MARCHUT, ALEXANDER JOSEPH. Simulation of Polyglutamine Aggregation With An
In-termediate Resolution Protein Model. (Under the direction of Carol K. Hall)
The pathological manifestation of nine hereditary neurodegenerative diseases including
Huntington’s disease is the presence within the brain of aggregates of disease-specific proteins
that contain polyglutamine tracts longer than a critical length. The molecular level mechanisms
by which these proteins aggregate are still unclear. In an effort to shed light on this
impor-tant phenomenon, we are investigating the aggregation of model fibril-forming peptides using
molecular-level computer simulation. A simplified model of polyglutamine, the protein that is
known to form fibrils (ordered aggregates of proteins in beta-sheet conformations) in the brains
of victims of Huntington’s disease, has been developed. This model accounts for the most
im-portant types of intra- and inter-molecular interactions - hydrogen bonding and hydrophobic
interactions - while allowing the folding process to be simulated in a reasonable time frame. The
model utilizes discontinuous potentials such as hard spheres and square wells in order to take
advantage of discontinuous molecular dynamics (DMD), a fast simulation technique that is very
computationally efficient.
DMD is used to examine the folding and aggregation of systems of model polyglutamine
peptides ranging in size from isolated peptides to 96 peptides. Our model peptides form
amor-phous aggregates at low temperatures, structures with significant beta sheet character at
interme-diate temperatures, and random coils at high temperatures.
The effects of hydrophobicity on the multi-chain systems are studied by varying the strength
interac-nular structures that are made up of beta sheets starting from random configurations of random
coils. This result is interesting because annular structures were recently found in experiments
on polyglutamine aggregation and because of Perutz’s prediction that polyglutamine would form
water-filled nanotubes. Our results not only provide insight into the mechanisms of Huntington’s
disease but also demonstrate one possible application of biomolecules in nanoscience.
The effect of chain length on the behavior of our model peptides was examined by
simulat-ing the foldsimulat-ing of isolated polyglutamine peptides 16, 32, and 48 residues long and the foldsimulat-ing
and aggregation of systems of twenty-four model polyglutamine peptides 16, 32, 36, 40, and
48 residues long. Although the isolated polyglutamine peptides did form some alpha and beta
backbone - backbone hydrogen bonds they did not have as many of these bonds as they would
have if they had folded into a complete alpha helix or beta sheet. In one of the simulations on
the isolated polyglutamine peptide 48 residues long we observed a structure that resembles a
beta helix. In our multi-peptide simulations we found that the temperature at which the model
peptides undergo the transition from amorphous aggregates to ordered aggregates increases with
increasing chain length. Furthermore, the temperature at which the model peptides undergo the
transition from ordered aggregates to random coils also increases with increasing chain length.
Our finding that the stability of the ordered aggregates increases as the peptide chain length
in-creases may help to explain the experimentally observed relation between polyglutamine tract
length and aggregation in vitro and disease progression in vivo. We have also observed in our
simulations that the optimal temperature for the formation of beta sheets increases with chain
dominated by relatively ordered beta sheet structures at chain lengths greater than 36. Our
find-ing of this critical chain length of 36 glutamine residues is interestfind-ing because a critical chain
W
ITH
A
N
I
NTERMEDIATE
R
ESOLUTION
P
ROTEIN
M
ODEL
by
ALEXANDER J. MARCHUT A dissertation submitted to the Graduate Faculty
of North Carolina State University in partial fulfillment of the requirements for the
Degree of Doctor of Philosophy
CHEMICAL ENGINEERING
Raleigh NC 27695-7905
January 5, 2006
APPROVED BY:
Carol K. Hall Robert M. Kelly
Chair of Advisory Committee
Biography
Alexander J. Marchut was born at the Hospital of the University of Pennsylvania in
Philadel-phia, Pennsylvania on April 28, 1978 to Laura and Leslie Marchut. His sister, Amber, was born
there on January 28, 1980. His family moved to Maplewood, New Jersey in 1981 where he
graduated from Columbia High School in 1996. He attended the University of Pennsylvania
from 1996 to 2000 and graduated with a Bachelor of Science in Chemical Engineering. In
Au-gust 2000, he moved to Raleigh, North Carolina to pursue his doctorate degree in Chemical
Acknowledgments
I would like to take this opportunity to acknowledge the many people who have made it
possible to perform the research described in this thesis.
I would like to thank Professor Carol Hall, my advisor. Her enthusiasm and insight were
invaluable in starting this project and advancing it to its current state. I am grateful for her
guidance, both professional and personal, and for the funding she provided for my research and
presentations.
I would like to thank Dr. Anne V. Smith and Dr. Hung D. Nguyen for their parts in
devel-oping the models and codes that I have built upon to create the model and code that I have used
in studying protein folding and aggregation. None of the work presented in this thesis would
have been possible without their groundbreaking research. I would also like to thank Dr.
An-drew J. Schultz for helpful discussions and advice on developing the code and making it more
computationally efficient.
I would also like to thank Professor John Cavanagh, Professor John van Zanten, and
Pro-fessor Robert Kelly for serving on my committe and for their helpful discussions and advice on
my project. I would also like to thank the faculty and staff of NCSU’s Chemical and
Biomolec-ular Engineering department for everything they’ve done for me over the past five years. Special
thanks go to Mrs. Sandra Bailey, the department’s graduate secretary.
I am grateful to have had the opportunity to be a trainee in the NIH Molecular Biotechnology
Training Program at North Carolina State University. Funding from the National Institutes of
Health made this research possible. The High-Performance Computing center at NCSU and the
appreciated. I also thank the system administrators at NCSU and Purdue University, Eric Sills
and William Whitson, for their rapid response to problems I encountered when running on their
systems.
I thank my fellow system administrators of the Hall group network, Julie McCormick, Brian
Attwood, Andrew Schultz, Aysa Akad, Arthi Jayaraman, Victoria Wagoner, and Amit Goyal for
their efforts to maintain the health and performance of our computer network. I also thank all
the members of the Hall group for their support and friendship.
Finally, I am grateful to my friends and family whose love and undying support has kept me
going through the good and the bad. I would like to thank my mother, Laura Vlasits, for always
being supportive no matter what the situation and my father, Leslie Marchut, for encouraging
my scientific endeavors and curiousity from the time I was a small child to this very moment.
I would like to thank my sister, Amber Marchut, for her love, friendship, and understanding. I
would like to thank all the friends I’ve had over the years for their camaraderie. Last, and far
from least, I would like to thank my fiancee, Sabrina Tachdjian, for being absolutely wonderful
Table of Contents
Page
List of Figures vii
Chapter 1 Introduction 1
1.1 Motivation . . . 1
1.2 Objective . . . 2
1.3 Approach . . . 3
Chapter 2 Spontaneous Formation of Annular Structures Observed In Molecular Dynamics Simulations of Polyglutamine Peptides 6 2.1 References . . . 13
2.2 Figures . . . 16
Chapter 3 Solvent Effects on the Aggregation of Model Polyglutamine Peptides 20 3.1 Introduction . . . 20
3.2 Methods . . . 27
3.2.1 Model Forces . . . 27
3.2.2 Discontinuous Molecular Dynamics . . . 30
3.3 Results . . . 33
3.5 Acknowledgements . . . 40
3.6 References . . . 41
3.7 Figures . . . 52
Chapter 4 Effects of chain length on the aggregation of model polyglutamine pep-tides: Molecular dynamics simulations 63 4.1 Introduction . . . 63
4.2 Methods . . . 69
4.2.1 Model Forces . . . 69
4.2.2 Discontinuous Molecular Dynamics . . . 73
4.3 Results . . . 75
4.4 Discussion . . . 83
4.5 Acknowledgements . . . 85
4.6 References . . . 86
4.7 Figures . . . 98
Chapter 5 Future Work 108 5.1 Seeded Aggregation Simulations . . . 108
5.2 Further Development of Polyglutamine Model . . . 109
5.3 Development of Models for All Sidechains . . . 111
5.4 Modeling Changes in pH . . . 112
5.5 References . . . 114
List of Figures
Page
Chapter 1 Introduction 1
Chapter 2 Spontaneous Formation of Annular Structures Observed In Molecular
Dynamics Simulations of Polyglutamine Peptides 6
2.1 Four-sphere representation of a polyglutamine sidechain. Hard spheres are
white. Square wells representing hydrophobic interactions are shown in a
hon-eycomb pattern. Square wells representing hydrogen bond acceptors have thick
horizontal stripes and square wells representing hydrogen bond donors have thin
vertical stripes. Covalent bonds are shown in bold and pseudo bonds to constrain
bond angles are thin lines. . . 17
2.2 A, close up of the tube formed in our simulations of 24 polyglutamine 16mers.
B, AFM image from the supplementary information of Wacker et al. showing
annular structures formed from a mutant huntingtin fragment containing a
polyg-lutamine tract 53 residues long (arrows indicate spherical oligomeric structures;
arrowhead indicates small annular structure). . . 18
2.3 A, Close up of the side of the tube formed from 24 polyglutamine 16mers with
Chapter 3 Solvent Effects on the Aggregation of Model Polyglutamine Peptides 20
3.1 Four-sphere representation of a polyglutamine sidechain. Hard spheres are
white. Square wells representing hydrophobic interactions are shown in a
hon-eycomb pattern. Square wells representing hydrogen bond acceptors have thick
horizontal stripes and square wells representing hydrogen bond donors have thin
vertical stripes. Covalent bonds are shown in bold and pseudo bonds to constrain
bond angles are thin lines. . . 54
3.2 A, Snapshot of the whole system of 24 polyglutamine 16mers at R=0.125,
T*=0.155. B, Close up of the tube formed from 24 polyglutamine 16mers at
R=0.125, T*=0.155 . . . 55
3.3 A: Snapshot of large diameter tube formed by 24 polyglutamine 16mers at
R=0.125, T*=0.185. B: Snapshot of connected tubes morphology formed by 24
polyglutamine 16mers at R=0.125, T*=0.185. C: Snapshot of tube connected to
sheet morphology formed from 24 polyglutamine 16mers at R=0.125, T*=0.165 56
3.4 Beta intermolecular backbone / backbone hydrogen bonds vs. T* for the 24
polyglutamine 16mers system . . . 57
3.5 Fraction peptides in beta sheets vs. T* for the 24 polyglutamine 16mers system 58
3.6 Number of beta regions vs. T* for the 24 polyglutamine 16mers system . . . . 59
3.7 Fraction peptides in beta sheets vs. T* for the 48 polyglutamine 16mers system 60
3.8 Intermolecular hydrogen bonds vs. T* at R=0.075 for the 24 polyglutamine
3.9 Intermolecular hydrogen bonds vs. T* at R=0.05 for the 24 polyglutamine
16mers system . . . 62
Chapter 4 Effects of chain length on the aggregation of model polyglutamine
pep-tides: Molecular dynamics simulations 63
4.1 Four sphere representation of a polyglutamine sidechain. Hard spheres are white,
square wells representing hydrophobic interactions are shown in a honeycomb
pattern, square wells representing hydrogen bond acceptors have thick horizontal
stripes and square wells representing hydrogen bond donors have thin vertical
stripes. Covalent bonds are shown in bold and pseudo bonds to constrain bond
angles are thin lines. . . 100
4.2 Normalized number of backbone - backbone alpha and beta hydrogen bonds for
the isolated polyglutamine peptides 16, 32, and 48 residues long vs. T*. . . 101
4.3 A: A beta hairpin formed by a polyglutamine peptide 16 residues long, B: A
partial alpha helix formed by a polyglutamine peptide 32 residues long, C: A
three-stranded beta sheet formed by a polyglutamine peptide 32 residues long,
D: A three-stranded beta sheet formed by a polyglutamine peptide 48 residues
long. . . 102
4.4 A: Snapshot of beta helix configuration formed by a polyglutamine peptide 48
residues long, B: Snapshot of the same beta helix with the sidechains removed
4.5 A: Snapshot of the tube formed from 24 polyglutamine 16mers at T*=0.145.
B: Snapshot of the tube formed from 24 polyglutamine 32mers at T*=0.155.
C: Snapshot of the ring structure formed from 24 polyglutamine 36mers at
T*=0.125. D: Snapshot of the ring structure formed from 24 polyglutamine
40mers at T*=0.165. . . 103
4.6 Snapshot of the ring structure formed from 24 polyglutamine 40mers at
T*=0.165. The sidechains and peptides not involved in the ring structure have
been removed for clarity. . . 103
4.7 Normalized number of beta backbone - backbone hydrogen bonds vs. T* for
polyglutamine peptides 16, 32, 36, 40, and 48 residues long. . . 104
4.8 Beta regions vs. T* for polyglutamine peptides 16, 32, 36, 40, and 48 residues
long. . . 105
4.9 Phase diagram for polyglutamine peptide self assembly in temperature-chain
length space. . . 106
4.10 Normalized number of intermolecular beta backbone - backbone hydrogen bonds
for 48 polyglutamine 16mers, 24 polyglutamine 32mers and 16 polyglutamine
48mers at constant packing fraction. . . 107
Chapter 5 Future Work 108
5.1 Snapshot of the annular structure formed from 24 polyglutamine 40mers at
T*=0.165 with sidechains and peptides not involved in the annular structure
5.2 Snapshot of slablike fibrils formed by 24 polyglutamine 16mers with van der
C
HAPTER
1
I
NTRODUCTION
1.1
Motivation
Aggregation of polyglutamine-containing proteins in the brain is a cause or an associated
symptom of nine hereditary neurodegenerative diseases including Huntington’s disease.
Aggre-gates of these proteins have been found not only in the brains of victims but also in cells made to
express the genes that cause the diseases. It has been demonstrated that the proteins that cause
the diseases will only do so when the polyglutamine sequence is longer than a certain critical
value. Likewise, polyglutamine-containing proteins aggregate in vitro and in vivo only when
the polyglutamine sequence is longer than a critical value. The aggregation of these proteins is
known to occur by a nucleation and growth mechanism which speeds up as the length of the
polyglutamine sequence increases. This may explain why the diseases manifest themselves in
their victims at earlier ages as the length of the polyglutamine sequence increases. Although
the causative link between the aggregation of polyglutamine-containing proteins and the
polyg-lutamine diseases has not yet been firmly established, protein aggregation is widely believed to
play some sort of role.
aggregate is still unclear. It is believed that these proteins misfold and then interact with other
misfolded proteins to aggregate. The aggregates contain a significant amount of one of the more
common structural motifs in proteins - beta-sheets. One possible molecular mechanism for the
formation of aggregates starts with the formation of a nucleus containing a very small amount of
beta-sheet structure which grows rapidly as more and more proteins in a beta-sheet conformation
are added on.
In principle, important information about the formation and structure of
polyglutamine-containing protein aggregates can be found by computer simulation because of the molecular
detail that such simulations can provide. With appropriate techniques and fast enough
comput-ers, it is possible to watch the molecules as they misfold and aggregate and to study the events
and conditions that lead to misfolding and aggregation. Theories describing the molecular
mech-anisms by which proteins misfold and aggregate can be tested. Useful information that cannot
be obtained by experiments can be provided by computer simulation. Thus computer simulation
has great potential to expand our knowledge of protein folding and aggregation.
1.2
Objective
The objective of this study is to improve our understanding of the processes by which
polyglutamine-containing proteins misfold and aggregate. Since it is impossible to simulate the
folding of a single protein in atomistic detail given the limits of current computational
technol-ogy, I have used an intermediate resolution model. I have modified the intermediate resolution
protein model developed by Smith, Nguyen and Hall, known as PRIME, to represent
essential features of the forces responsible for protein folding, such as the hydrophobic effect
and hydrogen bonding, yet can still be used to simulate large systems at long timescales in a
reasonable time. It stands at the crossroads between the so-called “all-atom” models that
ac-count for the motions of every atom on the protein being simulated (as well as every solvent
atom) and more coarse-grained models that simply represent amino acid residues or a groups
of amino acid residues as beads on a lattice. This model is computationally efficient like the
coarse-grained models yet it represents protein geometry and interactions relatively realistically
like the all-atom models.
1.3
Approach
Polyglutamine aggregation phenomena have been investigated by performing simulations
on systems of polyglutamine chains modeled by PRIME. We have examined the folding of an
isolated peptide and the folding and/or aggregation of many peptides starting from the fully
de-natured state. These phenomena have been studied for different chain lengths and system sizes at
different temperatures, peptide concentrations, and solvent conditions. A system of twenty-four
polyglutamine chains sixteen residues long at a concentration of 5 mM spontaneously formed
large beta sheets which curved to form tube-like annular structures that resemble beta barrels.
We examined the self assembly of these structures at relative hydrophobicity, R, (which we define
as the hydrophobic interaction strength divided by the hydrogen bonding interaction strength) of
0.005, 0.075, 0.10, and 0.125 in order to find a reasonable value for this parameter, which is
the only adjustable parameter in the model. We concluded that the relative hydrophobicity value
per-formed simulations of isolated polyglutamine peptides 16, 32, and 48 residues long and systems
of twenty-four model polyglutamine peptides 16, 32, 36, 40, and 48 residues long in order to
determine what effects polyglutamine chain length would have on the folding and / or
aggrega-tion of these model peptides. Although the isolated polyglutamine peptides did form some alpha
and beta backbone - backbone hydrogen bonds they did not have as many of these bonds as they
would have if they had folded into a complete alpha helix or beta sheet. In one of the simulations
on the isolated polyglutamine peptide 48 residues long we observed a structure that resembles a
beta helix. In our multi-peptide simulations we observed that the temperature at which the model
peptides undergo the transition from amorphous aggregates to ordered aggregates increases with
increasing chain length. Furthermore, the temperature at which the model peptides undergo the
transition from ordered aggregates to random coils also increases with increasing chain length.
Our finding that the stability of the ordered aggregates increases as the peptide chain length
in-creases may help to explain the experimentally observed relation between polyglutamine tract
length and aggregation in vitro and disease progression in vivo. We have also observed in our
simulations that the optimal temperature for the formation of beta sheets increases with chain
length up to 36 glutamine residues but not beyond. Equivalently, at fixed temperature we find
a transition from a region dominated by random coils at chain lengths less than 36 to a region
dominated by relatively ordered beta sheet structures at chain lengths greater than 36. Our
find-ing of this critical chain length of 36 glutamine residues is interestfind-ing because a critical chain
length of 37 glutamine residues has been observed experimentally.
Chapters 2 through 4 are adapted from the following publications:
Observed in Molecular Dynamics Simulations of Polyglutamine Peptides” Computational
Biol-ogy and Chemistry, submitted.
Chapter 3: A. J. Marchut and C. K. Hall “Solvent Effects on the Aggregation of Model
Polyglutamine Peptides” Biophysical Journal, submitted.
Chapter 4: A. J. Marchut and C. K. Hall “Effects of Chain Length on the Aggregation of
C
HAPTER
2
S
PONTANEOUS
F
ORMATION OF
A
NNULAR
S
TRUCTURES
O
BSERVED
I
N
M
OLECULAR
D
YNAMICS
S
IMULATIONS OF
P
OLYGLUTAMINE
P
EPTIDES
Annular structures have been observed experimentally in vitro in aggregates of
polyglutamine-containing proteins and other proteins associated with diseases of the brain1–3. Aggregation of
proteins that contain expanded polyglutamine tracts has been implicated in nine hereditary
neu-rodegenerative diseases including Huntington’s disease4,5. The first experimental observation of
annular structures within aggregates of huntingtin, the protein with an expanded polyglutamine
tract that causes Huntington’s disease, was published by Wacker et al.1 who used atomic force
microscopy (AFM) to examine aggregates formed from a mutant huntingtin fragment. They
found that when these proteins contained polyglutamine tracts of 53 residues in length they
formed annular structures nested within larger annular structures, but when the proteins
con-tained polyglutamine tracts of 20 residues in length they did not aggregate. Similar structures
have also been found in aggregates of mutantα-synuclein, the protein that causes Parkinson’s
microscopy.
Here we report the observation of annular structures in molecular-level simulations of large
systems of model polyglutamine peptides. A system of twenty-four polyglutamine chains sixteen
residues long at a concentration of 5 mM spontaneously formed large beta sheets which curved
to form tube-like annular structures that resemble beta barrels. The structures were formed over
the course of a slow-cooling simulation in which a random configuration of random coils at a
high temperature was quenched to the temperature of interest. These simulations took an average
of 100 hours on a 2.2 GHz AMD Athlon(TM) XP 3200+ workstation.
To our knowledge these are the first molecular simulations of the aggregation of large
systems of polyglutamine peptides in nearly atomistic detail. Starikov et al.6 examined the
structure of a 40-residue polyglutamine protein using three different all-atom simulation codes,
CHARMM, AMBER, and OPLSAA. The protein folded into a beta sheet when CHARMM was
used and into a compact random coil when the two other packages were used. Finke et al.7
devel-oped a united-atom model for polyglutamine in which each residue is represented by two
beads-one for the backbbeads-one atoms and another for the sidechain atoms. They parameterized their model
so that it would agree with experiments that they conducted on CI2 proteins with polyglutamine
insertions. They then used their model to simulate a single CI2 protein with polyglutamine
in-sertions and concluded that isolated short polyglutamine peptides should exist as random coils.
Burke et al.8modeled a copolymer of polyglutamine and polyproline as beads on a string where
each bead represented three glutamine residues or three proline residues. Their glutamine beads
were freely jointed but their proline beads were subject to angular constraints causing them to act
beads and three to twenty glutamine beads and found that their models formed the phases
ex-pected for diblock copolymers including spheres and cylinders. Although their glutamine chains
formed amorphous structures similar to random coils and structures with one turn similar to
hair-pins, they could not truly monitor secondary structure formation because of their low-resolution
representation. The work reported here is novel in comparison to these approaches in that it
rep-resents glutamine residues in more detail than the models of Burke et al.8 and Finke et al.7 and
looks at the aggregation of many more polyglutamine molecules than the studies by Starikov et
al.6and Finke et al.7. Our more detailed representation of the glutamine residues permits us to
monitor the secondary structures that form and the inter- and intra-molecular hydrogen bonding
and hydrophobic interactions
This work was accomplished by extending the PRIME model9–14 (PRotein Intermediate
resolution ModEl) to polyglutamine. PRIME is an off-lattice, unbiased, intermediate-resolution
protein model9–12based on an amino acid representation of between three to seven united atoms
depending on the residue being modeled. This model is designed to be used with discontinuous
molecular dynamics (DMD)15–17, an extremely fast alternative to traditional molecular
dynam-ics. PRIME has realistic united atom diameters, bond lengths and bond-angle constraints and has
the ability to interact both intra- and inter-molecularly via hydrogen bonding and hydrophobic
interaction potentials. Each amino acid residue is composed of a three-sphere backbone
com-prised of united atoms NH, CαH, and C=O, and a sidechain represented by between zero (in the
case of glycine) and four spheres (in the case of glutamine). All backbone bond lengths and bond
angles are fixed at their ideal values; the distance between consecutive Cαatoms is fixed so as to
the energy function as a potential of mean force.
All forces are modeled by either hard-sphere or square-well potentials. The excluded
vol-umes of the united atoms are modeled using hard-sphere potentials with realistic diameters.
Interactions between hydrophobic sidechain beads are represented by a square-well potential of
depthHP and range 1.5σRwhereσRis the side chain bead diameter. Hydrogen bonding
be-tween amide hydrogen atoms and carbonyl oxygen atoms on the same or neighboring chains is
represented by a square-well potential of depthHBbetween NH and C=O united atoms
when-ever: the virtual hydrogen and oxygen atoms (whose location can be calculated at any time)
are separated by 4.2 ˚Aand the nitrogen-hydrogen and carbon-oxygen vectors point towards each
other within a fairly generous tolerance. For more details on our hydrogen bonding method see
articles by Nguyen et al.13and Ding et al.18. We define the reduced temperature, T* askT /HB
and the relative hydrophobicity, R asHP/HB. Thus far, we have found that the formation of
annular structures is best at R=0.075.
The polyglutamine sidechain is modeled with the same level of detail as the PRIME model
backbone. Each sidechain is represented by four spheres as depicted in figure 4.1. Spheres 1
and 2 (as labeled in figure 4.1) each represent a methylene group and have hard cores that are
surrounded by square wells to mimic excluded volume and hydrophobic interactions. Sphere 3
represents a carbonyl group; it has a hard core to mimic excluded volume and a
directionally-dependent square well to mimic a hydrogen bond acceptor. Sphere 4 represents an amine group;
it has a hard core to mimic excluded volume and a directionally-dependent square well to mimic
a hydrogen bond donor. The four spheres all have their bond angles constrained by a system of
modeled in the same way as the methyl, carbonyl, and amine beads in the polyalanine PRIME
model10,11,13and as such, have exactly the same diameters, bond lengths, pseudobond lengths,
interaction strengths, and interaction ranges.
Systems containing twenty-four chains were slowly quenched from a random configuration
of high temperature random coils at T*=0.5 to the temperature of interest which ranged from
T*=0.075 to T*=0.205. The number of particles and box volume were picked to give the desired
concentration, 5 mM. Between three and five simulations were run at every data point.
Simula-tions were run for 30 billion DMD events to ensure that a stable structure was formed. At low T*,
the system formed a collapsed state that was relatively disordered. As T* increased, the system
formed large beta sheets which curved to form tube-like annular structures because the system
had more flexibility than the systems at low T*, giving it the freedom to find more ordered states.
At high T*, the peptides were in random coil configurations.
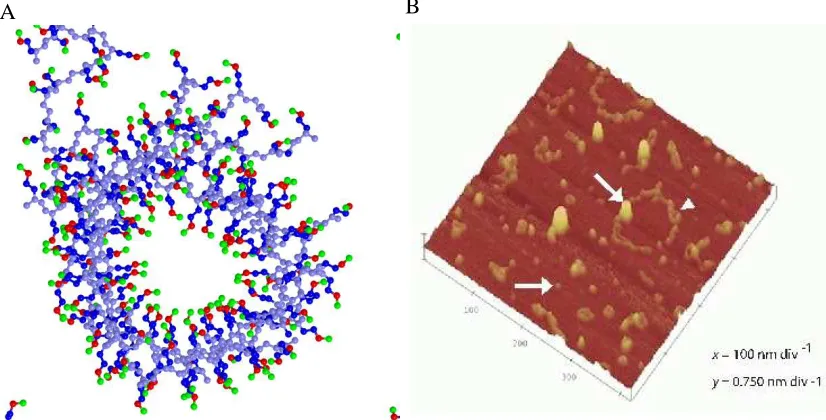
A snapshot of one of the tube structures and an AFM image from the supplementary
infor-mation of Wacker et al.1 is shown in figure 2.2. Our simulation results are similar to the small
annular oligomer indicated by the arrowhead in figure 2.2 (B). Figure 2.3 (A) shows that these
nanotubes were made up of beta sheets that curve. Peptides on the front face of the tube are
shown in bold colors; the peptides on the back face of the tube are shown in light colors. A
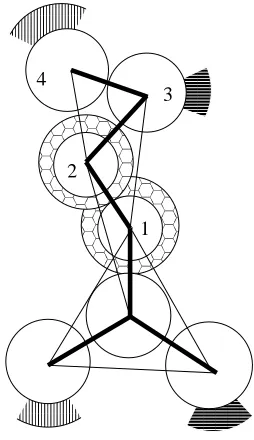
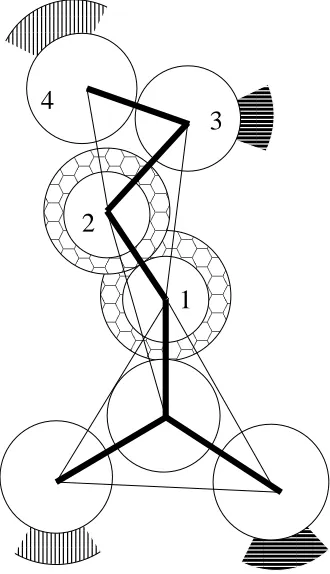
schematic of the configuration showing the various dimensions is presented in figure 2.3 (B).
The tube in figure 2.2 (A) has an inner diameter, I.D., of 17-18 angstroms, an outer diameter,
O.D., of 41 to 43 angstroms and a height, h, of 30 to 45 angstroms. There are eleven peptides
in the tube and they are oriented at an angle, θ, of 40-45 degrees with respect to the tube axis
between pointing into the tube and out of the tube. We have also observed larger tubes
contain-ing as many as 22 peptides with inner diameters of 32-39 angstroms, outer diameters of 55-62
angstroms, and heights of 43-49 angstroms.
Although our results are in qualitative agreement with the findings of Wacker et al.1 that
tubular protofibrils are found in aggregates of mutant huntingtin fragments containing expanded
polyglutamine tracts, some quantitative differences exist. While Wacker et al.1 found annular
structures ranging in diameter from 5-65 nm, the structures we have observed thus far have
diameters ranging from 4-6 nm. Wacker et al.1 did not find annular structures in aggregates of
proteins with a polyglutamine tract 20 residues in length, while we have found annular structures
in polyglutamine peptides 16 residues in length. A possible explanation for the latter difference
is that the folded state of huntingtin proteins with polyglutamine tracts 20 residues in length
is stable but a 20 residue tract of polyglutamine by itself does not have a stable folded state. In
other words the rest of the huntingtin protein surrounding the polyglutamine tract acts to stabilize
what would otherwise be an unstable and aggregation prone sequence. This is consistent with
the observation that polyglutamine does not fold to a unique native state. Due to the limitations
of current computers, simulations of the aggregation of multiple huntingtin proteins with long
polyglutamine tracts are impossible to perform in sufficient detail to resolve these questions.
Our results are interesting not only because of the recent discovery of tubular protofibrils in
experiments on aggregation of mutant huntingtin fragments containing expanded polyglutamine
tracts1but also because Perutz et al. predicted that polyglutamine forms water filled nanotubes19.
Perutz et al. examined x-ray scattering data onD2Q15K2and found that the scattering was
tube having an outer diameter of 31 angstroms and an inner diameter of 11.8 angstroms which
is slightly smaller than the simulated structures we typically find for polyglutamine chains 16
residues long. Finally, the peptides in the tubes that Perutz et al. envisioned were nearly
perpen-dicular to the tube axis; in contrast the tubes that we see are at an angle of about 45 degrees with
the tube axis.
In conclusion, we have found spontaneous formation of annular structures made up of beta
sheets in simulations of systems of model polyglutamine peptides starting from random
config-urations. These results are in qualitative agreement with the experimental findings of Wacker et
al.1and with the theoretical predictions of Perutz et al.19. We are currently performing
simula-tions of longer chains that are closer to the critical repeat length for Huntington’s disease in order
to better understand the connection between polyglutamine tract length and aggregate structure.
We would like to thank Dr. Andrew J. Schultz for his help in speeding up our discontinuous
molecular dynamics simulations. This work was supported by a National Institutes of Health
Molecular Biology Training Program fellowship and National Institutes of Health Grant
2.1
References
[1] Wacker, J. L., Zareie, M. H., Fong, H., Sarikaya, M., and Muchowski, P. J., Hsp70 and
hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning
monomer. Nature Structural and Molecular Biology, 11, 1215 (2004).
[2] Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., and Jr, P. T. L.,
Alpha-synuclein, especially the parkinson’s disease-associated mutants, forms pore-like
annular and tubular protofibrils. J. Mol. Biol., 322, 1089 (2002).
[3] Ding, T. T., Lee, S.-J., Rochet, J.-C., and Peter T. Lansbury, J., Annular alpha-synuclein
protofibrils are produced when spherical protofibrils are incubated in solution or bound to
brain-derived membranes. Biochemistry, 41, 10209 (2002).
[4] Wanker, E. E., Protein aggregation and pathogenesis of huntington’s disease: Mechanisms
and correlations. Biol. Chem., 381, 937 (2000).
[5] Bates, G., Huntingtin aggregation and toxicity in huntington’s disease. Lancet, 361, 1642
(2003).
[6] Starikov, E. B., Lerach, H., and Wanker, E. E., Folding of oligoglutamines: A theoretical
approach based upon thermodynamics and molecular mechanics. Journal of Biomolecular
Structure and Dynamics, 17, 409 (1999).
[7] Finke, J. M., Cheung, M. S., and Onuchic, J. N., A structural model of polyglutamine
deter-mined from a host-guest method combining experiments and landscape theory. Biophysical
[8] Burke, M. G., Woscholski, R., and Yaliraki, S. N., Differential hydrophobicity drives
self-assembly in huntington’s disease. Proc. Natl. Acad. Sci. USA, 100, 13928 (2003).
[9] Smith, A. V. and Hall, C. K., Bridging the gap between homopolymer and protein models:
A discontinuous molecular dynamics study. J. Chem. Phys., 113, 9331 (2000).
[10] Smith, A. V. and Hall, C. K., Alpha-helix formation: Discontinuous molecular dynamics
on an intermediate resolution model. Proteins: Structure, Function, and Genetics, 44, 344
(2001).
[11] Smith, A. V. and Hall, C. K., Assembly of a tetrameric alpha-helical bundle: Computer
simulations on an intermediate resolution protein model. Proteins: Structure, Function
and Genetics, 44, 376 (2001).
[12] Smith, A. V. and Hall, C. K., Protein folding versus aggregation: Computer simulations on
an intermediate resolution protein model. Journal of Molecular Biology, 312, 187 (2001).
[13] Nguyen, H. D., Marchut, A. J., and Hall, C. K., Solvent effects on the conformational
transition of a model polyalanine peptide. Protein Science, 13, 2909 (2004).
[14] Nguyen, H. D. and Hall, C. K., Molecular dynamics simulations of spontaneous fibril
for-mation by random-coil peptides. Proc. Natl. Acad. Sci. USA, 101, 16180 (2004).
[15] Alder, B. J. and Wainwright, T. E., Studies in molecular dynamics I. General method. J.
Chem. Phys., 31, 459 (1959).
[16] Rapaport, D. C., Molecular dynamics study of a polymer chain. J. Chem. Phys., 71, 3299
[17] Bellemans, A., Orban, J., and Belle, D. V., Molecular dynamics of rigid and non-rigid
necklaces of hard disks. Mol. Phys., 39, 781 (1980).
[18] Ding, F., Borreguero, J. M., Buldyrey, S. V., Stanley, H. E., and Dokholyan, N. V.,
Mecha-nism for the alpha-helix to beta-hairpin transition. Proteins, 53, 220 (2003).
[19] Perutz, M. F., Finch, J. T., Berriman, J., and Lesk, A., Amyloid fibers are water-filled
2.2
Figures
Page
2.1 Four-sphere representation of a polyglutamine sidechain. Hard spheres are
white. Square wells representing hydrophobic interactions are shown in a
hon-eycomb pattern. Square wells representing hydrogen bond acceptors have thick
horizontal stripes and square wells representing hydrogen bond donors have thin
vertical stripes. Covalent bonds are shown in bold and pseudo bonds to constrain
bond angles are thin lines. . . 17
2.2 A, close up of the tube formed in our simulations of 24 polyglutamine 16mers.
B, AFM image from the supplementary information of Wacker et al. showing
annular structures formed from a mutant huntingtin fragment containing a
polyg-lutamine tract 53 residues long (arrows indicate spherical oligomeric structures;
arrowhead indicates small annular structure). . . 18
2.3 A, Close up of the side of the tube formed from 24 polyglutamine 16mers with
1 3 4 2
A B
θ
h
O.D.
I.D.
s
A
B
C
HAPTER
3
S
OLVENT
E
FFECTS ON THE
A
GGREGATION OF
M
ODEL
P
OLYGLUTAMINE
P
EPTIDES
3.1
Introduction
The pathological manifestation of nine hereditary neurodegenerative diseases, including
Huntington’s disease, is the presence within the brain of aggregates of disease-specific proteins
that contain polyglutamine tracts longer than a critical length1,2. In the case of Huntington’s
the associated protein is called Huntingtin. Although the causative link between the aggregation
of polyglutamine-containing proteins and these so called “polyglutamine diseases” has not yet
been firmly established, protein aggregation is widely believed to play a key role. Evidence
pointing towards such a link is the observation that the diseases occur only when the protein’s
polyglutamine tract is longer than a certain critical length3–13,1 and that the proteins aggregate
in vitro and in vivo only when their polyglutamine tract is longer than a critical value14–21. Furthermore, the aggregation proceeds via a nucleation and growth mechanism that speeds up
as the length of the polyglutamine stretch increases16,22–24; this may explain why the diseases
increases.
The molecular mechanisms by which the proteins implicated in the polyglutamine diseases
aggregate is still unclear. In order to shed light on these mechanisms and to improve our
under-standing of the processes by which polyglutamine-containing proteins misfold and aggregate, we
have conducted molecular dynamics simulations of the aggregation of model polyglutamine
pep-tides. Although some work has been done on simulating isolated polyglutamine molecules25,26
and dimers of polyglutamine27, to our knowledge these are the first molecular simulations of the
aggregation of large systems of polyglutamine peptides.
Our simulations have been conducted using an intermediate-resolution protein model
de-veloped in our lab, PRIME, which is extended here to represent polyglutamine. PRIME was first
introduced by Smith and Hall28–31and later improved by Nguyen et al.32; it has the advantage
that it captures the essential features of the forces responsible for protein folding, such as the
hydrophobic effect and hydrogen bonding, yet can still be used to simulate large systems at long
timescales in a reasonable time. It stands at the crossroads between the “all-atom” models that
account for the motions of every atom on the protein being simulated as well as every solvent
atom and the more coarse-grained models that simply represent amino acid residues or a groups
of amino acid residues as beads on a lattice.
Proteins containing polyglutamine tracts are thought to form beta-sheets that are stabilized
by hydrogen bonds not only between backbone atoms but also between atoms on glutamine
sidechains33. This was first suggested by Max Perutz based on atomic level models that showed
that if polyglutamine were folded into an antiparallel beta sheet, the sidechain groups would be
group on the next strand34. In this structure the sidechains lie above and below the plane of the
beta sheet so that every hydrogen bond donor on one sidechain is matched with a hydrogen bond
acceptor on the next sidechain34.
Polyglutamine has been found to form beta sheets by a variety of experimental techniques
including circular dichroism, electron microscopy, and x-ray diffraction35,36. Bevivino et al.37
used infrared spectroscopy, circular dichroism, and electron microscopy to examine the structure
of an ataxin-3 protein with an expanded polyglutamine tract and found that the polyglutamine
regions adopt beta sheet structures. They also showed, using atomic level molecular modeling,
that the strands of the beta sheet need to be parallel in order to match every hydrogen bond donor
with a hydrogen bond acceptor. Tanaka et al.38used circular dichroism, infrared spectroscopy,
and electron microscopy to show that glutamine repeats adopt beta sheet structures when inserted
into myoglobin. They proposed that the beta sheets are antiparallel and that when the
polyglu-tamine sequence becomes too long, the beta sheets move to the surface of the protein where
they can more readily form intermolecular aggregates with corresponding beta sheets on other
molecules. Scherzinger et al.39 used electron microscopy to study the structure of aggregates
formed from Huntingtin containing an expanded polyglutamine tract and found that these
aggre-gates are fibrillar. Cooper et al.40stained protein fragments that contain expanded polyglutamine
tracts with Congo Red dye and examined them with polarized light microscopy to show that
they have beta-sheet conformations. Chen et al.24used circular dichroism to show that synthetic
polyglutamine peptides exist in beta-sheet conformations.
Based on analysis of x-ray crystallographic data, Perutz et al.41 recently suggested that
nanotube idea led naturally to the following explanation for the dependence of polyglutamine
aggregation on tract length. Polyglutamine tracts of forty residues or longer could form two turns
within the nanotube structure; these would be stablized through hydrogen bonding between the
turns. This double-turn structure could then serve as a nucleus for growth of a nanotube.
Polyglu-tamine tracts shorter than forty residues would not be able to adopt the whole two-turn structure;
structures formed by such peptides would be less stable since they would have fewer hydrogen
bonds41. This prediction is controversial and the x-ray diffraction patterns have recently been
reinterpreted42 to suggest that the peptides are in a stacked beta sheet conformation similar to
the classical fibril structure43. Recently, Sharma et al.44collected new x-ray diffraction data on
polyglutamine aggregates and suggested that polyglutamine beta sheets stack to form slablike
fibrils.
Annular structures have been observed experimentally in aggregating systems of
polyglu-tamine and other proteins associated with diseases of the brain. Wacker et al.45used atomic force
microscopy to examine aggregates formed from huntingtin containing a polyglutamine tract 53
residues in length and found annular structures that were themselves made up of smaller annular
structures. Lashuel et al.46 used electron microscopy to examine mutantα-synuclein proteins
that are linked to Parkinson’s disease and found annular structures. Ding et al.47used atomic
force microscopy to examineα-synuclein and also found annular structures.
The beta-sheets formed by polyglutamine-containing proteins assemble via a nucleated
pro-cess in which the rate limiting step is the formation of the first small section of beta-sheet
struc-ture, after which the beta-sheet grows quickly16,22–24. Experimental support for this
proteins increases. This is consistent with a nucleation-dependent mechanism, because
nu-cleus formation is more probable at higher concentrations16,18. Furthermore, aggregation of
polyglutamine-containing proteins is preceded by a so-called lag time, the time it takes for the
nucleus to assemble. After that, aggregation proceeds rapidly as the nucleus grows16,48,24. The
lag time depends on the concentration of the aggregating protein- the higher the concentration
the more likely the nucleus is to form and the smaller the lag time16. The lag time also depends
on the length of the polyglutamine tract; longer polyglutamine peptides tend to have shorter lag
times48. Nucleation of polyglutamine aggregates can also be “seeded” by adding small amounts
of aggregated polyglutamine1,16,24.
Simulations have been used to study the folding of isolated polyglutamine peptides and the
dimerization of small systems of two polyglutamine peptides. Starikov et al.25 examined the
structure of a 40-residue polyglutamine protein using three different all-atom simulation codes,
CHARMM, AMBER, and OPLSAA. The protein folded into a beta sheet when CHARMM was
used and into a compact random coil when the two other packages were used. The simulated
beta sheet exhibited inter-strand hydrogen bonding between donors and acceptors on the
back-bone but more intra- than inter-strand hydrogen bonding between sites on the sidechains. In the
compact random coil they found many sidechain-sidechain hydrogen bonds. They suggested
that polyglutamine chains longer than a critical chain length fold into beta hairpins which then
aggregate if the concentration is high enough for nucleation.
Finke et al.26 developed a united-atom model for polyglutamine in which each residue is
represented by two beads- one for the backbone atoms and another for the sidechain atoms.
CI2 proteins with polyglutamine insertions. The strengths of the various interactions in their
model were adjusted so that the simulated change in stability between the CI2 protein and the
CI2 protein with a polyglutamine insertion would agree with the change in stability determined
experimentally. They then used their model to simulate a single CI2 protein with polyglutamine
insertions and concluded that isolated short polyglutamine peptides should exist as random coils.
Stork et al.27used fully atomistic CHARMM simulations with explicit water molecules to
measure the stability of various configurations of polyglutamine 56 and 60 residues long as well
as of two polyglutamine chains each 36 residues long. They started their simulations in two of
the conformations that polyglutamine has been postulated to adopt- the nanotube and the
beta-helix. The nanotube configuration is the configuration suggested by Perutz that was discussed
earlier. The beta helix is similar to the nanotube except that the helix is shaped like a triangle
(as opposed to being circular) with straight portions separated by three sharp turns. Stork et
al. determined the stability of these configurations based on whether the peptides unfolded or
remained in a particular configuration. They found that none of the tube configurations were
stable but that a beta-helical peptide with three helical repeats of 18 residues and a dimer of two
beta-helical petides each with two helical repeats of 18 residues were stable.
In this paper we describe the extension of PRIME32,49–51 (previously used to describe
polyalanine) to the description of polyglutamine and present our findings on the aggregation
of a systems of twenty-four and forty-eight polyglutamine peptides that are sixteen residues
long. PRIME uses discontinuous potentials in order to take advantage of discontinuous
molecu-lar dynamics (DMD), a simulation technique that is very fast compared to traditional molecumolecu-lar
at high temperatures and slowly cooled to the simulation temperature. Simulations are performed
in the canonical ensemble with fixed number of particles, system volume, and temperature. The
number of particles and system volume are picked to give a fixed concentration of 5mM. The
effects of hydrophobicity on the system are studied by varying the strength of the hydrophobic
interaction from 12.5 percent to 5 percent of the strength of the hydrogen bonding interaction.
We monitor the aggregation of the polyglutamine peptides over a wide range of temperatures.
Aggregation and aggregate structure are monitored by calculating the number of beta hydrogen
bonds, the percent of the peptides in beta sheets, and the number of beta regions in the system.
Highlights of our results include the following. At intermediate values of temperature we
observed the spontaneous formation of beta sheets and annular structures made up of beta sheets.
These annular structures resemble nanotubes and beta barrels. The number of peptides in beta
sheets decreases with increasing temperature. At the very highest temperatures the system does
not fold or aggregate- it remains a system of isolated random coils. At low temperatures
amor-phous aggregates are formed.
The organization of this paper is as follows. In Section II the peptide model and the
simula-tion method are discribed. In Secsimula-tion III we present our results on the formasimula-tion of beta sheets,
the formation of annular structures made up of beta sheets, and the effects of hydrophobicity on
the aggregation of our model polyglutamine peptides. Section IV contains a discussion of our
3.2
Methods
3.2.1 Model Forces
In this work, we extend the protein model, PRIME (PRotein Intermediate resolution ModEl)
to polyglutamine. PRIME was originally developed by Smith and Hall28–31, inspired by the work
of Takada et al.52and improved upon by Nguyen et al.32. This model is designed to be used with
a simulation technique known as discontinuous molecular dynamics (DMD)53–55, an extremely
fast alternative to traditional molecular dynamics. DMD simulations are appropriate for systems
with discontinuous potentials such as hard sphere and square well potentials where the forces
operating on the molecules change only at specific points in time and space when the particles
collide. In contrast, the forces in traditional molecular dynamics simulations (which are based
on continuous potentials) change at all points in space and time. DMD simulations are much
faster than traditional molecular dynamics simulations because Newton’s laws can be solved
analytically instead of by numerically integrating the potentials. Furthermore, the time course of
the simulation is not restricted to small equally-spaced time steps as is the case with traditional
molecular dynamics; instead the entire system can advance in time to the next discontinuity in
the potential, which is referred to as an “event.”
PRIME is an off-lattice, unbiased, intermediate-resolution protein model28–32 which has
thus far been applied mainly to polyalanine. Each amino acid residue is composed of a
three-sphere backbone comprised of united atoms NH, CαH, and C=O. An alanine sidechain is
rep-resented by one sphere. PRIME has realistic bond lengths and bond-angle constraints and has
the ability to interact both intra- and inter-molecularly via hydrogen bonding and hydrophobic
the distance between consecutive Cα atoms is fixed so as to maintain the interpeptide bond in
the trans configuration. The sidechains are held in positions relative to the backbone so that all
residues are L-isomers. The effect of solvent is factored into the energy function as a potential
of mean force.
All forces are modeled by discontinuous potentials. The excluded volumes of the united
atoms are modeled using hard-sphere potentials with realistic diameters. Covalent bonds are
maintained between adjacent spheres along the backbone by imposing hard-sphere repulsions
whenever the bond lengths attempt to move outside of the range between (1-δ)` and (1+δ)`
where`is the bond length andδ is a tolerance which we set equal to 2.5 percent. Ideal
back-bone bond angles, Cα-Cαdistances and residue L-isomerization are achieved by imposing
pseu-dobonds which also fluctuate within a tolerance of 2.5 percent. Interactions between hydrophobic
sidechains are represented by a square-well potential of depthHP and range 1.5σR whereσR
is the side chain united atom diameter. Hydrophobic sidechains must be separated by at least
one intervening residue in order to interact. Hydrogen bonding between amide hydrogen atoms
and carbonyl oxygen atoms on the same or neighboring chains is represented by a square-well
potential of depthHBbetween NH and C=O united atoms whenever: (1) the virtual hydrogen
and oxygen atoms (whose location can be calculated at any time) are separated by 4.2 ˚A(the sum
of the NH and C=O well widths), (2) the nitrogen-hydrogen and carbon-oxygen vectors point
to-wards each other within a fairly generous tolerance, (3) neither the NH nor the C=O are already
involved in a hydrogen bond with a different partner, and (4) the NH and C=O are separated
by at least one intervening residue along the chain. We allow hydrogen bonds between groups
of separations along the chain of hydrogen bonding pairs observed in structures from the
pro-tein data bank by Stickle et al.56. In order to satisfy the requirement that the nitrogen-hydrogen
and carbon-oxygen vectors point towards each other, we have used the approach of Nguyen et
al.32 but the parameter set of Ding et al.57. This parameter set allows formation of hydrogen
bonds at all temperatures while still reproducing the hydrogen bond angle and distance
distri-butions of Smith and Hall29. The reason that we did not use the parameter set of Nguyen et
al.32is that beta sheets formed in simulations with that parameter set at temperatures just below
the beta sheet / random coil transition temperature were unphysical in that they had very few
hydrogen bonds. This happened in our polyglutamine simulations but not in the polyalanine
simulations of Nguyen and Hall50,49,51 because the additional hydrophobic bead on the
polyg-lutamine sidechains held the polygpolyg-lutamine beta strands together in a beta sheet like structure at
higher temperatures and this parameter set makes it harder for hydrogen bonds to form at high
temperatures. Since beta sheets with few hydrogen bonds are physically unrealistic, we used
the parameter set of Ding et al.57. We define the reduced temperature, T* as kT /HB and the
relative hydrophobicity, R asHP/HB.
PRIME is extended to polyglutamine by adopting a more complex sidechain representation.
We considered a variety of approaches including a two-sphere representation but eventually
set-tled on a four-sphere sidechain representation since this gives the most faithful representation
of the geometry of glutamine sidechains. The four-sphere representation allows us to model
the polyglutamine sidechain with the same level of detail as the PRIME model backbone. Each
sidechain is represented by four spheres as depicted in figure 4.1. Spheres 1 and 2 (as labeled in
wells to mimic excluded volume and hydrophobic interactions. Sphere 3 represents a carbonyl
group; it has a hard core to mimic excluded volume and a directionally-dependent square well
to mimic a hydrogen bond acceptor. Sphere 4 represents an amine group; it has a hard core to
mimic excluded volume and a directionally-dependent square well to mimic a hydrogen bond
donor. The four spheres all have their bond angles constrained by a system of pseudobonds. The
methyl, carbonyl, and amine groups on the polyglutamine sidechain are modeled in the same way
as the methyl, carbonyl, and amine groups in the polyalanine PRIME model and as such, have
exactly the same sizes, bond lengths, pseudobond lengths, interaction strengths, and interaction
ranges.
3.2.2 Discontinuous Molecular Dynamics
DMD simulations are conducted as follows. Each sphere of the model protein chain is
as-signed a random initial position that does not violate any of the size constraints or asas-signed bond
lengths and angles. It is also assigned an initial velocity chosen at random from a
Maxwell-Boltzmann distribution at a specified reduced temperature, T*. The simulation proceeds
accord-ing to the followaccord-ing schedule: identify the first event, move forward in time until that event
occurs, calculate new velocities for the pair of spheres involved in the event and calculate any
changes in system energy resulting from hydrogen bond events or hydrophobic interactions, find
the second event, and so on. Types of events include excluded volume events, covalent bond
events, pseudobond events, square-well hydrogen bond events, and square-well hydrophobic
in-teraction events. An excluded volume event occurs when the surfaces of two hard-spheres collide
to move outside of their assigned bond length and the two particles feel an infinite repulsion that
forces them back into their assigned bond length. Square-well events include capture,
well-bounce, and well-dissociation “collisions” when a sphere enters, attempts to leave, or leaves the
square-well of another sphere. For more details on DMD simulations with square-well potentials,
see articles by Alder and Wainwright53and Smith et al.58.
The simulations were performed in the canonical ensemble, which means that the number
of particles, volume, and temperature are held constant. The number of particles and box volume
were picked to give the desired concentration. Between three and five simulations were run at
every data point. Simulations of twenty-four chains were run for 30 billion DMD events and
sim-ulations of forty-eight chains were run for 80 billion DMD events to ensure that a stable structure
was formed. Properties were averaged over the last 2 billion DMD events and error bars represent
the standard deviation between all the runs at a given state point. Periodic boundary conditions
were used to eliminate artifacts due to simulation box walls. The Andersen thermostat59 was
used to maintain constant temperature; in this method all the particles experience random,
infre-quent events where they are given a new velocity selected randomly from a Maxwell-Boltzmann
distribution centered at the simulation temperature. These events are referred to as “ghost
colli-sions” or collisions with “ghost particles.”
Systems containing twenty-four and forty-eight chains at a concentration of 5 mM were
slowly quenched from a random configuration of high temperature random coils at T*=0.5 to
the temperature of interest which ranged from T*=0.075 to T*=0.205. Simulations at relative
hydrophobicity R=0.05, 0.075, 0.1, and 0.125 were performed in order to learn how the strength
number of beta backbone - backbone hydrogen bonds which we define as hydrogen bonds in
which both the donor and acceptor backbone dihedral angles,φandψ, adjacent to the hydrogen
bond lie in the range 0 ≤ ψ ≤ 180 and −180 ≤ φ ≤ −30. This range for the dihedral
angles is the same as that used by Nguyen et al.32. We also monitored the number of backbone
- backbone, sidechain - sidechain, and sidechain - backbone hydrogen bonds as well as the
fraction of peptides in beta sheets and the number of beta regions (defined below). The fraction
of peptides in beta sheets was calculated based on the following definitions. A beta sheet is
defined to be a structure in which two or more peptides share a number of beta hydrogen bonds
between them that is greater than or equal to half the chain length (eight hydrogen bonds in the
case of our 16-residue chains). This definition was motivated by the following. If two peptides
are perfectly aligned, the number of hydrogen bonds between them could equal the chain length.
Although the N and C spheres associated with each residue could both form hydrogen bonds, the
direction in which the residues face alternates along the chain from facing the peptide in question
to facing away from the peptide in question. Thus the maximum possible number of hydrogen
bonds between them would equal the chain length. The fraction of peptides in beta sheets is the
number of peptides involved in beta sheet structures divided by the total number of peptides in
the system. We were also interested in the contiguousness of the beta hydrogen bonds so we
defined a beta region to be a section in an aggregate where two neighboring peptides share at
least five beta hydrogen bonds in a row. Since the residues alternate between facing towards
and away from each other, the term “in a row” does not include the two hydrogen bonds formed
when the residues in question face away from each other. Thus five in a row means hydrogen
number along the chain.
3.3
Results
DMD simulations were performed on systems containing 24 and 48 16-residue
polyglu-tamine chains to see if they would form fibrils. We chose a chain length of sixteen since Nguyen
and Hall49found that systems containing polyalanine chains sixteen residues long that were
ini-tially in random coil conformations spontaneously formed fibrils as the simulations progressed.
We found that as our simulations progressed the polyglutamine peptides folded into beta sheets
that tended to curve. When there were enough peptides in the system, the beta sheets rolled up
into a tube-like structure that resembled a classic beta barrel. Snapshots of one of the resulting
tube structures are shown in figure 3.2. The tube in this figure contains eleven peptides and has
an inner diameter of 17-18 angstroms, an outer diameter of 41 to 43 angstroms and a height of
30 to 45 angstroms.
We observed a variety of tube morphologies in our simulations including tubes with large
diameters (figure 3.3 A), tubes that are connected to each other (figure 3.3 B), and tubes
con-nected to beta sheets (figure 3.3 C). The large tube shown in figure 3.3 A has an inner diameter
of 32-39 angstroms, an outer diameter of 55-62 angstroms, and a height of 43-49 angstroms.
We examined the self assembly of these structures at relative hydrophobicity values of
R=0.005, 0.075, 0.10, and 0.125 in order to find a reasonable value for this parameter, which
is the only adjustable parameter in the model. It is difficult to assign a value to this parameter
a priori because we are not only coarse graining away detail about the hydrogen bond donors
level structure of polyglutamine aggregates, besides that they are made up of beta sheets (at this
point a controversy exists as to whether these beta sheets form ring like structures41,45 or
slab-like fibrils42,44), it would be useful to know the value of the relative hydrophobicity at which the
system self assembles into the most ordered beta sheets. This value could then be used in future
simulations.
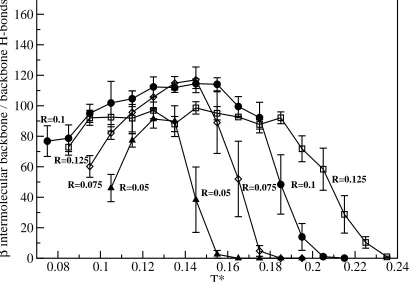
Figure 3.4 shows the number of beta intermolecular backbone - backbone hydrogen bonds
as a function of T* at the four different values of the relative hydrophobicity. At all R the system
shows the following behavior as a function of the reduced temperature. At low T*, the system
has a small number of beta intermolecular hydrogen bonds because it is in a collapsed state that
is relatively disordered. As T* increases, the system forms more and more beta intermolecular
hydrogen bonds because it has more flexibility, which gives it the freedom to find more ordered
states. At high T*, the number of beta intermolecular hydrogen bonds decreases as temperature
increases because entropy takes over and the peptides become increasingly random coil-like.
The value of the relative hydrophobicity, R, affects the self assembly of the peptides as
follows. At the highest values of the relative hydrophobicity (R=0.125), the system remains in a
beta sheet conformation for higher values of T* than it does at all the other values of the relative
hydrophobicity. It also has fewer beta intermolecular hydrogen bonds at low temperatures than
systems at intermediate values of the hydrophobicity (R=0.1 and R=0.075). As the hydrophobic
interaction decreases from R=0.125 to R=0.05, the T* at which the system transitions from beta
sheets to random coils decreases. Additionally, as the hydrophobic interaction decreases from
R=0.1 to R=0.05, the low value of T* at which the peptides undergo a transition from beta sheets
into beta sheets becomes narrower as the hydrophobic interaction strength decreases. At low
values of the hydrophobic interaction (R=0.05) the peptides do not self-assemble into beta sheets
as well as they do at higher values of R because they do not have as many beta intermolecular
backbone - backbone hydrogen bonds as the systems at higher values of R. At high values of
the hydrophobic interaction (R=0.125) the peptides do not self-assemble in to beta sheets as well
as they do at lower values of R because they get caught in kinetic traps more easily due to the
strength of the hydrophobic attraction.
In order to quantify the structure of the aggregates that are formed, we have determined
the fraction of peptides that are in beta sheets and the number of beta regions in the system.
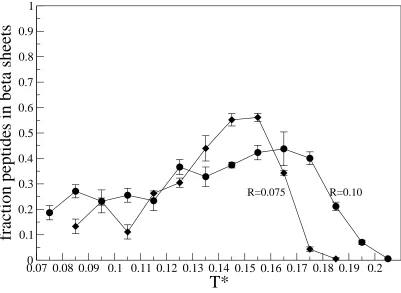
Figure 3.5 shows the fraction of peptides that are in beta sheets versus T* for all the hydrophobic
interaction strengths studied. Figure 3.6 shows the number of beta regions versus T* for all the
hydrophobic interaction strengths studied.
The trends seen in figure 3.4 are also seen in figures 3.5 and 3.6. At low values of T*,
the system is kinetically trapped in amorphous structures and hence all measures of beta sheet
formation are low. As T* increases, all of the curves go through a maximum at an optimum
temperature for beta sheet formation. As T* increases further, the system undergoes a transition
to random coils because entropy is taking over. As the hydrophobicity increases, the temperature
at which the transition from beta sheets to random coils occurs increases and the temperature at
which the transition from beta sheets to amorphous aggregates occurs decreases. This trend is
not followed however, at the highest value of the relative hydrophobicity, R=0.125. In this case
the fraction of peptides in beta sheets is generally lower than at the other values of the relative