, with these effects being conserved in higher eukaryotes, (Wasko & Kaeberlein,
2014). A recent high-throughput study of replicative lifespan (RLS) extension in S. cerevisiae
40
Caenorhabditis elegans were 5 times more likely to influence lifespan in yeast, implying a
41
42
2.4.2 Sirtuins and ERCs
The first group of proteins found to extend lifespan in yeast were the Sirtuins. Sirtuins are a group of protein deacetylases that deacetylate their targets in response to signalling pathways. For example, the namesake of the group, Sir2, deacetylates histones to silence certain genetic loci (Finkel et al., 2009). Only Sir2 is highly conserved across species, having homologues in bacteria, yeast, flies, worms and mammals (Brachmann et al., 1995). Over-expression of Sir2 robustly extends replicative lifespan in budding yeast, and a null mutant significantly reduced RLS, an effect which has been replicated in chronological lifespan studies from worms and flies (Kaeberlein et al., 1999; Brian K Kennedy et al., 1997; Rogina & Helfand, 2004; Tissenbaum & Guarente, 2001). This lifespan-extending effect is thought to be due to the activity of Sir2 in promoting genomic stability via chromatin silencing, preventing extensive recombination of DNA and limiting global transcription levels (Dang et al., 2009). As yeast mother cells age, reduced levels of Sir2 caused increased histone acetylation, opening up chromatin and causing increased levels of recombination and a global rise in transcription rate that may lead to an aberrant proteome (Dang et al., 2009).
43
an active mechanism, but a passive one, in which ERCs bound or unbound to NPCs are prevented from moving into the daughter nucleus due to the geometry of the bud neck and limited timespan of mitosis (Gehlen et al., 2011). This passive mechanism might also explain how cytoplasmic structures are partitioned asymmetrically, in cases were their ac-tive transport to the daughter cell has not been selected for throughout evolution in a kind of “default” segregation to the mother cell. The observation that other centromere-lacking DNA constructs such as plasmids are also retained in the mother cell seems to support this idea, as they may lack the sequence or epigenetic features required for NPC binding. Fur-ther support comes from a study demonstrating that a relaxing of the diffusion barrier, caused by mild heat shock, can allow a more symmetrical partitioning of ERCs, extends lifespan of mother cells (Baldi et al., 2017) and is reliant on changes in the activity of protein kinase A (PKA) and target of rapamycin (TOR) signalling pathways, which have also been shown to influence lifespan (see below).
The number of ERCs can be influenced by lifespan-extending genetic changes as well as environmental factors. For example, deletion of the Fob1 gene, involved in DNA replication of rDNA regions, extends replicative lifespan and appears to reduce recombination in the rDNA locus, thereby reducing ERCs (Defossez et al., 1999). rDNA instability can also be decreased through dietary restriction (DR) in yeast, or by inhibiting the downstream sig-nalling target or DR, TOR (Ha & Huh, 2011; Riesen & Morgan, 2009). Taken together it is clear that at least in yeast, ERCs play some role in the progression of replicative ageing.
2.4.3 Asymmetric inheritance of protein aggregates
44
Liu, 2014). Cells lacking Hsp104 fail to form these compartments, leading to a breakdown in inheritance of aggregated proteins, with smaller aggregates passing through the bud neck to the daughter cell (Spokoini et al., 2012). Sir2 has also been implicated in the asym-metric partitioning of aggregated proteins highlighting a potential role in ageing for these aggregates (Sampaio-Marques et al., 2012).
Whether aggregates are actively or passively retained in mother cells remains to be proven. Modelling of aggregate motility suggested that similarly to ERCs, the geometry of the bud-ding yeast cell prevents the inheritance of aggregates due to their slow diffusion rate. Bud neck diameter and mitosis time were found to be key factors in the model of aggregate inheritance (Zhou et al., 2011) although this modelling was based on an average of the diffusion coefficients of observed aggregates. For larger aggregates such as JUNQ and IPOD that are attached to organelles, evidence of active transport supporting retention in mother cells has been reported. Large scale genetic screens identified the actin cable nu-cleating polarisome machinery as an essential component for protein aggregate retention (Liu et al., 2010; Tessarz et al., 2009) and Sir2 may be affecting the function of actin when regulating retention. Protein aggregates have also been shown to localise with actin in live cell imaging experiments (Liu et al., 2011). Multiple studies have also shown that aggregates may associate with various organelles and could be co-trafficked to ensure their retention (Kaganovich et al., 2008; S. B. Miller et al., 2015; Specht et al., 2011; Zhou et al., 2014).
45
2.4.4 Dietary restriction and downstream signalling pathways
Dietary restriction (DR), in which the caloric intake of an organism is restricted to below usual levels, acts through a number of signalling pathways. DR has been shown to increase replicative lifespan in yeast and lifespan in a number of multicellular organisms (B. K. Kennedy et al., 2007; Omodei & Fontana, 2011). The downstream effects of DR are thought to prolong lifespan in different ways, including a reduction in protein synthesis and asso-ciated misfolding of proteins (Kaeberlein, 2013), a general increase in autophagy of dam-aged cellular components (Johnson et al., 2013) and changes in mitochondrial respiration (Hempenstall et al., 2012). DR appears to work by influencing nutrient sensing signalling pathways within the cell, to decrease activities associated with cell growth and increase activities associated with the response to stress.
An increase in mitochondrial aerobic respiration compared to anaerobic respiration ap-pears to be conserved across species, although there is contradictory evidence in yeast, in that a lack of mitochondrial DNA does not remove the impact of DR on replicative life span (Hempenstall et al., 2012; Kaeberlein et al., 2005). An increase in aerobic respiration appears to contradict the long held theory that reactive oxygen species (ROS) from aerobic respi-ration are one of the main drivers of cellular ageing, causing damage to many components of the cell (Harman, 2006). It may be that a breakdown of the asymmetrical inheritance of damage could extend mother lifespan while reducing overall fitness, although how this would work in multicellular organisms remains unclear. Another possibility is that the as-sociated upregulation of autophagy compensates for the increase in ROS, as autophagy normally diminishes with age (Cuervo, 2008). Surprisingly, an increase in autophagy alone does not increase RLS in yeast (Wasko & Kaeberlein, 2014).
46
homolog,TORC1 regulates global mRNA translation rates as well as autophagy and the response to stress and an ortholog of the yeast substrate of TORC1, the serine threonine protein kinase, Sch9 has similar activity in multicellular organisms (Jorgensen et al., 2002; Stanfel et al., 2009; Urban et al., 2007). Inhibition of TOR signalling can also increase mito-chondrial biogenesis leading to increased respiration (Wasko & Kaeberlein, 2014).
Dietary restriction also inhibits signalling via Protein Kinase A, a cyclic AMP dependent kinase complex (Lin et al., 2000; Toda et al., 1987). PKA signalling is similar to TOR, in that it negatively regulates the stress response, including stress responsive transcription factors. Reducing PKA signalling genetically mimics the effect of dietary restriction in yeast (Lin et al., 2000), and has a corresponding effect on replicative lifespan which cannot be enhanced by dietary restriction in the same strain. Interestingly, both PKA and TOR signalling have been found to be important in the regulation and assembly of mRNP granules including p-bodies and stress granules during the response to stress in yeast (Ramachandran et al., 2011; Sfakianos et al., 2018; Vindry et al., 2017).
The underlying pathways of ageing appear to be conserved from single-celled eukaryotes such as budding yeast to multicellular eukaryotes. Factors such as genome instability, nu-trient signalling, mitochondrial dysfunction and proteostasis all appear to play roles in con-tributing to the ageing process. Interestingly the mechanisms by which ageing can be in-terrupted, including TOR and PKA signalling also promote the activity of stress responsive elements such as p-bodies within the cell. Although the interventions that increase RLS in yeast (and general lifespan in higher eukaryotes) do not necessarily equate to improve-ments in selective fitness, they may not be of great importance in the treatment and/or prevention of neurodegenerative diseases that appear later in life. Understanding the re-lationship between the ageing process and the stress response, including the function of p bodies, will help us develop a systems level view of eukaryotic ageing, the beginnings of which are outlined in figure 10.
2.5
Objectives
47
environment. Signalling pathways, in which interventions can lengthen lifespan in model organisms, control the transcriptional and post-transcriptional response to stress, including the formation and regulation of cytoplasmic mRNP granules. The full functionality of these mRNP granules remains elusive. They may play a role in mRNA decay and/or storage, and more recently they have been implicated in the transport of mRNA in dividing cells. Few data have been gathered on the changes that may occur to p-bodies during the ageing process.
Saccharomyces cerevisiae represents an attractive model organism for the study of ageing
and age-related effects on organelles/cellular compartments such as p-bodies. The easy of creating genetic modifications such as introducing fluorescent protein fusions makes live cell imaging possible and many other genetic tools have been developed to aid re-search in this organism. Recently, age related rere-search in yeast has been greatly facilitated by the introduction and use of microfluidic devices capable of selectively retaining yeast mother cells while removing the daughters. This allows cells to be monitored for the dura-tion of their replicative lifespan.
This study aimed to take advantage of the recent advances in fluorescence microscopy and microfluidics as powerful tools for use in the investigation of the impact of age on the
48
localisation and function of p-bodies in the budding yeast Saccharomyces cerevisiae.
49
50
3.1
Molecular Cloning
Multiple plasmids were constructed for this work using a number of molecular cloning methods, making it unfeasible to describe the construction of each in detail. Instead a detailed summary of each technique is presented as well as the necessary oligonucleotide primers and plasmid descriptions (Appendix).
3.1.1 DNA Manipulation
Restriction Cloning
For plasmids constructed using traditional restriction cloning, DNA fragments were gener-ated in one of 2 ways. Either the fragment was directly excised from an existing plasmid using restriction enzymes, or the fragment was amplified from a template using PCR with oligonucleotide primers containing restriction sites, which were then digested.
For directly excised fragments, 1 μg of the source plasmid was incubated with the required restriction enzymes (New England Biolabs, ThermoFisher Scientific) and suitable buffer so-lution at a temperature and time according to the manufacturer’s recommendations in a total volume of 40 μl. After incubation the reaction mixture was mixed with a 6X DNA Loading Dye (New England Biolabs) at a ratio of 5:1. Agarose gels were prepared using 1% w/v agarose (Sigma) dissolved in 1X TAE Buffer (see appendix for composition) containing 0.5μg/ml ethidium bromide. DNA fragments were loading in their entirety into the wells of the agarose gels and electrophoretic separation was performed at 100V constant voltage for 45 minutes in a horizontal electrophoresis gel tank (Bio-Rad) alongside a marker of standard dsDNA lengths. After separation, DNA fragments of the correct size were excised from the gel using gel excision pipette tips under UV light illumination and the DNA was purified using a spin column-based DNA recovery kit (Zymo Research). Finally, the eluted DNA concentration was measured using an automated UV-Vis Spectrophotometer (Nanodrop 2000 - Thermo Scientific).
51
frame with the existing ORF. PCR amplification was performed using a high fidelity proof reading polymerase (Q5 - New England Biolabs) with buffer and nucleotide concentrations as well as thermal profiles according to the manufacturers recommendations in an auto-mated thermocycler (T100 - Bio-Rad) at a total volume of 50 μl. After amplification PCR products were purified using a spin column based DNA purification kit (Zymo Research) and DNA concentration was measured via a spectrophotometer as with restriction digest fragments.
Destination plasmids were digested, isolated and purified in the same manner as restriction fragments, but were further treated with alkaline phosphatase before isolation to remove phosphate groups from the DNA ends to prevent re-annealing of the plasmid. 1 μl alkaline phosphatase (Calf intestinal – New England Biolabs) was added to the digestion reaction mixture and incubated at 37°C for 30 minutes after which the isolation and purification steps proceeded as previously.
After generation, DNA fragments were annealed by incubating with T4 DNA ligase and a corresponding buffer system (New England Biolabs) for 20 minutes at room temperature, after which they were placed on ice. Fragments were ligated in a molar ratio of 3 parts
insert to 1-part destination plasmid forming a total of 100ng of DNA. 5 μl of the ligation
mixture was used directly as the DNA for bacterial transformation (see Bacteria Transfor-mation).
Gibson assembly
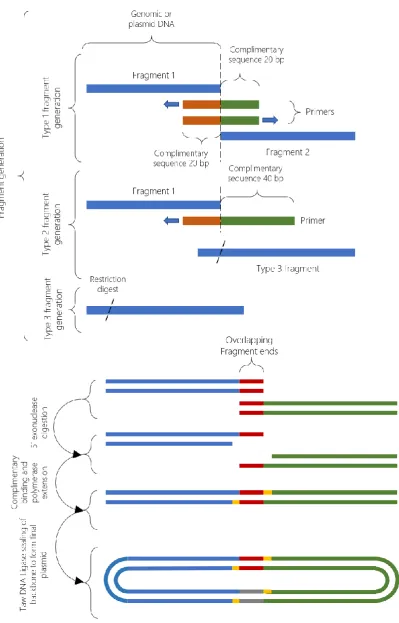
For plasmids constructed via Gibson assembly, there were again 2 methods of fragment generation. Gibson assembly requires overlapping ends between fragments; the required overlap can be introduced on both, or a single fragment depending on the convenience of adding overlaps via PCR. When joining multiple fragments from different sources, oli-gonucleotide primers for the PCR fragments can be designed that include the necessary overlaps, making it possible to include restriction only fragments in the assembly as well. For an overview of the assembly process see Figure 11.
52 (Type 2) and restriction fragments (Type 3).
Figure 11: An overview of the Gibson assembly cloning process used to create plasmids in this study. Gibson assembly allowed the ligation of multiple fragments in a single reaction to simplify and speed up the cloning process. 3 fragment types were generated as outline
in the upper panel. These fragments underwent 5’ digestion, followed by overlapping
53
For Type 1 fragments, 40 nucleotide primers were designed according to standard design principles and included 20 bp tails, complementary to the primer sequence of the adjacent PCR fragment in the assembly so that the primers for adjacent fragments are reverse com-plements of each other. Fragments were amplified from template DNA using a high-fidelity proofreading polymerase (Expand High-Fidelity PCR System – Roche) with buffer and nu-cleotide concentrations as well as thermal profiles according to the manufacturers recom-mendations in an automated thermocycler (T100 - Bio-Rad) at a total volume of 50 μl. After amplification PCR products were purified using a spin column-based DNA purification kit (Zymo Research) and DNA concentration was measured using an automated UV-Vis Spec-trophotometer (Nanodrop 2000 - Thermo Scientific).
For Type 2 fragments, 60 nucleotide primers were designed according to standard design principles and included 40 bp tails, complementary to the DNA sequence of the adjacent DNA fragment in the assembly. PCR was performed as with type 1 fragments.
For Type 3 fragments, restriction digest and gel extraction were performed identically to the process for restriction cloning.
After fragment generation, assembly was performed using 2X Gibson assembly master mix (New England Biolabs). Fragments were mixed with ddH2O in a ratio according to the manufacturer’s recommendations to a total volume of 10 μl. The fragment mixture was then mixed with 10 μl of 2X Gibson assembly master mix and incubated at 50°C for 60 minutes before placing on ice prior to bacterial transformation.
Bacterial Transformation
Bacterial transformation with plasmid DNA was performed using chemically competent Escherichia coli. Top10 Competent cells were created using the following protocol:
54
To prepare competent cells, 250ml SOB was inoculated with a single 1 ml seed stock and incubated at room temperature until the culture reached OD600 0.3, approximately 14 hours (usually overnight). Cells were centrifuged at 3000 rpm in pre-chilled centrifuge tubes at 4˚C for 10 minutes. Supernatant was discarded, and cells were resuspended in a total volume of 80 ml CCMB80 buffer (see appendix for composition) before incubating on ice for 20 minutes. Cells were again centrifuged as previously and resuspended in a total volume of 10 ml CCMB80 buffer. Chilled CCMB80 buffer was added until an OD of 1 – 1.5 was achieved when 50 μl cells were mixed with 200 μl SOB media. Competent cells were aliquoted in 50 μl volumes in Eppendorf tubes and stored at -80˚C. Competence was tested using 1 μl of standard pUC19 plasmid (Invitrogen).
To transform chemically competent cells with plasmid DNA, 1 50 μl aliquot of cells per transformation was thawed on ice. 1 - 5 μl of ligation/Gibson mixture, derived from a mix-ture of the previously described fragment types, was added to the cells before incubation on ice for 30 minutes. Cells were then heat shocked for 45 – 60 seconds at 42˚C in a dry heat block (Eppendorf) before returning to ice for 2 minutes. 250 μl SOB liquid media was added per tube and then incubated at 37˚C for 2 hours in a shaking incubator at 200 rpm. Cells were plated on appropriate antibiotic containing media (see appendix for details) at 1 μl, 10 μl and 289 μl volumes on 3 separate plates and incubated overnight at 37˚C.
Clone Screening
Bacterial clones were screened in multiple ways to ensure the correct plasmid sequence was present. Where suitable primers were available, cloned were screened using colony PCR. To further confirm that plasmids were correct, or where colony PCR was not suitable, plasmids were purified using a commercial miniprep kit and digested with restriction en-zymes and compared to expected digestion profiles. Finally, purified plasmids were sent for DNA sequencing with suitable primers to a commercial DNA sequencing service (GATC, Cologne, Germany).