PAIRWISE OXIDATIVE DEAROMATIZATION AND N-HYDROXYCARBAMATE DEHYDROGENATION: MOLECULAR COMPLEXITY VIA AN ACYLNITROSO DIELS–ALDER CYCLOADDITION CASCADE
INSPIRED BYTETRODOTOXIN
Steffen N. Good
A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry.
Chapel Hill 2018
Approved by:
Jeffrey S. Johnson
Sidney M. Wilkerson-Hill
Michel R. Gagné
Alexander J. M. Miller
ABSTRACT
Steffen N. Good: Development and Application of an Oxidative Dearomatization/Acylnitroso Diels–Alder Cycloaddition Cascade Toward the Total Synthesis of (±)-Tetrodotoxin
(Under the direction of Jeffrey S. Johnson)
I. Highly Functionalized Tricyclic Oxazinanones via Pairwise Oxidative Dearomatization and N-Hydroxycarbamate Dehydrogenation: Molecular Diversity Inspired by Tetrodotoxin. Benzenoids in principle represent attractive and abundant starting materials for the preparation of
substituted cyclohexanes; however, the synthetic tools available for overcoming the considerable aromatic
energies inherent to these building blocks limit the available product types. Drawing inspiration from the
complex natural product tetrodotoxin, we demonstrate access to heretofore unknown heterotricyclic
structures by leveraging oxidative dearomatization of 2-hydroxymethyl phenols with concurrent
N-hydroxycarbamate dehydrogenation using a common oxidant. The pairwise-generated, mutually reactive
species then participate in a second stage acylnitroso DielsAlder cycloaddition. The reaction chemistry of
the derived [2.2.2]-oxazabicycles, bearing four orthogonal functional groups and three stereogenic centers,
is shown to yield considerable diversity in downstream products.
II. Oxidative Dearomatization/Acylnitroso Diels–Alder Cycloaddition Cascade: An Approach Toward the Total Synthesis of (±)-Tetrodotoxin
Tetrodamine, a common conceptual intermediate en route to tetrodotoxin, represents a challenging
cycloaddition cascade to access a highly functionalized cyclohexanone intermediate bearing structural and
ACKNOWLEDGEMENTS
Firstly, I would like to thank my family for their support. I would especially like to thank my mother,
Sherry Good. We may not always agree, but I have tremendous respect for the sacrifices you have made
for my brothers and I. I would certainly not be who I am today without you. I would also like to thank my
older brother, Justin Good. You were a role model to me growing up and I aspired to be more like you in
many ways. My interest in chemistry, and science in general, stems in part from you.
I would like to thank Ashley Kretsch. You have helped me grow as a person, challenging me and
providing different perspectives. Whether I had a good day or a bad day, you always made me smile.
Although I am sincerely thankful for our time together spent exploring new places and new foods, I am most
thankful for the little things – four years of bad jokes, walks around the neighborhood, workout dates,
discussing chemistry on the bus, and impromptu Sunday debates. Simply put, you have made my graduate school experience “pretty good.”
I would also like to thank the Johnson group members and alumni with whom I overlapped for
helping me develop as a chemist. Whether it was helping me prepare for my preliminary candidacy exam,
helping me with revisions to my manuscript/presentations, or discussing chemistry over a cold beer,
feedback from the group was invaluable. I would like to especially thank Kendrick Smith, Blane Zavesky,
and Jacob Robins. Kendrick Smith: for your willingness to discuss potential ideas and for being an
incredible literature resource. Blane Zavesky: for helpful discussion regarding the synthesis of tetrodotoxin.
Jacob Robins: for taking over the synthesis of tetrodotoxin. I have no doubt you will be able to finish it.
Finally, I would like to acknowledge my PI, Jeff Johnson. I am sincerely grateful to have learned
and developed under your guidance. The freedom you allow your students to explore and develop their
projects fosters an atmosphere of independent learning and problem solving while your knowledge and
expertise is an invaluable resource. Your practical approach to challenging/significant problems in synthetic
TABLE OF CONTENTS
LIST OF TABLES...ix
LIST OF FIGURES AND SCHEMES...x
LIST OF ABBREVIATIONS AND SYMBOLS...xii
CHAPTER ONE HIGHLY FUNCTIONALIZED TRICYCLIC OXAZINANONES VIA PAIRWISE OXIDATIVE DEAROMATIZATION AND N-HYDROXYCARBAMATE DEHYDROGENATION: MOLECULAR DIVERSITY INSPIRED BY TETRODOTOXIN...………1
1.1 Introduction...…1
1.2 Background...1
1.2.1 History and Retrosynthetic Analysis of Tetrodotoxin……….1
1.3 Results and Discussion………...3
1.3.1 Acylnitroso Diels–Alder Cycloaddition Optimization………..3
1.3.2 Development of a One-pot Oxidation/Acylnitroso Diels–Alder Cycloaddition………..5
1.3.3 Substrate Scope and Dienophile Facial Selectivity………...6
1.3.4 Secondary Transformations of the [2.2.2]-cycloadducts....………..7
1.4 Conclusions ………..8
1.5 Experimental Details………....8
REFERENCES………...29
CHAPTER TWO OXIDATIVE DEAROMATIZATION/ACYLNITROSO DIELS–ALDER CYCLOADDITION CASCADE: AN APPROACH TOWARD THE TOTAL SYNTHESIS OF (±)-TETRODOTOXIN...32
2.1 Introduction…....………....………...…...32
2.1.1 Previous Syntheses of Tetrodotoxin………...32
2.2 Synthesis of a Functionalized Benzenoid Starting Material....………...34
2.3.1 Dihydroxylation of the [2.2.2]-Bicycle Scaffold...…...………...38
2.3.2 Installation of the Glycolate in the [2.2.2]-Bicycle Scaffold……….39
2.3.3 Ring Opening of the Epoxide and Ketone Reduction in the [2.2.2]-Bicycle Scaffold…….42
2.3.3 Reductive N–O bond cleavage...………...43
2.4 Functionalization of the Cyclohexane Scaffold……….44
2.4.1 Protection of the syn-1,2-Diol and Amine Functionality in the Cyclohexane Scaffold…...44
2.4.2 Installation of the Glycolate Functionality in the Cyclohexane Scaffold………...46
2.4.3 Reductive Epoxide Opening Ketone Reduction in the Cyclohexane Scaffold...48
2.5 Conclusions………...52
2.6 Experimental Details………..52
LIST OF TABLES
LIST OF FIGURES AND SCHEMES
Scheme 1-1 Synthetic Strategy for Tetrodotoxin and Proposed Pairwise Oxidation/Cycloaddition...1
Scheme 1-2 Intramolecular Installation of Nitrogen at C8a..………..…....2
Scheme 1-3 Proposed Pairwise Oxidation/Cycloaddition Cascade………..…...3
Scheme 1-4 Chemoselective Reactions of Heterocycloadducts………7
Scheme 2-1 Intramolecular Installation of the Nitrogen at C8a………33
Scheme 2-2 Oxidation/Cycloaddition Cascade – Development of Three Stages……….34
Scheme 2-3 First Generation Synthesis of Salicyl Alcohol 2.16………..34
Scheme 2-4 Synthesis of Salicyl Alcohol 2.16from 2,3-Dimethylphenol………...35
Scheme 2-5 Synthesis of Salicyl Alcohol 2.16from 2,3,6-Trimethylphenol………...35
Scheme 2-6 Proposed Exhaustive Bromination/Hydrolysis...………36
Scheme 2-7 BenzylicBromination/Acetoxylation of 2,3,6-Trimethylphenol...…...………...36
Scheme 2-8 Second Generation Synthesis of Salicyl Alcohol 2.16………...…...37
Scheme 2-9 Application of the Oxidative Dearomatization/Acylnitroso Diels–Alder Cycloaddition Toward Tetrodotoxin...…...………...38
Figure 2-1 Functional Group Manipulations Needed to Access Tetrodamine from the [2.2.2]-Bicycle Scaffold...…...………...38
Scheme 2-10 Alkene Dihydroxylation of the [2.2.2]-Bicycle Scaffold………39
Scheme 2-11 Proposed Installation of the Glycolate via Nucleophilic Addition....………..39
Scheme 2-12 Nucleophilic Addition to Unsaturated Aldehyde 2.40………..40
Scheme 2-13 Nucleophilic Addition to Dioxolane Aldehyde 2.43………..41
Scheme 2-14 Proposed Installation of the Glycolate via Dihydroxylation of a 1,1-Di-X Alkene…………41
Scheme 2-15 SmI2 Promoted Reductive Opening of the Epoxyketone in the [2.2.2]-Bicycle Scaffold...………..42
Scheme 2-16 Pd/C Catalyzed Hydrogenolysis of Triol 2.37………...43
Scheme 2-17 Failed Reductive Cleavage of the N–O Bond in [2.2.2]-Bicycle Analogs……….44
Scheme 2-18 Functional Group Manipulations Required to Access Tetrodamine from the Cyclohexane Scaffold………...………44
Scheme 2-19 Chemoselective N-Boc Protection of Lactol 2.63...……….45
Scheme 2-21 Chemoselective N-Cbz Protection and 1,2-Diol Dioxolane Protection of lactol 2.63...….46
Scheme 2-22 Attempted Installation of the Glycolate Functionality via Reduction of Lactol 2.73………46
Scheme 2-23 Oxidative Cleavage and Vinyl addition to Lactol 2.73……….47
Scheme 2-24 Vinyl Addition to the bis-Dioxolane Cyclohexane Scaffold……….47
Scheme 2-25 Installation of the Glycolate Functionality via Ozonolysis in the Cyclohexane Scaffold………...48
Figure 2-2 Analogs Investigated for the Epoxide Opening and Ketone Reduction in the Cyclohexane Scaffold………...48
Scheme 2-26 SmI2 Promoted Reductive Opening of TES Epoxyketone 2.92……….49
Scheme 2-27 SmI2 Promoted Reductive Opening of Oxazolidinone Epoxyketone 2.93………...49
Scheme 2-28 SmI2 Promoted Reductive Opening of Glycolate Lactols 2.85 and 2.89...………....50
Scheme 2-29 Lactol Cleavage via Protection of Glycolate 2.85 and 2.89………50
Scheme 2-30 Proposed Lactol Reduction and Cp2TiCl Promoted Reductive Epoxide Opening....…….51
Scheme 2-31 Reduction of Glycolate Lactols 2.85 and 2.89………..51
LIST OF ABBREVIATIONS AND SYMBOLS
[M] generic metal
°C degrees Centigrade
µL microliter
13C NMR carbon nuclear magnetic resonance
1H NMR proton nuclear magnetic resonance
2,2-DMP 2,2-dimethoxypropane
4-MeOC6H4 4-methyoxyphenyl
ABNO 9-azabicyclo[3.3.1]nonane N-oxyl
Ac acyl
Ac2O acetic anhydride
AcOH acetic acid
AIBN azobisisobutyronitrile
ANDA acylnitroso Diels–Alder
BH3•THF borane tetrahydrofuran complex
BnEt3NCl N-benzyl-N,N,N-triethylammonium chloride
Boc tert-butoxycarbonyl
Boc2O di-tert-butylcarbonate
Br bromine atom
Br2 molecular bromine
C carbon atom
C6H6 benzene
cat. catalytic
catBH catechol borane
CBr4 carbon tetrabromide
Cbz benzyloxycarbonyl
CbzCl benzyl chloroformate
CH2(CN)2 malononitrile
CH2Cl2 methylene chloride
CH3NO2 nitromethane
CHCl3 chloroform
CN– cyanide
CO2 carbon dioxide
conv conversion
CuCl2 copper(II) chloride
D deuterium atom
d doublet
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
dd doublet of doublets
ddd doublet of doublet of doublets
decomp decomposition
DMAP 4-(dimethylamino)pyridine
DMF dimethylformamide
DMP Dess-Martin periodinane
dq doublet of quartets
dr diastereomeric ratio
equiv equivalents
Et2O diethyl ether
EtOAc ethyl acetate
EtOH ethanol
g gram
h hours
H hydrogen atom
H+ proton or generic acid source
H2O water
HCl hydrochloric acid
HCO2Na sodium formate
hept heptet
HONH2•HCl hydroxylamine hydrochloride
Hz hetz
I2 molecular iodine
IBX 2-iodoxybenzoic acid
iPr2NEt diisopropylethylamine
iPr2NH diisopropylamine
K2CO3 potassium carbonate
KCN potassium cyanide
KOH potassium hydroxide
L liter
LA Lewis acid
LiAlH4 lithium aluminum hydride
LiOtBu lithium tert-butoxide
m multiplet
mCPBA meta-chloroperoxybenzoic acid
Me methyl
Me2NH•BH3 borane dimethylamine complex
Me2S dimethylsulfide
Me3SO•I trimethylsulfoxonium iodide
MeCN acetonitrile
MeNO2 nitromethane
MeOH methanol
Mg magnesium atom
MgO magnesium(II) oxide
MHz megahertz
min minutes
mL milliliter
Mo(CO)6 molybdenum hexacarbonyl
mol. sieves molecular sieves
Ms methanesulfonyl
Na sodium metal
Na2S2O3 sodium thiosulfate
Na2SO4 sodium sulfate
NaBH3CN sodium cyanoborohydride
NaBH4 sodium borohydride
NaCN sodium cyanide
NaH sodium hydride
NaHCO3 sodium bicarbonate
NaIO4 sodium periodate
NaOAc sodium acetate
NaOH sodium hydroxide
NBS N-bromosuccinimide
nBu4NIO4 N,N,N,N-tetrabutylammonium periodate
nBuLi n-butyllithium
NH4Cl ammonium chloride
NiCl nickel(I) chloride
NMO N-methylmorpholine N-oxide
NOE nuclear Overhauser effect
Nu nucleophile
O oxygen
O3 ozone
OsO4 osmium tetroxide
Pd(OH)2 palladium(II) hydroxide
Pd/C palladium on carbon
Ph phenyl
PhI(OAc)2 (diacetoxyiodo)benzene
p-O2NC6H4COCl para-nitrophenyl chloroformate
PPh3 triphenylphosphine
ppm parts per million
q quartet
Rh rhodium
rt room temperature
s singlet
sec seconds
SmI2 samarium(II) iodide
t triplet
TBAF N,N,N,N-tetrabutylammonium fluoride
TBDPSCl tert-butyldiphenylchlorosilane
TBS tert-butyldimethylsilyl
TBSCl tert-butyldimethylchlorosilane
tBuNH2•BH3 borane tert-butylamine complex
tBuOH tert-butanol
tBuPh2Si tert-butyldiphenylsilyl
TEA triethylamine
TESCl triethylchlorosilane
TESOTf triethylsilyl trifluoromethanesulfonate
TFA trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography
TMS trimethylsilyl
TPAP N,N,N,N-tetrapropylammonium perruthenate
TsOH para-toluenesulfonic acid
TTX tetrodotoxin
UV ultraviolet
Zn Zinc metal
ZnCl2 zinc(II) chloride
1Chapter One: Highly Functionalized Tricyclic Oxazinanones via Pairwise Oxidative
Dearomatization and N-Hydroxycarbamate Dehydrogenation: Molecular Diversity Inspired by Tetrodotoxin*
1.1 Introduction
The latent functionality embedded within the benzene nucleus offers tantalizing opportunities to
chemists seeking to deploy the parent aromatic core structure as a point of departure for synthesizing chiral,
functionalized cyclohexanes. The key challenge thwarting the realization of this potential is the design not
only of transformations that overcome the formidable aromatic stabilization energy associated with
benzenoids,1-7 but also a suitable second stage reaction that takes advantage of the functionality, often of
tenuous stability, revealed in the first stage. The work described here uses the cyclohexane substructure
of the complex natural product tetrodotoxin (TTX, 1.1, Scheme 1-1) as an inspiration to develop a new oxidative dearomatization cascade sequence for the de novo creation of heterotricyclic compounds
presenting four orthogonal functional groups. We demonstrate that considerable product diversity is
available from this new platform.
1.2. Background
1.2.1 History and Retrosynthetic Analysis of Tetrodotoxin
Tetrodotoxin (1.1), a potent neurotoxin first isolated from puffer fish, has garnered significant attention since its structure was first elucidated in 1964 due to its unique biological properties and densely
functionalized structure (Scheme
1-1
).8 (−)-TTX inhibits voltage gated sodium ion channels and isconsequently a leading molecular probe for further study in this area.9
Additionally, (−)-TTX has been under
study as a treatment for chemotherapy-induced neuropathic pain.10 Since the first synthesis of (±)-TTX by
Kishi in 1972, there have been five total syntheses, each enabled by the development of creative tactics to
access the densely functionalized core.11-19 While the recent chemoenzymatic formal synthesis of (–)-TTX
by Hudlicky required only 22 steps,20 the synthesis of TTX remains challenging; all previous syntheses
require >30 steps. “Tetrodamine” (1.2,Scheme 1-1) is a common conceptual target in
Scheme 1-1. Synthetic Strategy for Tetrodotoxin and Proposed Pairwise Oxidation/Cycloaddition
tetrodotoxin syntheses.11-18 The C6–C7–C8–C8a stereotetrad comprises a challenging functional group
constellation. The C7/C8 syn-diol is suggestive of a cis alkene difunctionalization; direct dihydroxylation of
a suitably substituted benzene has been suggested as a potential entry point for tetrodotoxin20 and OsO4
-catalyzed alkene functionalization was realized en route to 1.2.18 Perhaps more nettlesome is the
C6/C8a syn-1,4-aminocyclohexanol where both heteroatoms reside at fully substituted stereogenic
centers (Scheme
1-1
). As such, the challenging installation of nitrogen at C8a has inspired a number of creative approaches typically utilizing intramolecular strategies (Scheme 1-2).11-14,16,18 In consideringretrosynthetic approaches, the presence of a C7/C8 alkene and antithetic reconnection of the C6/C8a amino alcohol (Scheme 1-1, red arrows) would create a retron for an acylnitroso Diels–Alder
cycloaddition.21 We envisioned oxidative remodeling of an achiral benzenoid could be triggered by Adler
oxidation of the corresponding salicyl alcohol 1.4.22 Trapping the resulting spiroepoxydienone 1.6 with a
concurrently generated dienophile 1.5 might trigger a second stage hetero-Diels–Alder cycloaddition (Scheme 1-3). An enabling feature of the envisioned strategy is that acylnitroso 1.5 and epoxydienone 1.6 could in principle be prepared by oxidation of N-hydroxycarbamate 1.323 and salicyl
alcohol 1.4, respectively, with a common oxidant.24 Development of this methodology would allow for the
Scheme 1-3. Proposed Pairwise Oxidation/Cycloaddition Cascade
rapid preparation of rigid, bicyclic products with four orthogonal functional groups from simple, aromatic
feedstock while potentially allowing for an expeditious, flexible synthesis of the tetrodamine substructure
and ultimately (±)-TTX and its structural congeners.25
1.3 Results and Discussion
1.3.1 Acylnitroso Diels–Alder Cycloaddition Optimization
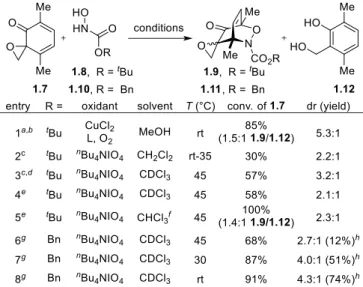
We began by investigating the proposed acylnitroso Diels–Alder cycloaddition (Table 1-1). The
known dimerization of spiroepoxydienones was a concern;26 therefore, spiroepoxydienone 1.7, a compound
bears an analogous substitution pattern to an idealized substrate for elaboration to TTX. Treating
spiroepoxydienone 1.7 with N-hydroxycarbamate 1.8 under Cu(II)-catalyzed aerobic oxidation conditions28
afforded a 1.5:1 mixture of the desired Diels–Alder cycloadduct 1.9 (5.4:1 dr) and salicyl alcohol 1.12 (Table 1-1, entry 1). Formation of alcohol 1.12 only occurred in the presence of 1.8, suggesting the in situ reduction of 1.7 could not be prevented under these reaction conditions. Stoichiometric quantities of tetra-n -butylammonium periodate (nBu4NIO4) in CH2Cl2 afforded cycloadduct 1.9 (2.3:1 dr) in low conversion
without any observable formation of 1.12 (entry 2). Repeating the reaction in CDCl3 at 45 °C showed trace
conversion to 1.9, but full consumption of 1.8 after 1 h. Based on these observations, we speculated that decomposition of the transient acylnitroso species, which has a lifetime on the order of 1 ms at infinite
dilution,23 was likely the source of the poor reactivity. Accordingly, we proposed the reactivity could be
enhanced by maintaining an excess of spiroepoxydienone 1.7 relative to the acylnitroso species; therefore, an additional 2 equiv of nBu4NIO4 were added followed by the dropwise addition of 1.8 (2 equiv) over 2 h,
affording cycloadduct 1.9 (3.2:1 dr) in 57% conversion
Table 1-1. Optimization of the Acylnitroso Diels–Alder Cycloaddition
(entry 3). When the reaction was conducted in CHCl3 (entry 5), phenol 1.12 was observed in similar
quantities relative to the Cu(II)-catalyzed aerobic oxidation conditions (entry 1). The variance in reactivities
by ethanol, typically present in CHCl3 as a stabilizer. Indeed, a similar effect was observed when methanol
was used as the solvent (entry 1). In light of these observations, we avoided these and similar reaction
media in subsequent experiments. Further optimization showed increased conversion of
1.7 using Cbz analog 1.10, but cycloadduct 1.11 was isolated in only 12% yield (entries 5, 6). Repeating the reaction at cooler temperatures resulted in increased conversions and isolated yields of 1.11 (entries 7, 8). The diminished yields at elevated temperatures indicated possible thermal instability of the cycloadducts. Specifically, retro Diels–Alder cycloaddition followed by incomplete recombination and
decomposition of the acylnitroso species was hypothesized. This hypothesis was later confirmed via a
crossover experiment (vide infra).
1.3.2 Development of a One-pot Oxidation/Acylnitroso Diels–Alder Cycloaddition
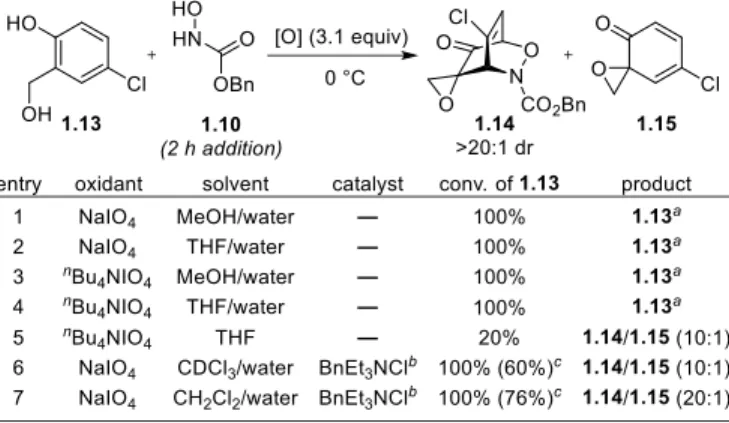
To explore the viability of a one-pot Adler oxidation/acylnitroso cycloaddition sequence, we added
a solution of 1.10 over 2 h to a mixture of salicyl alcohol 1.13 and NaIO4 in methanol/water (Table 1-2, entry
1). Full conversion of phenol 1.13 to spiroepoxydienone 1.15 was observed after 15 min, but complete reduction of 1.15 back to 1.13 was observed after 1 h. Screening nBu4NIO4 as the oxidant and
Table 1-2. Optimization of the One-pot Adler Oxidation/Acylnitroso Cycloaddition Protocol
removing methanol as a solvent returned identical results (entries 2-4). Excluding water from the reaction
afforded cycloadduct 1.14 (>20:1 dr), albeit in low conversion (entry 5). These results indicated that excess water was promoting the Adler oxidation while simultaneously promoting the reduction of intermediate
spiroepoxydienone in the presence of the acylnitroso species in water-miscible solvents. Collectively, these
(entry 6, 7). Indeed, using CH2Cl2 as the co-solvent afforded 1.14 in excellent yield with complete
consumption of 1.13 (entry 7).
1.3.3 Substrate Scope and Dienophile Facial Selectivity
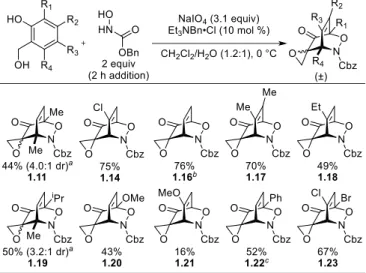
We next probed the scope of the corresponding one-pot procedure (Table 1-3). Readily available
electron-rich and electron-poor salicyl alcohols29-31 were tolerated, affording the derived cycloadducts in
good yield. For most substrates, the cycloadduct is initially formed with >20:1 dr; however, cycloadducts
1.11 and 1.19 were isolated as a mixture of equilibrating diastereomers (determined by chemical derivatization and selective 1D NOESY NMR analysis; see experimental details (Section 1.4) for details).
Table 1-3. Scope of the One-Pot Oxidative Dearomatization/Acylnitroso Cycloaddition Cascade
An X-ray diffraction study of bicyclic dihydrooxazine 1.9 (Table 1-1) revealed a regiochemical preference for the illustrated isomer, which matched that needed for extrapolation to TTX,27 and acylnitroso approach
from the methylene face of the epoxide of 1.7. The observed facial selectivity was surprising as C–C dienophile approach from the oxygen face of spiroepoxydienones is well documented.26 By contrast, an
X-ray diffraction study of dihydrooxazine 1.14 revealed acylnitroso approach from the oxygen face of the epoxide. We hypothesize that acylnitroso approach from the oxygen face of the epoxide is kinetically favored while the “methylene endo” product is the more thermodynamically stable diastereomer. This
hypothesis is additionally supported by the diminished pre-equilibration diastereoselectivities observed for
experiment between 1.19 and Boc–NO confirmed a retro-Diels–Alder/recombination sequence, providing a likely pathway for isomerization of the dihydroxazine cycloadducts (see section 1.5 for experimental
details of the crossover experiment and thermal isomerization). 1.3.4 Secondary Transformations of the [2.2.2]-cycloadducts
The reactivity of cycloadduct 1.16 was explored in a number of orthogonal transformations that independently probed each functional group (Scheme 1-4). Reduction of the ketone afforded alcohol 1.24 in good yield while ring-opening of the epoxide was achieved by treating ketone 1.16 with bromodimethylsulfonium bromide,32 yielding bromohydrin 1.25. Gram-scale dihydroxylation of 1.16 gave
Scheme 1-4. Chemoselective Reactions of Heterocycloadducts
diol 1.26 with high diastereoselection and matched stereochemistry needed for elaboration toward tetrodamine 1.2. Subsequent one-pot oxazinane reduction33 and epoxide ring-opening34 afforded
yield.35 Reductive fragmentation36 of epoxyoxime 1.29 afforded primary alcohol 1.30 and thence the
crystalline bis-(p-nitrobenzoate) 1.31. Corey-Chaykovsky epoxidation37 afforded bis-epoxide 1.32.
1.4 Conclusions
In summary, we developed a one-pot oxidative dearomatization/acylnitroso Diels-Alder
cycloaddition cascade sequence for the rapid preparation of rigid [2.2.2]-bicyclic products bearing four
orthogonal functional groups from simple, aromatic feedstocks. The synthetic utility of these new
cycloadducts was demonstrated through an array of chemoselective transformations.
1.5 Experimental Details Methods
General: Infrared (IR) spectra were obtained using a Jasco 460 Plus Fourier transform infrared spectrometer. Proton and carbon magnetic resonance spectra (1H NMR and 13C NMR) were recorded on
a Bruker model Avance 400 (1H NMR at 400 MHz), Bruker Avance III 500 (1H NMR at 500 MHz and 13C
NMR at 126 MHz), or a Bruker Avance III 600 (1H NMR at 600 MHz and 13C NMR at 151 MHz) spectrometer
with solvent resonance as the internal standard (1H NMR: CDCl3 at 7.26 ppm, (CD3)2CO at 2.05 ppm,
CD3OD at 3.31 ppm; 13C NMR: CDCl3 at 77.0 ppm, CD2Cl2 at 53.5 ppm, (CD3)2CO at 206 ppm, CD3OD at
49 ppm). 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, br s = broad singlet,
d = doublet, t = triplet, q = quartet, hept = heptet, dd = doublet of doublets, ddd = doublet of doublet of
doublets, dq = doublet of quartets, m = multiplet), coupling constants (Hz), and integration. Mass spectra
were obtained using a Thermo LTqFT mass spectrometer with electrospray introduction and external
calibration unless otherwise noted. All samples were prepared in methanol. Analytical thin layer
chromatography (TLC) was performed on Sorbent Technologies 250 µm glass-backed Silica Gel TLC
plates. Visualization was accomplished with UV light, KMnO4, ceric ammonium molybdate (CAM), and/or
Seebach’s stain followed by heating. Purification of the reaction products was carried out by flash
chromatography using Siliaflash-P60 silica gel (40-63μm) purchased from Silicycle. Unless otherwise
noted, all reactions were carried out under an atmosphere of dry nitrogen with magnetic stirring. Yield refers
to isolated yield of analytically pure material unless otherwise noted. Yields are reported for a specific
experiment and as a result may differ slightly from those found in figures, which are averages of at least
Materials:
General: Solvents were not dried prior to use unless specified as “anhydrous.” Anhydrous tetrahydrofuran (THF), diethyl ether (Et2O), and dichloromethane (CH2Cl2) were dried by passage through a column of
neutral alumina under nitrogen prior to use. Anhydrous dimethylformamide (DMF) was purchased from
Sigma-Aldrich and was used as received. Ethyl acetate (EtOAc) was purchased from Fisher Scientific and
was used as received.
Experimental Procedures:
5,8-Dimethyl-1-oxaspiro[2.5]octa-5,7-dien-4-one (1.7):A solution of NaIO4 (773 mg, 3.61 mmol) in water
(5 mL) was added to a solution of 2-hydroxy-3,5-dimethylbenzyl alcohol31 (500 mg, 3.29 mmol) in MeOH
(20 mL) at rt. The mixture was stirred for 1 h at rt, resulting in the formation of a precipitate. The reaction
was diluted with water (40 mL) and the aqueous layer was extracted with diethyl ether (3 × 40 mL). The
combined extracts were washed with brine (60 ml), dried over Na2SO4, filtered, and concentrated via rotary
evaporation. The crude material as purified via flash chromatography on silica gel (EtOAc:hexanes 20:80)
to afford 1.7 as a yellow oil (401 mg, 81%). Analytical data for 1.7:1H NMR (400 MHz, CDCl
3) δ 6.92 (d, J
= 6.3 Hz, 1H), 6.24 (d, J = 6.3, 1, 1H), 3.22 (d, J = 8.3 Hz, 1H), 3.16 (d, J = 8.3 Hz, 1H), 1.90 (s, 3H), 1.80
(s, 3H); 13C NMR (151 MHz, CDCl
3) δ 196.4, 144.7, 138.9, 131.7, 123.8, 58.7, 58.7, 16.1, 15.1; IR (thin
film, cm-1) 2980, 2948, 2921, 1662, 1640; TLC (EtOAc:hexanes 20:80): Rf = 0.41; HRMS (ESI+): Calcd.
for C9H11O2: ([M+H]): 151.0759, Found: 151.0756.
5-Isopropyl-8-methyl-1-oxaspiro[2.5]octa-5,7-dien-4-one (1.33): NaIO4 (39 mg, 0.18 mmol) was added
to a solution of 2-hydroxy-3-isopropyl-6-methylbenzyl alcohol30 (20 mg, 0.11 mmol) in MeOH/H2O (1:1, 2
mL). The reaction was stirred at rt under air for 1 h then diluted with water (3 mL). The aqueous layer was
extracted with diethyl ether (3 × 3 mL) and the combined extracts were washed with brine, dried over
chromatography on silica gel (EtOAc:hexanes 10:90 to 20: 80) to afford 1.33 as a yellow oil (20 mg, 67 %). Analytical data for 1.33:1H NMR(400 MHz, CDCl
3) δ 6.86 (d, J = 6.5 Hz, 1H), 6.28 (dq, J = 6.5, 1.5 Hz, 1H),
3.20 (d, J = 8.2 Hz, 1H), 3.13 (d, J = 8.2 Hz, 1H), 2.91 (hept, J = 6.9 Hz, 1H), 1.79 (d, J = 1.5 Hz, 3H), 1.07
(d, J = 2.7 Hz, 3H), 1.06 (d, J = 2.7 Hz, 3H); 13C NMR (151 MHz, CDCl
3) δ 195.3, 144.3, 141.6, 135.5,
123.9, 59.0, 58.6, 26.1, 21.8, 21.6, 16.1; IR (thin film, cm-1) 2961, 2872, 1662, 1641, 1581; TLC
(EtOAc:hexanes 20:80): Rf = 0.45; HRMS (ESI+): Calcd. for C11H15O2: ([M+H]): 179.1072, Found:
179.1065.
Preparation of [2.2.2]-Bicyclic Products:
General Procedure A:
A solution of NaIO4 (663 mg, 3.10 mmol) in water (10 mL) was added to a mixture of the salicyl alcohol
(1.00 mmol) and N-benzyl-N,N,N-triethylammonium chloride (23 mg, 0.10 mmol) in CH2Cl2 (7.5 mL) at 0
°C while stirring vigorously. After 15 min, a solution of BnO2CNHOH (2.00 mmol) in CH2Cl2 (5 mL) was
added over 2 h via syringe pump, maintaining the reaction temperature between 0-5 °C. After complete
addition, the reaction was stirred for 10 min. The phases were partitioned and the aqueous phase was
extracted with CH2Cl2 (2 × 10 mL). The combined extracts were washed with brine (15 mL), dried over
Na2SO4, filtered, and concentrated by rotary evaporation (the rotary evaporation water bath was maintained
at ≤ 30 °C). The crude product was purified via flash chromatography on silica gel. All cycloadducts were
stored at -20 °C.
Note: The acylnitroso Diels–Alder cycloadducts are generally not thermally stable above 30 °C due to
appreciable retro-Diels–Alder and subsequent decomposition of the acylnitroso species and isomerization
of the spiroepoxydienones to the corresponding salicyl aldehydes. For this reason, the melting points of
crystalline adducts are not reported.
purified by flash chromatography on silica gel (EtOAc:hexanes 10:90 to 20:80) to afford 1.11 as an equilibrating mixture of diastereomers as a yellow oil (1.5:1 dr determined by 1H NMR, 137 mg, 44%). Note
1: the reported 1.5:1 dr refers to the ratio observed at the time the 1H NMR was taken and was calculated
by comparison of the integration of the resonances at δ 6.72 (minor diastereomer) and δ 6.69 (major
diastereomer). An equilibrium (≈4:1 dr) is reached after a few hours at rt. The equilibration can be slowed
but not stopped upon storage at -20 °C (neat); however, the mixture of diastereomers is not thermally stable
and will slowly degrade upon prolonged storage at -20 °C. Note 2: the title compound elutes slightly after
benzyl alcohol (a byproduct from hydrolysis of the intermediate acylnitroso). Separation of from benzyl
alcohol can be achieved by holding the eluent at EtOAc:hexanes 15:85 until benzyl alcohol elutes. Only
definitively discernible peaks are reported in the 1H NMR spectra for each isomer. Analytical data for 1.11:
Major isomer,1H NMR(600 MHz, CDCl
3) δ 6.69 (d, J = 8.2 Hz, 1H), 6.21 (d, J = 8.2 Hz, 1H), 5.20 (d, J =
12.3 Hz, 2H), 5.15 (d, J = 12.3 Hz, 1H), 3.12 (d, J = 5.7 Hz, 1H), 3.09 (d, J = 5.7 Hz, 1H), 1.67 (s, 3H), 1.64
(s, 3H); Minor isomer,1H NMR (600 MHz, CDCl3) δ 6.72 (d, J = 8.3 Hz, 1H), 6.21 (d, J = 8.3 Hz, 1H), 5.13 (d, J = 12.4 Hz, 1H), 3.19 (d, J = 5.6 Hz, 1H), 3.05 (d, J = 5.6 Hz, 1H), 1.76 (s, 3H), 1.62 (s, 3H); Combined 13C NMR (151 MHz, CDCl
3) δ 196.6, 193.9, 160.0, 159.6, 144.0, 143.1, 135.5, 135.3, 130.1, 129.2, 128.6,
128.6, 128.5, 128.4, 128.3, 127.9, 127.8, 82.6, 82.2, 68.3, 68.2, 65.3, 64.3, 58.7, 55.8, 55.4, 49.1, 48.9,
15.9, 14.6; IR (thin film, cm-1) 2994, 2937, 1757, 1725, 1271, 1243; TLC (EtOAc:hexanes 30:70): Rf = 0.30;
HRMS (ESI+): Calcd. for C17H18NO5: ([M+H]): 316.1185, Found: 316.1184.
Benzyl 7-chloro-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate (1.14): The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 20:80) to afford 1.14 as a yellow oil (246 mg, 76%) which solidified upon storage. The title compound was recrystallized by slow evaporation from
MTBE/hexanes to afford crystals suitable for X-ray crystallographic analysis. Analytical data for 1.14:1H NMR (400 MHz, CDCl3) δ 7.38 (s, 5H), 6.54 (d, J = 6.5, 1H), 5.26 (d, J = 12.0 Hz, 1H), 5.21 (d, J = 12.0 Hz,
film, cm-1) 3031, 1761, 1716, 1222; TLC (EtOAc:hexanes 30:70): Rf = 0.29; HRMS (ESI+): Calcd. for
C15H12ClNNaO5: ([M+Na]): 344.0302, Found: 344.0294.
Benzyl 3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate (1.16):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 20:80 to 40:60) to afford 1.16 as a yellow oil that slowly solidified upon storage to afford a white amorphous solid (202 mg, 70%). Analytical data for 1.16:1H NMR (400 MHz, CDCl3) δ 7.42 – 7.32 (m, 5H), 6.91 (ddd, J = 8.1, 6.0, 1.7 Hz, 1H), 6.66 (ddd, J = 8.1, 6.0, 2.2
Hz, 1H), 5.25 (d, J = 12.1 Hz, 1H), 5.17 (d, J = 12.1 Hz, 1H), 4.97 (dd, J = 6.0, 1.7 Hz, 1H), 4.80 (dd, J =
6.0, 2.2 Hz, 1H), 3.30 (d, J = 5.9 Hz, 1H), 3.01 (d, J = 5.9 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 194.4,
158.4, 137.3, 134.9, 128.7, 128.6, 128.4, 128.2, 78.6, 68.9, 59.9, 53.5, 51.4; IR (thin film, cm-1) 3274, 1758,
1716, 1220; TLC (EtOAc:hexanes 50:50, 2% v/v TEA/Et2O neutralized plate): Rf = 0.34; HRMS (ESI+):
Calcd. for C15H13NNaO5: ([M+Na]): 310.0686, Found: 310.0692.
Benzyl 7,8-dimethyl-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate (1.17):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 20:80) to afford 1.17 as a yellow oil (240 mg, 76%) which slowly crystallized upon storage. Analytical data for 1.17:1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 5.22 (d, J = 12.1 Hz, 1H), 5.17 (d, J = 12.1 Hz, 1H), 4.72 (s, 1H), 4.48 (s, 1H), 3.26
(d, J = 5.7 Hz, 1H), 3.01 (d, J = 5.7 Hz, 1H), 1.88 (s, 3H), 1.86 (s, 3H); 13C NMR (151 MHz, CDCl
3) δ 194.9,
158.6, 140.2, 135.1, 128.7, 128.6, 128.6, 128.4, 83.4, 68.6, 64.4, 53.8, 50.5, 17.0, 15.1; IR (thin film, cm-1)
3310, 1751, 1716, 1241, 1226; TLC (EtOAc:hexanes 30:70): Rf = 0.34; HRMS (ESI+): Calcd. for
C17H18NO5: ([M+H]): 316.1185, Found: 316.1176.
desired. Analytical data for 1.18:1H NMR (400 MHz, CDCl
3) δ 7.36 (s, 5H), 6.17 (br s, 1H), 5.25 (d, J =
12.1 Hz, 1H), 5.16 (d, J = 12.1 Hz, 1H), 4.95 (d, J = 6.2 Hz, 1H), 4.57 (s, 1H), 3.28 (d, J = 5.9 Hz, 1H), 3.00
(d, J = 5.9 Hz, 1H), 2.52 – 1.93 (m, 2H), 1.03 (t, J = 7.3 Hz, 3H); 13C NMR (151 MHz, CDCl
3) δ 194.4, 158.5,
135.1, 128.7, 128.6, 128.5, 117.8, 117.7, 79.1, 68.7, 63.5, 53.6, 50.7, 27.6, 10.3; IR (thin film, cm-1) 2968,
1757, 1716, 1224; TLC (EtOAc:hexanes 30:70): Rf = 0.34; HRMS (ESI+): Calcd. for C
17H18NO5: ([M+H]):
316.1185, Found: 316.1175.
Benzyl 4-isopropyl-1-methyl-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate (1.19):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 10:90 to 15:85) to afford 1.19 as a mixture of equilibrating diastereomers (yellow oil, 3.4:1 dr determined by 1H NMR, 144 mg, 45%). Note:
the diastereomer ratio was determined by comparison of the integrations of the resonances at δ 6.81 (minor
diastereomer) and δ 6.77 (major diastereomer). Only distinguishable peaks are reported for each
diastereomer in the 1H NMR spectra. Analytical data for 1.19: Major isomer, 1H NMR (500 MHz, CDCl
3) δ
6.77 (d, J = 8.4 Hz, 1H), 6.32 (d, J = 8.4 Hz, 1H), 5.17 (d, J = 12.0 Hz, 1H), 5.11 (d, J = 12.0 Hz, 1H), 3.07
(d, J = 5.7 Hz, 1H), 3.04 (d, J = 5.7 Hz, 1H), 1.66 (s, 3H), 1.03 (d, J = 6.9 Hz, 3H); Minor isomer, 1H NMR (500 MHz, CDCl3) δ 6.81 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 8.5 Hz, 1H), 5.16 (d, J = 12.1 Hz, 1H), 5.10 (d, J
= 12.1 Hz, 1H), 3.13 (d, J = 5.7 Hz, 1H), 3.01 (d, J = 5.7 Hz, 1H), 1.76 (s, 3H), 1.01 (d, J = 6.9 Hz, 3H);
combined 13C NMR (126 MHz, CDCl
3) δ 196.0, 193.1, 160.0, 160.0, 144.8, 143.9, 135.2, 135.1, 128.4,
128.4, 128.4, 128.3, 128.3, 128.3, 126.4, 126.2, 87.4, 87.1, 68.3, 68.3, 65.0, 63.9, 56.1, 55.6, 48.8, 48.7,
27.2, 26.8, 17.1, 17.0, 16.6, 16.6, 15.9, 15.7; IR (thin film, cm-1) 3066, 3033, 2967, 2881 1755, 1724, 1268;
TLC (EtOAc:hexanes 15:85): Rf = 0.25; HRMS (ESI+): Calcd. for C19H21NNaO5: ([M+Na]): 366.1318,
Found: 366.1315.
Benzyl 4-methoxy-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate(1.20):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 20:80 to 40:60) to afford 1.20 as a yellow oil (136 mg, 43%). Analytical data for 1.20:1H NMR (600 MHz, CDCl
= 8.9, 5.6 Hz, 1H), 6.51 (dd, J = 8.9, 1.9 Hz, 1H), 5.21 (s, 2H), 4.72 (dd, J = 5.7, 1.9 Hz, 1H), 3.73 (s, 3H),
3.31 (d, J = 5.9 Hz, 1H), 3.02 (d, J = 5.9 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 191.14, 158.8, 137.2, 135.0,
128.8, 128.6, 128.6, 128.3, 102.4, 68.8, 59.9, 53.9, 53.9, 51.2; IR (thin film, cm-1) 1770, 1716, 1259, 1220;
TLC (EtOAc:hexanes 30:70): Rf = 0.17; HRMS (ESI+): Calcd. for C16H16NO5: ([M+H]): 318.0978, Found:
318.0977.
Benzyl 7-methoxy-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate(1.21):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 20:80 to 50:50) to afford 1.21 as a yellow oil (50 mg, 16%) Analytical data for 1.21:1H NMR (400 MHz, CDCl
3) δ 7.43 – 7.31 (m, 5H), 5.28 (d, J =
12.1 Hz, 1H), 5.21 – 5.12 (m, 2H), 5.07 (d, J = 7.0 Hz, 1H), 4.63 (d, J = 3.2 Hz, 1H), 3.60 (s, 3H), 3.30 (d,
J = 5.9 Hz, 1H), 3.09 (d, J = 5.9 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 192.9, 163.0, 158.3, 135.2, 128.5,
128.5, 128.5, 89.3, 79.7, 68.7, 62.9, 56.2, 53.7, 50.5; IR (thin film, cm-1) 3260, 1757, 1716, 1624, 1226;
TLC (EtOAc:hexanes 30:70): Rf = 0.17; HRMS (ESI+): Calcd. for C16H16NO5: ([M+H]): 318.0978, Found:
318.0974.
Benzyl 3-oxo-4-phenyl-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate (1.22):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (acetone:pentane 10:90 to 20:80) to afford 1.22 as a yellow foam (165 mg >20:1 dr, 45%). Note 1: The title compound isomerizes readily at rt (8.3:1 dr after 40 min in
CDCl3). The isomerization is slowed significantly if the title compound is stored neat at -20 °C (>20:1 dr
after 8 months). The diastereomer ratio was determined by comparison of the integration of the resonances at δ 4.86 (major diastereomer) and δ 4.69 (minor diastereomer). Analytical data for 1.22: 1H NMR (500
MHz, CDCl3) δ 7.61 – 7.54 (m, 2H), 7.49 – 7.39 (m, 3H), 7.38 – 7.31 (m, 5H), 7.02 (dd, J = 8.4, 5.9 Hz, 1H),
6.66 (dd, J = 8.4, 2.1 Hz, 1H), 5.24 (d, J = 12.3 Hz, 1H), 5.20 (d, J = 12.3 Hz, 1H), 4.86 (dd, J = 5.9, 2.1 Hz,
1H), 3.36 (d, J = 5.9 Hz, 1H), 3.07 (d, J = 5.9 Hz, 1H); 13C NMR(126 MHz, CDCl
3) δ 193.5, 158.8, 135.2,
(acetone:pentane 20:80): Rf = 0.25; HRMS (ESI+): Calcd. for C21H18NO5: ([M+H]): 364.1185, Found:
364.1183.
Benzyl 4-bromo-7-chloro-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate(1.23):The title compound was prepared using General Procedure A and was purified by flash chromatography on silica gel (EtOAc:hexanes 15:85 to 40:60) to afford 1.23 as a yellow foam as a 2.3:1 mixture with the hydrate (265 mg, 66%). Note 1: the title compound streaks on the
TLC plate. If the plate is neutralized with a 2% TEA in Et2O solution the title compounds can be observed
a tight spot (EtOAc:hexanes 50:50). Note 2: the title compound is observed as a single product in the crude
1H NMR but forms a hydrate during purification. The hydrate could be stirred over 3Å molecular sieves in
CH2Cl2 for 5-6 h to afford the title ketone as a single diastereomer. Note 3: the compound is not thermally
stable and the spiroepoxydienone (from retro Diels–Alder) can be observed in the 1H and 13C NMR spectra
after stirring the hydrate with molecular sieves. Analytical data for 1.23:1H NMR (500 MHz, CDCl3) δ 7.38 (m, 5H), 6.60 (d, J = 2.8 Hz, 1H), 5.28 (d, J = 12.0 Hz, 1H), 5.25 (d, J = 12.0 Hz, 1H), 4.75 (d, J = 2.8 Hz,
1H), 3.39 (d, J = 5.8 Hz, 1H), 3.21 (d, J = 5.8 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 185.6, 157.5, 138.8,
134.5, 128.3, 128.7, 128.5, 127.2, 89.3, 69.6, 66.7, 52.4, 51.1; IR (thin film, cm-1); TLC (EtOAc:hexanes
50:50, TEA neutralized plate): Rf = 0.24; HRMS (ESI+): Calcd. for C16H15BrClNaNO6: ([M+CH3OH+Na]):
453.9669, Found: 453.9654.
Secondary Transformations:
Benzyl 3-hydroxy-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxiran]-7-ene-6-carboxylate (1.24): NaBH4 (4.6 mg, 0.12 mmol) was added to a solution of 1.16 (100 mg, 0.35 mmol) in THF(3.3 mL)at 0
°C. After stirring at 0 °C for 1 h, the reaction was quenched with saturated NH4Cl (5 mL). The aqueous
layer was extracted with EtOAc (3 × 5 mL), and the combined extracts were washed with brine (10 mL),
dried over Na2SO4, filtered, and concentrated by rotary evaporation. The crude residue was purified via
mixture of diastereomers as a colorless oil (71 mg, 71% combined, 1.7:1 dr determined by 1H NMR). The
diastereomer ratio was determined by comparison of the integration of the resonances at δ 4.88 (minor
diastereomer) and δ 4.85 (major diastereomer). Note: only distinguishable peaks are reported for each
diastereomer in the 1H NMR spectra. Analytical data for 1.24: Major diastereomer,1H NMR (600 MHz, CDCl3) δ 6.61 (t, J = 7.0 Hz, 1H), 5.20 (d, J = 12.2 Hz, 1H), 5.11 (d, J = 12.2 Hz, 1H), 4.85 (dt, J = 5.9, 1.7
Hz, 1H), 3.67 (dd, J = 8.2, 1.7 Hz, 1H), 3.04 (d, J = 4.5 Hz, 1H), 2.90 (d, J = 4.5 Hz, 1H); Minor diastereomer,
1H NMR (600 MHz, CDCl3) δ 6.70 (t, J = 7.0 Hz, 1H), 5.19 (d, J = 12.2 Hz, 1H), 5.10 (d, J = 12.2 Hz, 1H), 4.88 (ddd, J = 5.9, 4.4, 1.8 Hz, 1H), 4.12 (dd, J = 7.0, 4.4 Hz, 1H), 3.12 (d, J = 4.7 Hz, 1H), 2.78 (d, J = 4.7
Hz, 1H); Combined 13C NMR (151 MHz, CDCl
3) δ 158.2, 158.1, 135.3, 135.2, 131.1, 130.4, , 128.5, 128.4,
128.3, 128.2, 128.2, 77.2, 74.3, 68.6, 68.4, 68.3, 68.2, 67.9, 67.5, 65.2, 62.1, 58.4, 57.0, 55.3, 52.8, 49.1;
IR (thin film, cm-1) 3447, 1704, 1733, 1327, 1263, 1232, 1100, 1059; TLC (EtOAc:hexanes 60:40): Rf =
0.21; HRMS (ESI+): Calcd. for C15H16NO5: ([M+H]): 290.1029, Found: 290.1021.
Benzyl 8-(bromomethyl)-8-hydroxy-7-oxo-2-oxa-3-azabicyclo[2.2.2]oct-5-ene-3-carboxylate (1.25): Br2 (0.20 mL, 0.40 mmol) was added to a solution of SMe2 (0.04 mL, 0.54 mmol) in CH2Cl2 (1 mL) at 0 °C.
A precipitate formed immediately and the mixture was stirred at 0 °C for 15 min; epoxide 1.16 (30 mg, 0.10 mmol) was then added. The reaction was removed from the cold bath and stirred for 3 h. The reaction
was quenched with saturated sodium bisulfite (2 mL) and diluted with water (2 mL). The aqueous layer
was extracted with CH2Cl2 (3 × 3 mL). The combined extracts were washed with brine (5 ml), dried over
Na2SO4, filtered, and concentrated under vacuum to afford 1.25 as a yellow oil (38.4 mg, 86%). Analytical
data for 1.25:1H NMR (600 MHz, CDCl
3) δ 7.36 (m, 5H), 6.86 (ddd, J = 8.1, 5.9, 1.6 Hz, 1H), 6.61 (ddd, J
= 8.1, 6.1, 2.3 Hz, 1H), 5.36 (dd, J = 6.0, 2.2 Hz, 1H), 5.25 (d, J = 12.1 Hz, 1H), 5.19 (d, J = 12.1 Hz, 1H),
4.85 (dd, J = 6.1, 1.6 Hz, 1H), 3.79 (d, J = 11.7 Hz, 1H), 3.48 (br s, 1H), 3.17 (d, J = 11.7 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 196.5, 158.5, 136.8, 134.8, 128.7, 128.7, 128.4, 128.4, 77.5, 70.2, 69.07, 62.0, 35.8;
IR (thin film, cm-1) 3303, 2361, 1744, 1271, 1082; TLC (EtOAc:hexanes 40:60): Rf = 0.74; HRMS (ESI+):
Benzyl 7,8-dihydroxy-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxirane]-6-carboxylate (1.26):OsO4 (9 mg, 0.03 mmol, 1 mol %) was added to a mixture NMO (2.45 g, 20.9 mmol) of the alkene
1.16 (1.00 g, 3.48 mmol) in THF/acetone/H2O (5:5:1, 33 mL) at 0 °C. After warming gradually over 4 h, the
reaction was cooled to 0 °C and was diluted with water (20 mL). The reaction was quenched with saturated
sodium bisulfite (30 mL) and stirred for 30 min. The layers were partitioned and the aqueous layer was
extracted with EtOAc (3 × 30 mL). The combined extracts were washed with brine (60 mL), dried over
Na2SO4, filtered, and concentrated by rotary evaporation. The crude material was purified through a short
plug of silica gel (EtOAc:hexanes 70:30) to afford 1.26 as an off-white solid (1.12 g, 87%). Analytical data for 26:1H NMR (400 MHz, Acetone-d
6) δ 7.51 – 7.28 (m, 5H), 5.24 (d, J = 12.5 Hz, 1H), 5.20 (d, J = 12.4
Hz, 1H), 5.00 (d, J = 4.8 Hz, 1H), 4.95 (d, J = 5.1 Hz, 1H), 4.61 – 4.53 (m, 1H), 4.53 – 4.47 (m, 1H), 4.39
(d, J = 4.3 Hz, 1H), 4.37 (d, J = 4.7 Hz, 1H), 3.29 (d, J = 6.2 Hz, 1H), 3.21 (d, J = 6.2 Hz, 1H); 13C NMR (101 MHz, Acetone) δ 201.4, 156.7, 137.0, 129.4, 129.2, 129.1, 81.3, 68.7, 66.7, 65.8, 61.9, 56.9, 52.9; IR
(thin film, cm-1) 3341, 1758, 1710, 1249, 1051; TLC (EtOAc:hexanes 70:30): Rf = 0.23; HRMS (ESI+):
Calcd. for C15H15NO7Na: ([M+Na]): 344.0747, Found: 344.0735.
2-(chloromethyl)-2,4,5,6-tetrahydroxy-3-oxocyclohexan-1-aminium chloride (1.27): A vial was charged with 1.26 (100 mg, 0.31 mmol) and Pd/C (10% Pd on wet charcoal, 10 wt %) then purged with N2
for 10 min. MeOH (2.0 mL) and AcOH (0.09 mL, 1.56 mmol) were added and vial was purged with H2 for
10 min then stirred vigorously under a balloon of H2. After 30 min, the reaction was filtered through a 0.45
µm PTFE syringe filter. HCl in 1,4-dioxane (4 N, 2.00 mL) was added to the filtrate. After stirring for 15 min
The solid was washed with diethyl ether (2 × 10) and hexanes (2 ×10) then quickly transferred to a vial
and placed under vacuum for 1 d to afford 27 as a white solid (53 mg, 66 %). Note: The title compound is extremely hygroscopic and will quickly (≈5-10 min) turn from a white solid to a viscous brown oil if allowed
to stand on the fritted filter funnel. If this happens, the oil can be dissolved in methanol and re-triturated
with diethyl ether. Analytical data for 1.27:1H NMR (500 MHz, CD
3OD) δ 4.50 (d, J = 7.9 Hz, 1H), 4.20
(dd, J = 5.9, 3.4 Hz, 1H), 4.09 (d, J = 11.8 Hz, 1H), 3.95 – 3.88 (m, 2H), 3.83 (dd, J = 7.9, 3.4 Hz, 1H); 13C NMR (151 MHz, CD3OD) δ 206.0, 78.3, 77.0, 74.2, 69.0, 55.9; IR (thin film, cm-1) 3247, 2360, 1738, 1598,
1052; HRMS (ESI+): Calcd. for C7H13ClNO5: ([M-Cl]): 226.0483, Found: 226.0472.
Benzyl 6-oxo-4a,7a-dihydro-1H,6H-spiro[furo[3,2-c][1,2]oxazine-7,2'-oxirane]-1-carboxylate (1.28): mCPBA (75%, 36 mg, 0.15 mmol) was added to a mixture of 1.16 (30 mg, 0.10 mmol) and NaHCO3 (17
mg, 0.21 mmol) in CH2Cl2(1 mL)at rt. After stirring at rt for 16 h, the reaction was diluted with diethyl ether
(3 mL) and washed with saturated NaHCO3 (3 × 5 mL) and brine (5 mL). The organics were dried over
Na2SO4, filtered, and concentrated by rotary evaporation. The crude material was purified via flash
chromatography on silica gel (EtOAc:hexanes, 40:60) to afford 1.28 as a yellow oil (16 mg, 51%). Analytical data for 1.28:1H NMR (400 MHz, CDCl
3) δ 7.37 (m, 5H), 6.87 (d, J = 6.3 Hz, 1H), 5.71 – 5.53 (m, 1H), 5.32
– 5.19 (m, 4H), 3.31 (d, J = 6.3 Hz, 1H), 3.08 (d, J = 6.3 Hz, 1H); 13C NMR (101 MHz, CDCl
3) δ 170.5,
154.4, 147.9, 134.7, 128.7, 128.7, 128.2, 98.7, 69.1, 66.6, 52.5, 50.6, 49.6; IR (thin film, cm-1) 3312, 1732,
1508, 1238, 1117; TLC (EtOAc:hexanes 30:70): Rf = 0.29; HRMS (ESI+): Calcd. for C15H13NNaO6:
([M+Na]): 326.0641, Found: 326.0630.
(563 mg, 6.86 mmol) in THF(20 mL)at rt. After stirring at rt for 18 h, the reaction was diluted with water
(20 mL) and the layers were partitioned. The aqueous layer was extracted with EtOAc (3 × 20 mL) and the
combined extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated by rotary
evaporation. The crude material was purified via flash chromatography on silica gel (EtOAc:hexanes,
40:60 to 50:50) to afford 1.29 as a yellow foam (805.0 mg, 78%). Analytical data for 1.29:1H NMR (600 MHz, CDCl3) δ 7.95 (s, 1H), 7.40 – 7.30 (m, 5H), 6.81 – 6.74 (m, 1H), 6.64 (ddd, J = 8.0, 5.9, 1.9 Hz, 1H),
6.02 (dd, J = 5.9, 1.8 Hz, 1H), 5.23 (d, J = 12.2 Hz, 1H), 5.13 (d, J = 12.2 Hz, 1H), 4.61 (dd, J = 6.0, 1.9 Hz,
1H), 3.26 (d, J = 5.4 Hz, 1H), 3.03 (d, J = 5.4 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 158.1, 150.1, 135.2,
130.3, 128.6, 128.5, 128.4, 128.3, 68.5, 66.8, 58.9, 54.8, 52.5; IR (thin film, cm-1) 3367, 3066, 1715, 1265,
1235, ; TLC (EtOAc:hexanes 50:50): Rf = 0.33; HRMS (ESI+): Calcd. for C15H15N2O5: ([M+H]): 303.0981,
Found: 303.0972.
Benzyl 7-(hydroxyimino)-8-(hydroxymethyl)-2-oxa-3-azabicyclo[2.2.2]oct-5-ene-3-carboxylate (1.30): NaBH4 (8 mg, 0.20 mmol) was added to a solution of 1.29 (30 mg, 0.10 mmol) in anhydrous
MeOH(1 mL)at rt. After stirring at rt for 30 min, the reaction was quenched with saturated NH4Cl (5
mL). The aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined extracts were washed
with brine (10 mL), dried over Na2SO4, filtered, and concentrated by rotary evaporation. The
crude material was purified via flash chromatography on silica gel (EtOAc:hexanes, 70:30) to afford 1.30 as a yellow foam (102 mg, 51%). Analytical data for 1.30:1H NMR (600 MHz, CDCl
3) δ 8.07 (s, 1H), 7.39 –
7.30 (m, 5H), 6.88 – 6.66 (m, 1H), 6.54 (ddd, J = 8.0, 6.0, 1.9 Hz, 1H), 5.92 (dd, J = 6.0, 1.5 Hz, 1H), 5.22
(d, J = 12.2 Hz, 1H), 5.16 – 5.09 (m, 2H), 3.95 (dd, J = 11.0, 6.7 Hz, 1H), 3.89 – 3.81 (m, 1H), 2.75 (s, 1H),
2.62 (ddd, J = 7.9, 6.7, 2.9 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 158.8, 153.8, 135.9, 135.3, 128.7, 128.6,
128.5, 128.2, 68.4, 66.1, 62.7, 53.2, 40.3; IR (thin film, cm-1) 3365, 1732, 1716, 1698, 1272, 1074; TLC
(EtOAc:hexanes 70:30): Rf = 0.63; HRMS (ESI+): Calcd. for C15H17N2O5: ([M+H]): 305.1138, Found:
7-(((4-nitrobenzoyl)oxy)imino)-8-(((4-nitrobenzoyl)oxy)methyl)-2-oxa-3-azabicyclo[2.2.2]oct-5-ene-3-carboxylate (1.31):Anhydrous triethylamine (0.06 mL, 0.45 mmol) was added to a mixture of 1.30 (34 mg, 0.11 mmol), DMAP (1 mg, 0.01 mmol, 10 mol %), and p-nitrobenzoyl chloride (62 mg, 0.34 mmol) in
anhydrous THF (1 mL). The reaction was stirred under N2 for 2 d at rt. The reaction was diluted with water
(3 mL) and the aqueous layer was extracted with EtOAc (3 × 3 mL). The combined extracts were washed
with brine (5 mL), dried over Na2SO4, filtered, and concentrated by rotary evaporation. The crude residue
was purified by flash chromatography on silica gel (EtOAc:hexanes 15:85 to 20:80) to afford 1.31 as a crystalline yellow solid. Analytical data for 1.31:1H NMR (500 MHz, CDCl
3) δ 8.39 – 8.31 (m, 2H), 8.29 –
8.18 (m, 6H), 7.42 – 7.29 (m, 5H), 7.00 – 6.89 (m, 1H), 6.65 (ddd, J = 8.1, 6.1, 1.9 Hz, 1H), 6.02 (dd, J =
6.1, 1.5 Hz, 1H), 5.33 – 5.28 (m, 1H), 5.20 (d, J = 12.1 Hz, 1H), 5.17 (d, J = 12.1 Hz, 1H), 4.93 (dd, J =
11.2, 6.1 Hz, 1H), 4.72 (dd, J = 11.2, 9.2 Hz, 1H), 3.25 (ddd, J = 9.2, 6.0, 3.0 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 164.1, 161.3, 161.0, 150.9, 150.6, 135.0, 134.9, 133.6, 130.9, 130.8, 128.7, 128.7, 128.7, 128.3,
127.6, 123.9, 123.9, 123.9, 123.6, 68.8, 67.5, 64.3, 53.1, 38.2, 38.1; IR (thin film, cm-1) 1748, 1732, 1716,
1523; TLC (EtOAc:hexanes 50:50): Rf = 0.59; HRMS (ESI+): Calcd. for C29H22N4NaO11: ([M+Na]):
625.1183, Found: 625.1229.
Benzyl 5'-oxa-6'-azadispiro[oxirane-2,2'-bicyclo[2.2.2]octane-3',2''-oxiran]-7'-ene-6'-carboxylate (1.32): Anhydrous THF (1 mL) was added to a flame dried vial containing Me3SO+I-(23 mg, 0.12 mmol)
under N2. NaH (60 wt % in mineral oil, 4 mg, 0.11 mmol) was added in one portion. After stirring for 20
(0.5 mL) was added via syringe. After stirring at 0 °C for 2 h, the reaction was quenched with saturated
NH4Cl (5 mL) and diluted with diethyl ether (3 mL). The layers were partitioned and the aqueous layer was
extracted with diethyl ether (3 × 5 mL). The combined extracts were washed with brine (10 mL), dried over
Na2SO4, filtered, and concentrated by rotary evaporation. The crude material was purified via flash
chromatography on silica gel (EtOAc:hexanes, 15:85 to 20:80) to afford 1.32 as a white, amorphous solid (18.6 mg, 59%). The relative stereochemistry was determined by 2D-NOESY analysis. Analytical
data for 1.32:1H NMR (400 MHz, CDCl3) δ 7.41 – 7.29 (m, 5H), 6.76 (ddd, J = 8.1, 5.8, 1.9 Hz, 1H), 6.71 (ddd, J = 8.1, 5.4, 2.0 Hz, 1H), 5.24 (d, J = 12.2 Hz, 1H), 5.14 (d, J = 12.2 Hz, 1H), 4.58 (dd, J = 5.8, 2.0
Hz, 1H), 4.37 (dd, J = 5.4, 1.9 Hz, 1H), 2.94 (d, J = 4.6 Hz, 1H), 2.82 (d, J = 4.6 Hz, 1H), 2.80 (d, J = 4.7
Hz, 1H), 2.70 (d, J = 4.7 Hz, 1H); 13C NMR (151 MHz, CDCl
3) δ 158.0, 135.2, 132.1, 131.6, 128.6, 128.5,
128.5, 128.3, 75.3, 68.5, 58.8, 58.2, 57.5, 49.9, 48.2; IR (thin film, cm-1) 3065, 3031, 2925, 1716; TLC
(EtOAc:hexanes 60:40): Rf = 0.93; HRMS (ESI+): Calcd. for C16H16NO5: ([M+H]): 302.1029, Found:
302.1020.
Benzyl 7,8-dihydroxy-4-isopropyl-1-methyl-3-oxo-5-oxa-6-azaspiro[bicyclo[2.2.2]octane-2,2'-oxirane]-6-carboxylate (1.34): OsO4 (2 mg, 0.01 mmol, 1 mol %) was added to a mixture NMO (577 mg,
4.93 mmol) of the 1.19 (3.4:1 mixture of diastereomers, 300 mg, 0.82 mmol) in THF/acetone/H2O (5:5:1,
11 mL) at rt. The reaction was stirred for 2.5 h then placed in a water bath. The reaction was quenched
with saturated sodium bisulfite (10 mL) and stirred for 30 min (exothermic). The mixture was diluted with
water and the aqueous layer was extracted with diethyl ether (3 × 20 mL). The combined extracts were
washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated by rotary evaporation. The crude
material was purified by flash chromatography on silica gel (EtOAc:hexanes, 30:70 to 50:50) to afford 1.34 as an inseparable mixture of diastereomers (1.1:1 dr determined by 1H NMR) as viscous colorless oil (231
are reported. Analytical data for 1.34: Major isomer, 1H NMR (600 MHz, CDCl
3) δ 7.42 – 7.30 (m, 10H),
5.28 (d, J = 12.0 Hz, 1H), 5.23 – 5.12 (m, 3H), 4.41 – 4.34 (m, 3H), 4.14 (dd, J = 8.5, 6.7 Hz, 1H), 3.20 (d,
J = 6.0 Hz, 1H), 3.12 – 3.08 (m, 1H), 3.03 (d, J = 5.8 Hz, 1H), 2.97 (d, J = 5.9 Hz, 1H), 2.90 (d, J = 5.8 Hz, 1H), 2.92 – 2.83 (m, 3H), 2.50 (hept, J = 7.0 Hz, 1H), 2.37 (hept, J = 7.0 Hz, 1H), 1.60 (s, 6H), 1.05 (d, J =
7.0 Hz, 3H), 1.03 – 0.99 (m, 6H), 0.87 (d, J = 7.1 Hz, 3H); 13C NMR (151 MHz, CDCl
3) δ 203.5, 201.5,
157.9, 157.6, 135.2, 135.1, 128.7, 128.7, 128.6, 128.5, 128.5, 128.4, 128.3, 87.5, 85.9, 69.0, 69.0, 68.5,
68.4, 68.1, 67.3, 64.6, 64.3, 60.7, 59.3, 49.9, 47.6, 27.4, 25.5, 16.5, 16.3, 15.9, 15.3, 15.2, 14.1; IR (thin film, cm-1), 345, 2974, 1753, 1711, 1385, 1293, 1057; TLC (EtOAc:hexanes 50%): Rf = 0.37; HRMS (ESI+):
Calcd. for C19H23NNaO7: ([M+Na]): 400.1373, Found: 400.1367.
Benzyl
4'-isopropyl-2',2',7'-trimethyl-5'-oxotetrahydro-5'H-spiro[oxirane-2,6'-[4,7](epoxyimino)benzo[d][1,3]dioxole]-8'-carboxylate (1.35): PTSA (10 mol %, 5.0 mg, 0.027 mmol) was added to a solution of diol 1.34 (1.1:1 dr, 100 mg, 0.27 mmol) in acetone/2,2-DMP (1:1, 1.4 ml). The reaction was heated and stirred at 40 °C for 2.5 h. The reaction was cooled to rt then quenched with
saturated sodium bicarbonate (3 ml). The aqueous layer was extracted with ether (3 × 3 ml). The
combined extracts were washed with brine (5 ml), dried over Na2SO4, filtered, and concentrated by rotary
evaporation. The crude material was purified by flash chromatography on silica gel (EtOAc/hexanes
10:90 to 15:85 then ether) to afford 1.35 as an inseparable mixture of diastereomers as a yellow oil (1.2:1 dr, 95 mg, 86%). The diastereomer ratio was determined by comparison of the integration of the
resonances at δ 2.46 (major diastereomer) and δ 2.32 (minor diastereomer). Note: The combined 1H
and 13C spectra are reported. Analytical data for 1.35: 1H NMR (600 MHz, CDCl
3) δ 7.42 – 7.30 (m, 9H),
5.28 (d, J = 12.0 Hz, 1H), 5.23 – 5.14 (m, 3H), 4.59 – 4.53 (m, 3H), 4.39 (d, J = 6.8 Hz, 1H), 3.29 (d, J =
6.1 Hz, 1H), 3.01 (d, J = 6.1 Hz, 1H), 2.97 (d, J = 6.1 Hz, 1H), 2.86 (d, J = 6.1 Hz, 1H), 2.46 (hept, J = 7.0
Hz, 1H), 2.32 (hept, J = 7.0 Hz, 1H), 1.64 (s, 3H), 1.61 (s, 2H), 1.33 (s, 3H), 1.33 (s, 3H), 1.30 (s, 3H),
CDCl3) δ 202.0, 200.2, 157.8, 157.6, 135.3, 135.2, 128.7, 128.6, 128.5, 128.4, 128.2, 111.7, 111.1, 86.5,
84.8, 76.5, 76.0, 74.3, 74.1, 68.4, 68.4, 63.7, 63.7, 59.7, 58.3, 50.8, 48.0, 28.3, 25.9, 25.6, 25.3, 24.4,
24.1, 16.6, 16.3, 16.1, 16.0, 15.7, 15.1; IR (thin film, cm-1) 2978, 2940, 2360, 1756, 1723, 1383, 1268,
1211, 1081, 1051; TLC (EtOAc:hexanes 40:60): Rf = 0.77; HRMS (ESI+): Calcd. for C22H28NO7: ([M+H]):
1D-NOESY Determination of Diastereomers:
Selective 1D-NOESY Correlation Experiment - (1H, 600 MHz, CDCl3)
The mixture of isomers was determined to be diastereomers resulting from the facial approach of the
acylnitroso dienophile. The black spectrum (5) is the mixture of diastereomers. Selective excitation of
each epoxide resonance revealed a correlation to the bridgehead methyl in both diastereomers A and B
(spectrum 3 and 2 respectively) but only diastereomer A exhibited a correlation between the epoxide and
the acetonide methyl (4).
Diastereomer B – no observable epoxide correlation to acetonide methyl
Cross-over experiment:
A solution of tBuO2CNHOH (77 mg, 0.58 mmol) in CH2Cl2 (1 mL) was added via syringe pump over 2 h to
a mixture of 1.19 (3.4:1 dr, 20 mg, 0.06 mmol) and nBu4NIO4 (252 mg, 0.58 mmol) in CH2Cl2 (1 mL) at rt.
After complete addition, the reaction was diluted with MTBE (4 mL) and the mixture was filtered through a
plug of Celite®. The filtrate was concentrated by rotary evaporation and the crude residue was purified by
flash chromatography on silica gel (EtOAc:hexanes 10:90 to 20:80) to afford an inseparable mixture of 37 and 1.36 (mixture of diastereomers, no yield recorded). The 1H NMR spectrum of the mixture of products
matched an independently synthesized mixture of 1.33 and 1.36 (prepared using a modified General Procedure A in which BnO2CNHOH is exchanged for tBuO2CNHOH). The 1H spectrum of the
![Figure 2-1. Functional Group Manipulations Needed to Access Tetrodamine from the [2.2.2]-Bicycle Scaffold](https://thumb-us.123doks.com/thumbv2/123dok_us/8327595.2208409/55.918.215.710.180.291/figure-functional-manipulations-needed-access-tetrodamine-bicycle-scaffold.webp)
