Acta Cryst.(2001). E57, o463±o465 DOI: 101107/S1600536801006948 Norbert Nagelet al. C23H32N2O5
o463
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
Ramipril
Norbert Nagel,* Harald Schweitzer, HansjoÈrg Urbach, Winfried Heyse, Barbara MuÈller and Harald Berchtold
Aventis Pharma Deutschland GmbH, Global Pharmaceutical Development Analytical Sciences, Solid State Characterization, X-Ray Crystallography, D-65926 Frankfurt am Main, Germany
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.008 AÊ
Rfactor = 0.064
wRfactor = 0.085
Data-to-parameter ratio = 11.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of the antihypertensive and cardiovas-cular protective ramipril {systematic name: 1-[2-(1-ethoxy- carbonyl-3-phenylpropylamino)propionyl]octahydrocyclo-penta[b]pyrrole-2-carboxylic acid}, C23H32N2O5, has been determined. Within the crystal structure, strong hydrogen bonds connect the molecules into chains along the [100] direction. The preferred crystal growth along these chains causes a crystal morphology of very thin needles which previously prevented the crystal structure determination.
Comment
Ramipril, (I), is an ACE inhibitor, which can dramatically improve cardiovascular situations (Hallet al., 1997; Yusufet al., 2000). The active pharmaceutical ingredient is contained in numerous drugs with trade names such as Delix, Altace, Triace, Triatec, Delmuno, Unimax, Acovil and Vesdil. Crystals of ramipril adopt the morphology of long very thin needles and, during drug development in the eighties, all attempts to determine the crystal structure remained unsuccessful. The absolute con®guration (all S) was con®rmed by structure determinations of derivatives (Paulus, Geiger et al., 1987; Paulus, Henning & Urbach, 1987), taking into account thatl -alanine was one of the educts. In 1987, the last attempt to determine the crystal structure failed, since the collected diffraction data of the largest crystal found (0.7 0.05
0.03 mm) was too weak. A new data collection on the same crystal applying a rotating anode operated at 50 kV and 120 mA still yielded a data set with re¯ections still only observed up to about 1.0 AÊ, but the structure was immediately solved with direct methods.
Ramipril crystallizes in the orthorhombic space group
P212121 with one molecule in the asymmetric unit (Fig. 1). Since there are no atoms higher than oxygen present in the
Received 19 April 2001 Accepted 26 April 2001 Online 30 April 2001
organic papers
o464
Norbert Nagelet al. C23H32N2O5 Acta Cryst.(2001). E57, o463±o465crystal structure, it was not possible to recon®rm the absolute con®guration (Flack & Bernadinelli, 1999) and the absolute con®guration with ramipril in an all-S con®guration was chosen for re®nement. All bond lengths and angles (Table 1) are in the usual range of values (Orpenet al., 1994). The ®rst ®ve-membered ring (N01, C01±C04) is twisted about C01Ð C02 and the second ®ve-membered ring (C03±C07) is twisted about C5ÐC6. A Cremer & Pople puckering analysis (Cremer & Pople, 1975) yieldsQ= 0.275 AÊ and'= 61.48, and Q=
0.4407 AÊ and ' = 121.64, respectively. Despite the
coex-istence of a carboxylic acid group and an amino group, the neutral molecule and not the zwitterionic tautomer is found in the crystal. The carboxylic acid group adopts the frequently observed synplanar (s-cis) conformation (Leiserowitz, 1976; Gandour, 1981). An intramolecular hydrogen bond between the amino hydrogen H2 and the amido oxygen O03 is formed with contact distances N02 O03 2.829 (5) AÊ and H2 O03 2.43 (3) AÊ and an angle N02ÐH2 O03 114 (3). Within the
crystal structure, the molecules are connected in chains via
strong ±COOH NH± hydrogen bonds between the
carboxylic acid groups and the amino groups of adjacent molecules. The molecules within these chains are symmetry related by the 21screw axis parallel to the crystallographica axis. These hydrogen-bonded chains account for a preferred crystal growth in [100] direction and for the corresponding crystal morphology with the long needle axis coinciding with this direction. The intermolecular hydrogen bond exhibits distances O01 N02i 2.601 (5) AÊ and H1 N02i 1.58 (6) AÊ,
and an angle O01ÐH1 N02 168 (4) [symmetry code: (i)
x+1
2, 12ÿy, ÿz]. In addition, a short intermolecular distance C03ÐH03 O05iito the carbonyl oxygen of the ester group is found [C03 O5ii3.388 (6) AÊ, H03 O05 2.45 AÊ and C03Ð H03 O5ii161; symmetry code: (ii)3
2ÿx, ÿy, z+12] which
might be discussed as a weak intermolecular CÐH O
hydrogen bond (Desiraju, 1996; Desiraju & Steiner, 1998). The experimentally determined powder diffraction pattern of the sample agrees with the one calculated from the crystal structure model, which proves that the crystal structure determined here represents the crystalline phase of the bulk sample.
Experimental
The sample was recrystallized several times from ethanol±diisopropyl ether for further puri®cation. In the ®nal crystallization step, an ethanolic solution, which was almost saturated at 313 K, was slowly cooled to room temperature.
Crystal data C23H32N2O5
Mr= 416.51
Orthorhombic,P212121
a= 7.4845 (11) AÊ
b= 13.937 (2) AÊ
c= 22.012 (3) AÊ
V= 2296.0 (6) AÊ3
Z= 4
Dx= 1.205 Mg mÿ3
MoKradiation Cell parameters from 2316
re¯ections
= 2.4±18.2 = 0.09 mmÿ1
T= 293 (2) K Needle, colourless 0.700.050.03 mm
Data collection
Bruker SMART diffractometer
!scans
15 851 measured re¯ections 3291 independent re¯ections 1640 re¯ections withI> 2(I)
Rint= 0.166
max= 23.3
h=ÿ4!8
k=ÿ15!15
l=ÿ24!24 Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.064
wR(F2) = 0.085
S= 0.92 3291 re¯ections 279 parameters H atoms: see below
w= 1/[2(F
o2) + (0.0263P)2] whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.13 e AÊÿ3 min=ÿ0.14 e AÊÿ3
Absolute structure: Flack (1983) Flack parameter =ÿ2.0 (19)
Table 1
Selected geometric parameters (AÊ,).
O01ÐC08 1.311 (5)
O01ÐH1 1.04 (6)
O02ÐC08 1.202 (5)
O03ÐC09 1.226 (5)
O04ÐC13 1.334 (5)
O04ÐC14 1.463 (4)
O05ÐC13 1.194 (5)
N01ÐC09 1.347 (4)
N01ÐC01 1.466 (4)
N01ÐC04 1.467 (5)
N02ÐC10 1.472 (5)
N02ÐC12 1.482 (5)
N02ÐH2 0.77 (3)
C01ÐC08 1.518 (5)
C01ÐC02 1.534 (5)
C02ÐC03 1.515 (6)
C03ÐC05 1.537 (6)
C03ÐC04 1.546 (6)
C04ÐC07 1.523 (6)
C05ÐC06 1.513 (6)
C06ÐC07 1.505 (6)
C09ÐC10 1.529 (5)
C10ÐC11 1.532 (5)
C12ÐC13 1.513 (5)
C12ÐC16 1.527 (5)
C14ÐC15 1.485 (6)
C16ÐC17 1.524 (4)
C17ÐC18 1.506 (5)
C18ÐC19 1.374 (6)
C18ÐC23 1.381 (6)
C19ÐC20 1.378 (6)
C20ÐC21 1.365 (7)
C21ÐC22 1.378 (7)
C22ÐC23 1.371 (7)
C13ÐO04ÐC14 116.1 (4)
C09ÐN01ÐC01 126.3 (4)
C09ÐN01ÐC04 120.2 (4)
C01ÐN01ÐC04 113.4 (4)
C10ÐN02ÐC12 116.0 (3)
N01ÐC01ÐC08 111.5 (4)
N01ÐC01ÐC02 103.2 (3)
C08ÐC01ÐC02 113.5 (4)
C03ÐC02ÐC01 104.9 (4)
C02ÐC03ÐC05 117.6 (5)
C02ÐC03ÐC04 107.1 (4)
C05ÐC03ÐC04 102.8 (5)
N01ÐC04ÐC07 115.1 (4)
N01ÐC04ÐC03 103.5 (4)
C07ÐC04ÐC03 106.9 (4)
C06ÐC05ÐC03 105.0 (5)
C07ÐC06ÐC05 101.7 (5)
C06ÐC07ÐC04 105.9 (4)
O02ÐC08ÐO01 124.8 (4)
O02ÐC08ÐC01 124.0 (5)
O01ÐC08ÐC01 111.2 (4)
O03ÐC09ÐN01 122.6 (4)
O03ÐC09ÐC10 119.9 (4)
N01ÐC09ÐC10 117.3 (4)
N02ÐC10ÐC09 113.1 (4)
N02ÐC10ÐC11 109.0 (3)
C09ÐC10ÐC11 107.6 (3)
N02ÐC12ÐC13 107.4 (4)
N02ÐC12ÐC16 117.1 (3)
C13ÐC12ÐC16 108.3 (3)
O05ÐC13ÐO04 125.2 (5)
O05ÐC13ÐC12 125.2 (5)
O04ÐC13ÐC12 109.6 (5)
O04ÐC14ÐC15 110.5 (4)
C18ÐC17ÐC16 112.3 (3)
C19ÐC18ÐC23 117.9 (5)
C19ÐC18ÐC17 121.5 (5)
C23ÐC18ÐC17 120.6 (6)
C18ÐC19ÐC20 121.5 (5)
C21ÐC20ÐC19 120.1 (6)
C20ÐC21ÐC22 119.0 (7)
C23ÐC22ÐC21 120.8 (6)
C22ÐC23ÐC18 120.6 (6)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
O01ÐH1 N02i 1.04 (6) 1.58 (6) 2.601 (5) 168 (4)
N02ÐH2 O03 0.78 (3) 2.43 (3) 2.829 (5) 114 (3)
C03ÐH03 O05ii 0.98 2.45 3.388 (6) 161
Symmetry codes: (i)1
Data were originally collected to 542with a SMART 1 K CCD
area detector, but re¯ections were observed only up to about 1 AÊ/45
2. Re®nement was therefore performed with data up to 0.9 AÊ/472.
All H atoms were found in the electron-density difference map, but most of them were placed in idealized geometry with ®xed isotropic displacement parameters. Only the coordinates and isotropic displacement parameters of the H atom of the carboxylic acid group (H1) and of the H atom of the amino group (H2), which are involved in hydrogen bonds, were re®ned.
Data collection:SMART(Bruker, 1999); cell re®nement:SAINT+ (Bruker, 1999); data reduction:SAINT+; program(s) used to solve structure: SHELXS94 (Sheldrick, 1994); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL-PC XP(Bruker, 1998); software used to prepare material for publication:SHELXL97.
We thank K. Spieler for supporting the crystallization attempts and Professor B. SchoÈlkens for surveying the manuscript and his suggestions for improvement.
References
Bruker (1998). SHELXTL. Release 5.1. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (1999).SMART(Version 5.060) andSAINT+ (Version 6.01). Bruker AXS Inc., Madison, Wisconsin, USA.
Cremer, D. & Pople, J. A. (1975).J. Am. Chem. Soc.97, 1354±1358. Desiraju, G. R. (1996).Acc. Chem. Res.29, 441±449.
Desiraju, G. R. & Steiner, T. (1998).Chem. Commun.pp. 891±892. Flack, H. D. (1983).Acta Cryst.A39, 876±881.
Flack, H. D. & Bernadinelli, G. (1999).Acta Cryst.A55, 908±915. Gandour, R. D. (1981).Bioorg. Chem.10, 169±176.
Hall, A. S., Murray, G. D. & Ball, S. G. (1997).Lancet,349, 1493±1497. Leiserowitz, L. (1976).Acta Cryst.B32, 775±802.
Orpen, A. G., Brammer, L., Allen, F. H., Kennard, O., Watson, D. G. & Taylor, R. (1994).Structure Correlation, Vol. 2. edited by H.-B. BuÈrgi & J. D. Dunitz, Appendix A. Weinheim: VCH Publishers.
Paulus, E. F., Geiger, R., Henning, R, Teetz, V. & Urbach, H. (1987).Acta Cryst.C43, 938±941.
Paulus, E. F., Henning, R. & Urbach, H. (1987).Acta Cryst.C43, 941±945. Sheldrick, G. M. (1994).SHELXS94. University of GoÈttingen, Germany. Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Yusuf, S., Sleight, P., Pogue, J., Bosch, J., Davies, R. & Dagenais, G. (2000).N.
Engl. J. Med.342(3), 145±153.
Acta Cryst.(2001). E57, o463±o465 Norbert Nagelet al. C23H32N2O5
o465
organic papers
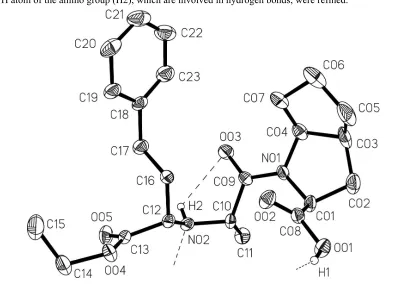
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o463–o465
supporting information
Acta Cryst. (2001). E57, o463–o465 [doi:10.1107/S1600536801006948]
Ramipril
Norbert Nagel, Harald Schweitzer, Hansj
ö
rg Urbach, Winfried Heyse, Barbara M
ü
ller and
Harald Berchtold
S1. Comment
Ramipril, (I), is an ACE inhibitor, which can dramatically improve cardiovascular outcomes (Hall et al., 1997; Yusuf et
al., 2000). The active pharmaceutical ingredient is contained in numerous drugs with trade names such as Delix, Altace,
Triace, Triatec, Delmuno, Unimax, Acovil and Vesdil. Crystals of ramipril adopt the morphology of long very thin
needles and during drug development in the eighties, all attempts to determine the crystal structure remained
unsuccessful. The absolute configuration (all S) was confirmed by structure determinations of derivatives (Paulus, Geiger
et al., 1987; Paulus, Henning & Urbach, 1987), taking into account, that L-alanine was one of the educts. In 1987, the last
attempt to determine the crystal structure failed, since the collected diffraction data of the largest crystal found (0.7 ×
0.05 × 0.03 mm) was too weak. A new data collection on the same crystal applying a rotating anode operated at 50 kV
and 120 mA still yielded a data set with reflections observed only up to about 1.0 Å, but the structure was solved with
direct methods, immediately.
Ramipril crystallizes in the orthorhombic space group P212121 with one molecule in the asymmetric unit (Fig. 1). Since
there are no atoms higher than oxygen present in the crystal structure, it was not possible to reconfirm the absolute
configuration (Flack & Bernadinelli, 1999) and the absolute structure with ramipril in ab all-S configuration was chosen
for refinement. All bond lengths and angles (Table 1) are in the usual range of values (Orpen et al., 1994). The first
five-membered ring (N01, C01–C04) is twisted on C01—C02 and the second five-five-membered ring (C03–C07) is twisted on C5
—C6. A Cremer & Pople puckering analysis (Cremer & Pople, 1975) yields Q = 0.275 Å and φ = 61.48°, and Q = 0.4407
Å and φ = 121.64°, respectively. Despite of the coexistence of a carboxylic acid group and an amino group, the neutral
molecule and not the zwitter ionic tautomer is found in the crystal. The carboxylic acid group adopts the frequently
observed synplanar (s-cis) conformation (Leiserowitz, 1976; Gandour, 1981). An intramolecular hydrogen bond between
the amino hydrogen H2 and the amido oxygen O03 is formed with contact distances N02···O03 2.829 (5) Å and H2···O03
2.43 (3) Å and an angle N02—H2···O03 114 (3)°. Within the crystal structure, the molecules are connected to chains via
strong –COOH···NH– hydrogen bonds between the caboxylic acid groups and the amino groups of adjacent molecules.
The molecules within these chains are symmetry related by the 21 screw axis parallel to the crystallographic a axis. These
hydrogen-bonded chains account for a preferred crystal growth in [10 0] direction and for the corresponding crystal
morphology with the long needle axis coinciding with this direction. The intermolecular hydrogen bond exhibits
distances O01···N02i 2.601 (5) Å and H1···N02i 1.58 (6) Å, and an angle O01—H1···N02 168 (4)° [symmetry code: (i) x +
1/2, 1/2 - y, -z]. In addition, a short intermolecular distance C03—H03···O05ii to the carbonyl oxygen of the ester group is
found [C03···O5ii 3.388 (6) Å, H03···O05 2.45 Å and C03—H03···O5ii 161°; symmetry code: (ii) 3/2 - x, -y, z + 1/2 which
supporting information
sup-2
Acta Cryst. (2001). E57, o463–o465
The experimentally determined powder diffraction pattern of the sample agrees with the one calculated from the crystal
structure model, which proves that the determined crystal structure represents the crystalline phase of the sample.
S2. Experimental
The sample was recystallized several times from ethanol–diisopropyl ether for further purification. In the final
crystallization step, an ethanolic solution, which was almost saturated at 313 K, was slowly cooled to room temperature.
S3. Refinement
Data was originally collected to 54° 2θ with a SMART 1 K CCD area detector, but reflections were observed only up to
about 1 Å/45° 2θ. Refinement was therefore done with data up to 0.9 Å/47° 2θ. All H atoms were found in the
electron-density difference map, but most of them were placed in idealized geometry with fixed isotropic displacement
parameters. Only the coordinates and isotropic displacement parameters of the H atom of the carboxylic acid group (H1)
[image:5.610.102.499.252.537.2]and of the H atom of the amino group (H2), which are involved in hydrogen bonds, were refined.
Figure 1
The molecular structure of ramipril (most H atoms omitted) showing anisotropic displacement ellipsoids at the 20%
probability level and the atom-numbering scheme. The dashed lines indicate hydrogen bonds.
1-[2-(1-ethoxycarbonyl-3-phenylpropylamino)propionyl]octahydrocyclopenta[b]- pyrrole-2-carboxylic acid
Crystal data
C23H32N2O5 Mr = 416.51
Orthorhombic, P212121 a = 7.4845 (11) Å
b = 13.937 (2) Å
c = 22.012 (3) Å
V = 2296.0 (6) Å3
Z = 4
F(000) = 896
Dx = 1.205 Mg m−3
Melting point: 109°C K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2316 reflections
supporting information
sup-3
Acta Cryst. (2001). E57, o463–o465
µ = 0.09 mm−1 T = 293 K
Needle, colourless 0.7 × 0.05 × 0.03 mm
Data collection
Bruker AXS four-circle diffractometer
Radiation source: rotating anode Graphite monochromator
ω scans
15851 measured reflections 3291 independent reflections
1640 reflections with I > 2σ(I)
Rint = 0.166
θmax = 23.3°, θmin = 2.4° h = −4→8
k = −15→15
l = −24→24
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.064 wR(F2) = 0.085 S = 0.92 3291 reflections 279 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0263P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.13 e Å−3
Δρmin = −0.14 e Å−3
Absolute structure: Flack (1983) Absolute structure parameter: −2.0 (19)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O01 0.8534 (4) 0.2836 (3) 0.11780 (14) 0.0676 (10) H1 0.939 (7) 0.336 (4) 0.101 (2) 0.109 (19)* O02 0.9727 (5) 0.1845 (2) 0.04956 (14) 0.0710 (10) O03 0.7016 (4) −0.0561 (2) −0.00152 (12) 0.0675 (10) O04 0.8473 (4) 0.2369 (2) −0.17916 (14) 0.0729 (10) O05 0.6397 (4) 0.1236 (2) −0.19571 (14) 0.0803 (11) N01 0.7448 (5) 0.0376 (2) 0.08079 (16) 0.0547 (10) N02 0.5954 (6) 0.1032 (3) −0.07252 (18) 0.0526 (12) H2 0.589 (5) 0.049 (2) −0.0805 (16) 0.039 (15)* C01 0.7351 (6) 0.1286 (3) 0.11407 (18) 0.0584 (12)
H01 0.6144 0.1553 0.1105 0.070*
C02 0.7694 (8) 0.0984 (4) 0.1801 (2) 0.0944 (17)
supporting information
sup-4
Acta Cryst. (2001). E57, o463–o465
H022 0.6579 0.0846 0.2008 0.113*
C03 0.8840 (9) 0.0091 (4) 0.1756 (2) 0.0900 (17)
H03 0.8515 −0.0358 0.2080 0.108*
C04 0.8450 (7) −0.0370 (3) 0.1131 (2) 0.0692 (15)
H04 0.7715 −0.0947 0.1180 0.083*
C05 1.0878 (10) 0.0224 (5) 0.1744 (3) 0.119 (2)
H051 1.1194 0.0833 0.1560 0.143*
H052 1.1366 0.0201 0.2152 0.143*
C06 1.1574 (8) −0.0604 (5) 0.1366 (3) 0.115 (2)
H062 1.2775 −0.0480 0.1220 0.138*
H061 1.1565 −0.1199 0.1594 0.138*
C07 1.0250 (7) −0.0628 (3) 0.0852 (2) 0.0769 (15)
H071 1.0207 −0.1262 0.0671 0.092*
H072 1.0575 −0.0168 0.0541 0.092*
C08 0.8697 (7) 0.2008 (3) 0.0902 (2) 0.0520 (12) C09 0.6838 (5) 0.0214 (3) 0.0242 (2) 0.0515 (13) C10 0.5754 (5) 0.1012 (3) −0.00601 (18) 0.0483 (12)
H10 0.6131 0.1632 0.0106 0.058*
C11 0.3784 (5) 0.0845 (3) 0.00971 (18) 0.0705 (14)
H111 0.3414 0.0232 −0.0056 0.106*
H112 0.3069 0.1341 −0.0084 0.106*
H113 0.3635 0.0859 0.0530 0.106*
C12 0.7651 (6) 0.1439 (3) −0.09581 (17) 0.0468 (11)
H12 0.7865 0.2047 −0.0747 0.056*
C13 0.7382 (8) 0.1659 (4) −0.1625 (2) 0.0581 (14) C14 0.8554 (8) 0.2576 (4) −0.2443 (2) 0.0966 (19)
H141 0.8798 0.3253 −0.2503 0.116*
H142 0.7409 0.2432 −0.2628 0.116*
C15 0.9973 (9) 0.1995 (4) −0.2738 (2) 0.125 (2)
H151 1.1101 0.2128 −0.2548 0.188*
H152 1.0037 0.2157 −0.3162 0.188*
H153 0.9697 0.1326 −0.2695 0.188*
C16 0.9326 (5) 0.0823 (3) −0.08925 (17) 0.0499 (12)
H161 1.0351 0.1199 −0.1018 0.060*
H162 0.9483 0.0666 −0.0466 0.060*
C17 0.9309 (6) −0.0108 (3) −0.12562 (19) 0.0674 (13)
H171 0.8300 −0.0495 −0.1129 0.081*
H172 0.9154 0.0041 −0.1683 0.081*
C18 1.1002 (7) −0.0677 (3) −0.1175 (2) 0.0586 (13) C19 1.2530 (9) −0.0455 (3) −0.14919 (19) 0.0693 (14)
H19 1.2520 0.0061 −0.1760 0.083*
C20 1.4077 (8) −0.0979 (5) −0.1421 (3) 0.0842 (17)
H20 1.5082 −0.0830 −0.1651 0.101*
C21 1.4137 (11) −0.1717 (5) −0.1014 (3) 0.100 (2)
H21 1.5191 −0.2057 −0.0952 0.119*
C22 1.2611 (11) −0.1951 (4) −0.0696 (3) 0.0953 (19)
H22 1.2632 −0.2462 −0.0425 0.114*
supporting information
sup-5
Acta Cryst. (2001). E57, o463–o465
H23 1.0043 −0.1611 −0.0560 0.099*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O01 0.080 (3) 0.060 (2) 0.062 (2) −0.0136 (19) 0.0148 (19) −0.009 (2) O02 0.086 (3) 0.058 (2) 0.069 (2) −0.0111 (18) 0.025 (2) −0.0078 (19) O03 0.088 (3) 0.0458 (18) 0.069 (2) −0.0039 (17) −0.0168 (17) −0.0041 (18) O04 0.089 (3) 0.079 (2) 0.051 (2) 0.003 (2) 0.005 (2) 0.0188 (18) O05 0.095 (3) 0.099 (3) 0.046 (2) −0.003 (2) −0.0097 (19) −0.013 (2) N01 0.079 (3) 0.046 (2) 0.039 (2) −0.005 (2) −0.006 (2) 0.006 (2) N02 0.063 (3) 0.041 (3) 0.054 (3) 0.002 (3) −0.006 (2) −0.010 (2) C01 0.085 (4) 0.054 (3) 0.037 (3) −0.011 (3) 0.012 (3) 0.000 (3) C02 0.151 (5) 0.081 (4) 0.051 (3) −0.013 (4) 0.012 (4) 0.002 (3) C03 0.138 (6) 0.090 (5) 0.042 (3) −0.023 (4) −0.022 (4) 0.020 (3) C04 0.082 (4) 0.061 (4) 0.064 (4) −0.011 (3) −0.012 (3) 0.018 (3) C05 0.153 (7) 0.118 (5) 0.088 (5) −0.020 (5) −0.069 (5) 0.009 (4) C06 0.120 (5) 0.110 (5) 0.114 (5) −0.006 (4) −0.049 (5) 0.038 (4) C07 0.084 (4) 0.068 (3) 0.079 (4) −0.004 (3) −0.022 (4) 0.024 (3) C08 0.059 (4) 0.054 (3) 0.043 (3) −0.003 (3) −0.007 (3) 0.000 (3) C09 0.056 (3) 0.045 (3) 0.053 (3) −0.007 (3) −0.001 (3) 0.013 (3) C10 0.059 (3) 0.053 (3) 0.033 (3) −0.007 (3) 0.001 (2) −0.006 (2) C11 0.055 (3) 0.083 (3) 0.073 (3) −0.005 (3) 0.006 (3) 0.001 (3) C12 0.053 (3) 0.045 (3) 0.043 (3) −0.001 (3) −0.005 (3) −0.010 (2) C13 0.071 (4) 0.062 (4) 0.042 (4) 0.020 (3) 0.005 (3) −0.003 (3) C14 0.115 (5) 0.113 (5) 0.062 (4) 0.018 (4) 0.010 (4) 0.031 (3) C15 0.140 (6) 0.166 (6) 0.069 (4) 0.015 (5) 0.021 (4) 0.003 (4) C16 0.053 (3) 0.052 (3) 0.045 (3) 0.007 (3) −0.006 (2) −0.001 (2) C17 0.069 (3) 0.069 (3) 0.064 (3) 0.014 (3) −0.010 (3) −0.015 (3) C18 0.063 (4) 0.050 (3) 0.062 (4) 0.011 (3) −0.007 (3) −0.024 (3) C19 0.079 (4) 0.060 (3) 0.069 (3) 0.014 (4) −0.003 (4) −0.021 (3) C20 0.072 (5) 0.077 (4) 0.104 (5) 0.003 (4) 0.002 (4) −0.041 (4) C21 0.106 (6) 0.065 (5) 0.128 (6) 0.027 (4) −0.038 (5) −0.035 (4) C22 0.110 (6) 0.062 (4) 0.113 (5) 0.021 (5) −0.015 (5) 0.004 (3) C23 0.091 (5) 0.069 (4) 0.088 (4) 0.011 (4) −0.002 (4) −0.014 (4)
Geometric parameters (Å, º)
O01—C08 1.311 (5) C09—C10 1.529 (5)
O01—H1 1.04 (6) C10—C11 1.532 (5)
O02—C08 1.202 (5) C10—H10 0.9800
O03—C09 1.226 (5) C11—H111 0.9600
O04—C13 1.334 (5) C11—H112 0.9600
O04—C14 1.463 (4) C11—H113 0.9600
O05—C13 1.194 (5) C12—C13 1.513 (5)
N01—C09 1.347 (4) C12—C16 1.527 (5)
N01—C01 1.466 (4) C12—H12 0.9800
supporting information
sup-6
Acta Cryst. (2001). E57, o463–o465
N02—C10 1.472 (5) C14—H141 0.9700
N02—C12 1.482 (5) C14—H142 0.9700
N02—H2 0.77 (3) C15—H151 0.9600
C01—C08 1.518 (5) C15—H152 0.9600
C01—C02 1.534 (5) C15—H153 0.9600
C01—H01 0.9800 C16—C17 1.524 (4)
C02—C03 1.515 (6) C16—H161 0.9700
C02—H021 0.9700 C16—H162 0.9700
C02—H022 0.9700 C17—C18 1.506 (5)
C03—C05 1.537 (6) C17—H171 0.9700
C03—C04 1.546 (6) C17—H172 0.9700
C03—H03 0.9800 C18—C19 1.374 (6)
C04—C07 1.523 (6) C18—C23 1.381 (6)
C04—H04 0.9800 C19—C20 1.378 (6)
C05—C06 1.513 (6) C19—H19 0.9300
C05—H051 0.9700 C20—C21 1.365 (7)
C05—H052 0.9700 C20—H20 0.9300
C06—C07 1.505 (6) C21—C22 1.378 (7)
C06—H062 0.9700 C21—H21 0.9300
C06—H061 0.9700 C22—C23 1.371 (7)
C07—H071 0.9700 C22—H22 0.9300
C07—H072 0.9700 C23—H23 0.9300
C08—O01—H1 114 (2) C09—C10—H10 109.1
C13—O04—C14 116.1 (4) C11—C10—H10 109.1
C09—N01—C01 126.3 (4) C10—C11—H111 109.5
C09—N01—C04 120.2 (4) C10—C11—H112 109.5
C01—N01—C04 113.4 (4) H111—C11—H112 109.5
C10—N02—C12 116.0 (3) C10—C11—H113 109.5
C10—N02—H2 102 (3) H111—C11—H113 109.5
C12—N02—H2 110 (3) H112—C11—H113 109.5
N01—C01—C08 111.5 (4) N02—C12—C13 107.4 (4)
N01—C01—C02 103.2 (3) N02—C12—C16 117.1 (3)
C08—C01—C02 113.5 (4) C13—C12—C16 108.3 (3)
N01—C01—H01 109.5 N02—C12—H12 107.9
C08—C01—H01 109.5 C13—C12—H12 107.9
C02—C01—H01 109.5 C16—C12—H12 107.9
C03—C02—C01 104.9 (4) O05—C13—O04 125.2 (5)
C03—C02—H021 110.8 O05—C13—C12 125.2 (5)
C01—C02—H021 110.8 O04—C13—C12 109.6 (5)
C03—C02—H022 110.8 O04—C14—C15 110.5 (4)
C01—C02—H022 110.8 O04—C14—H141 109.5
H021—C02—H022 108.8 C15—C14—H141 109.5
C02—C03—C05 117.6 (5) O04—C14—H142 109.5
C02—C03—C04 107.1 (4) C15—C14—H142 109.5
C05—C03—C04 102.8 (5) H141—C14—H142 108.1
C02—C03—H03 109.6 C14—C15—H151 109.5
supporting information
sup-7
Acta Cryst. (2001). E57, o463–o465
C04—C03—H03 109.6 H151—C15—H152 109.5
N01—C04—C07 115.1 (4) C14—C15—H153 109.5
N01—C04—C03 103.5 (4) H151—C15—H153 109.5
C07—C04—C03 106.9 (4) H152—C15—H153 109.5
N01—C04—H04 110.4 C17—C16—C12 115.0 (3)
C07—C04—H04 110.4 C17—C16—H161 108.5
C03—C04—H04 110.4 C12—C16—H161 108.5
C06—C05—C03 105.0 (5) C17—C16—H162 108.5
C06—C05—H051 110.7 C12—C16—H162 108.5
C03—C05—H051 110.7 H161—C16—H162 107.5
C06—C05—H052 110.7 C18—C17—C16 112.3 (3)
C03—C05—H052 110.7 C18—C17—H171 109.1
H051—C05—H052 108.8 C16—C17—H171 109.1
C07—C06—C05 101.7 (5) C18—C17—H172 109.1
C07—C06—H062 111.4 C16—C17—H172 109.1
C05—C06—H062 111.4 H171—C17—H172 107.9
C07—C06—H061 111.4 C19—C18—C23 117.9 (5)
C05—C06—H061 111.4 C19—C18—C17 121.5 (5)
H062—C06—H061 109.3 C23—C18—C17 120.6 (6)
C06—C07—C04 105.9 (4) C18—C19—C20 121.5 (5)
C06—C07—H071 110.6 C18—C19—H19 119.2
C04—C07—H071 110.6 C20—C19—H19 119.2
C06—C07—H072 110.6 C21—C20—C19 120.1 (6)
C04—C07—H072 110.6 C21—C20—H20 120.0
H071—C07—H072 108.7 C19—C20—H20 120.0
O02—C08—O01 124.8 (4) C20—C21—C22 119.0 (7)
O02—C08—C01 124.0 (5) C20—C21—H21 120.5
O01—C08—C01 111.2 (4) C22—C21—H21 120.5
O03—C09—N01 122.6 (4) C23—C22—C21 120.8 (6)
O03—C09—C10 119.9 (4) C23—C22—H22 119.6
N01—C09—C10 117.3 (4) C21—C22—H22 119.6
N02—C10—C09 113.1 (4) C22—C23—C18 120.6 (6)
N02—C10—C11 109.0 (3) C22—C23—H23 119.7
C09—C10—C11 107.6 (3) C18—C23—H23 119.7
N02—C10—H10 109.1
supporting information
sup-8
Acta Cryst. (2001). E57, o463–o465
C02—C03—C04—N01 12.1 (5) C16—C12—C13—O05 97.8 (5) C05—C03—C04—N01 −112.5 (5) N02—C12—C13—O04 153.3 (3) C02—C03—C04—C07 134.0 (4) C16—C12—C13—O04 −79.4 (4) C05—C03—C04—C07 9.4 (5) C13—O04—C14—C15 −89.9 (5) C02—C03—C05—C06 −149.4 (4) N02—C12—C16—C17 64.8 (4) C04—C03—C05—C06 −32.1 (5) C13—C12—C16—C17 −56.8 (5) C03—C05—C06—C07 42.6 (5) C12—C16—C17—C18 179.5 (4) C05—C06—C07—C04 −36.1 (5) C16—C17—C18—C19 −80.5 (5) N01—C04—C07—C06 130.8 (4) C16—C17—C18—C23 99.3 (5) C03—C04—C07—C06 16.6 (5) C23—C18—C19—C20 0.6 (6) N01—C01—C08—O02 −0.9 (6) C17—C18—C19—C20 −179.6 (4) C02—C01—C08—O02 115.1 (5) C18—C19—C20—C21 −2.3 (7) N01—C01—C08—O01 176.4 (4) C19—C20—C21—C22 2.7 (8) C02—C01—C08—O01 −67.6 (5) C20—C21—C22—C23 −1.4 (9) C01—N01—C09—O03 177.7 (4) C21—C22—C23—C18 −0.3 (8) C04—N01—C09—O03 3.3 (6) C19—C18—C23—C22 0.7 (7) C01—N01—C09—C10 −7.4 (6) C17—C18—C23—C22 −179.1 (4)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O01—H1···N02i 1.04 (6) 1.58 (6) 2.601 (5) 168 (4)
N02—H2···O03 0.78 (3) 2.43 (3) 2.829 (5) 114 (3)
C03—H03···O05ii 0.98 2.45 3.388 (6) 161