Reciprocal Mouse and Human Limb Phenotypes Caused by
Gain- and Loss-of-Function Mutations Affecting
Lmbr1
Richard M. Clark,* Paul C. Marker,*
,1Erich Roessler,
†Amalia Dutra,
‡John C. Schimenti,
§Maximilian Muenke
†and David M. Kingsley*
,**
*Department of Developmental Biology, Stanford University, Stanford, California 94305-5327,†Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892-1852,‡Cytogenetic and Confocal Microscopy Core,
National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892,§The Jackson Laboratory, Bar Harbor, Maine 04609 and**Howard Hughes Medical Institute, Stanford University, Stanford, California 94305-5327
Manuscript received March 20, 2001 Accepted for publication June 1, 2001
ABSTRACT
The major locus for dominant preaxial polydactyly in humans has been mapped to 7q36. In mice the dominantHemimelic extra toes(Hx) andHammertoe(Hm) mutations map to a homologous chromosomal region and cause similar limb defects. TheLmbr1gene is entirely within the small critical intervals recently defined for both the mouse and human mutations and is misexpressed at the exact time that the mouse
Hxphenotype becomes apparent during limb development. This result suggests thatLmbr1may underlie preaxial polydactyly in both mice and humans. We have used deletion chromosomes to demonstrate that the dominant mouse and human limb defects arise from gain-of-function mutations and not from haploinsufficiency. Furthermore, we created a loss-of-function mutation in the mouseLmbr1 gene that causes digit number reduction (oligodactyly) on its own andin transto a deletion chromosome. The loss of digits that we observed in mice with reducedLmbr1activity is in contrast to the gain of digits observed inHxmice and human polydactyly patients. Our results suggest that theLmbr1gene is required for limb formation and that reciprocal changes in levels ofLmbr1activity can lead to either increases or decreases in the number of digits in the vertebrate limb.
V
ERTEBRATE limb malformations that cause al. 1994;Tsukurovet al. 1994;Vargaset al. 1995; Zgur-icaset al. 1999). Despite this phenotypic variation, all changes in digit number are relatively common andinclude polydactyly (extra digits) and oligodactyly (too these mutations are dominantly inherited and highly penetrant, and recent mapping studies have localized few digits). Of these defects, preaxial polydactyly is the
most common in humans and includes forms of thumb many of the mutations to the same 7q36 region ( Heu-tinket al. 1994;Tsukurovet al. 1994;Hinget al. 1995; duplications, triphalangeal thumb, and index finger
du-plications on the anterior (preaxial) side of the limb Radhakrishnaet al. 1996;Vargaset al. 1998;Zguricas
et al. 1999; Dobbs et al. 2000). Therefore, a major lo-(TemtamyandMcKusick1978). While preaxial
poly-dactyly is frequently associated with additional defects at cus for triphalangeal thumb-polysyndactyly syndrome (TPTPS; OMIM 190605) at 7q36 is responsible for al-sites outside the limbs, a subset of families with inherited
preaxial polydactyly have limb-specific defects (Zguri- most all dominant preaxial polydactylies and polysyn-dactylies with defects restricted to the limbs.
cas et al. 1999). Limbs from patients harboring these
preaxial polydactyly mutations typically present with re- In the mouse, the dominantHemimelic extra toes(Hx) and Hammertoe (Hm) limb mutations are thought to placement of the thumb with one or more triphalangeal
elements (Canunet al. 1984;Cordeiroet al. 1986;Heu- be analogous to the human TPTPS mutations at 7q36 (Heutinket al. 1994;Tsukurovet al. 1994), and both
tinket al. 1994;Tsukurovet al. 1994;Hinget al. 1995;
Vargaset al. 1995;Radhakrishnaet al. 1996;Zguricas map to a mouse chromosome region homologous to 7q36 (Clark et al. 2000). Hx mice have limb defects et al. 1999;Dobbset al. 2000). Many of these polydactyly
mutations are also associated with additional distal limb that include preaxial polydactyly and radial and tibial defects that include soft tissue fusions (syndactyly) of hemimelia (Knudsen and Kochhar 1981; Masuyaet adjacent digits and/or radial or tibial dysplasia/aplasia al. 1995) that closely resemble the human limb pheno-(Canun et al. 1984; Cordeiroet al. 1986; Heutink et types.Hmmice do not have changes in digit number but have highly penetrant webbing between digits (Green
1964) similar to that observed in some polysyndactyly
Corresponding author:David M. Kingsley, Howard Hughes Medical mutations that have been mapped to 7q36 (Tsukurov Institute, Stanford University, Beckman Ctr., B300, 279 Campus Dr.,
et al. 1994). In crosses segregating Hx and Hm only Stanford, CA 94305-5327. E-mail: [email protected].
a single recombination was observed in 3664 meioses
1Present address:Department of Anatomy, University of California,
San Francisco, CA 94143-0452. (Sweet 1982). This extremely tight linkage suggests
that amplified the expected size fragments from commercial
that the mouse mutations affect neighboring genes or
genomic DNA (Clontech, Palo Alto, CA). Following sequence
alternatively may be different alleles of the same gene
verification of test amplicons, two primer pairs [HxF1b (5⬘-acc
with the observed recombination arising from an intra- tgttccaacacggctcgc-3⬘) and HxR1 (5⬘-actcccgcacttggctgtgg-3⬘) genic crossover. and the nested pair HxF1b (above) and HxR1b (5⬘-acacct
cgtcctgcccttcc-3⬘)] were used by the Physical Mapping Core
Recently,Heus et al. (1999) defined anⵑ450-kb
re-(National Human Genome Research Institute/National
Insti-gion on 7q36 that contains dominant polydactyly
muta-tutes of Health) to screen a human bacterial artificial
chromo-tions and identified a small set of genes that are
con-some (BAC) library (Incyte Genomics, Palo Alto, CA),
re-tained within the TPTPS critical region. In a parallel sulting in the identification of clone address 575h20. Direct study in the mouse, Clark et al. (2000) defined an sequencing of this clone as well as Southern blot hybridization
experiments verified that it contains exon 1 and 5⬘ flanking
ⵑ450-kb interval for the mouseHmandHxmutations
sequences ofLMBR1(data not shown).
and identified several genes within this critical region.
Fluorescentin situhybridization analysis:Slides with
chro-The mouse and human candidate genes are
ortholo-mosome metaphase spreads were incubated for 1 hr at 37⬚in
gous, confirming previous conjecture that the human 2⫻ SSC (0.3 m NaCl and 0.3 m sodium citrate) and then and mouse phenotypes arise by defects in similar genes dehydrated sequentially in 70, 80, and 90% ethanol.
Chromo-some DNA was denatured in 70% formamide, 2⫻SSC for 2
(Clarket al. 2000). However, extensive mutational
anal-min at 72⬚followed by dehydration in ethanol washes of 70,
ysis in both human and mouse has not identified lesions
80, 90, and 100%. Fluorescent in situhybridization (FISH)
in the coding sequences of any candidate genes (Heus
was performed with probes labeled with spectrum
orange-et al. 1999;Clark et al. 2000). dUTP (Vysis, Downers Grove, IL), essentially as described pre-The absence of coding region mutations raises the viously (Pinkel et al. 1986; Lichter et al. 1988). On each
slide, 100 ng of labeled DNA was applied. Nonunique and
possibility that the dominant mouse and human
muta-nonspecific DNA hybridization was blocked by preannealing
tions are regulatory alleles that disrupt expression of a
the probe with a 10-fold excess of human Cot1 DNA. Labeled
gene or genes in the interval.Clarket al. (2000) showed
and blocking DNAs were denatured at 75⬚ for 10 min and
that one of the genes within the mouse critical region, then preannealed at 37⬚for 15 min. The hybridization mixture called Limb region 1 (Lmbr1), is normally expressed in contained labeled DNA in 10 ml of 50% formamide, 2⫻SSC, developing limbs at the times that both theHxandHm and 10% dextran sulfate at pH 7.0. Slides were hybridized overnight at 37⬚. Post-hybridization washes were performed
phenotypes arise. More importantly,Lmbr1was
dynami-at 45⬚ as follows: (1) 50% formamide, 2⫻ SSC, 20 min; (2)
cally misexpressed in Hxlimbs at the exact time that
1⫻SSC, 10 min; and (3) 0.1⫻SSC, 10 min. Slides were
coun-Hxlimb morphology first appears. Expression changes terstained with propidium iodide-Antifade (Intergen, Pur-included a possible overexpression of the gene followed chase, NY) or 250 ng/ml 4⬘,6-diamidino-2-phenylindole (Boeh-by a dramatic decrease inLmbr1transcript levels at later ringer Mannheim, Indianapolis) with Antifade.
Design ofLmbr1targeting vector and homologous recombi-stages (Clarket al. 2000). The humanLmbr1ortholog
nation:The 5⬘ end of the Lmbr1gene is present on mouse
(LMBR1) is contained entirely within the human critical
BAC clone 136E36 from the 129 strain CITB mouse BAC II
region for TPTPS mutations at 7q36 (Heuset al. 1999),
library (Research Genetics, Huntsville, AL;Clarket al.2000,
and the striking correlation between the appearance of and our unpublished data). DNA from this BAC was digested defects in Hx mice and Lmbr1 misregulation suggests with restriction enzymes and subcloned into a plasmid vector, and an 8.3-kbBamHI fragment harboring the exon that
con-that the mouse and human limb mutations may be
al-tains the start site of theLmbr1open reading frame was isolated
leles of theLmbr1gene (Clarket al. 2000).
by hybridization with Lmbr1sequences. A 1.1-kbMluI/KpnI
Here we use deletion chromosomes in both mice and
fragment that contains this exon (seeresults) was replaced
humans to show that the dominant mouse and human by a PGKneo-positive selection cassette by cloning sequences limb phenotypes are likely to arise by gain-of-function flanking the 1.1-kb fragment into the pPNT vector. The pPNT mechanisms. In addition, we created a loss-of-function vector contains the herpes thymidine kinase gene for negative
selection.
allele of the Lmbr1 gene to test whether this gene is
Targeting of the endogenousLmbr1locus was performed
required for normal limb development. Mice with
re-in R1 embryonic stem (ES) cells (kre-indly provided by Janet
duced Lmbr1 function show distal limb reductions in- Rossant) as described previously (Joyner 1993). Construct cluding oligodactyly. These phenotypes are reciprocal linearized withNotI was electroporated into ES cells and posi-to those caused by the classical dominant mutations. tive selection was performed using G418 while Gancyclovir was used to select against nonhomologous integration events.
The complementary defects suggest that levels of
ex-Cells were grown on G418-resistant irradiated mouse
embry-pression of theLmbr1gene play a key role in controlling
onic fibroblasts. ES cell colonies with targeted integrations
the development of skeletal structures in the vertebrate were detected by nested PCR amplification of a 3.1-kb junction
limb. fragment created by homologous recombination between the
3⬘ arm of the replacement construct and the endogenous
Lmbr1 locus. Primers used for amplification were neo1 (5⬘ -gcagcctctgttccacataca-3⬘) and LK1 (5⬘ -tgagggagccagaggagtca-MATERIALS AND METHODS
3⬘) for primary PCR and neo2 (5⬘-gccaagttctaattccatcagaa-3⬘) and LK2 (5⬘-aaaatacaagaaaacctacagaatc-3⬘) for secondary PCR.
Isolation of human genomic bacterial artificial chromosome
clones:Primers were prepared on the basis of partial human Amplifications were performed with ampliTaq (Applied Bio-systems, Foster City, CA) with cycle conditions of 94⬚(3 min); expressed sequence tag (EST) sequences predicted to
by 6 cycles of 94⬚ (30 sec), 59⬚ (1 min), and 72⬚ (5 min); Hm limb phenotypes. Where necessary, inheritance of the
Lmbr1ATGallele was determined by typing with primers N1F and
followed by 72⬚ (15 min). Homologous recombinants were
verified by additional PCR with primers LK3 (5⬘-ggtaggggttatt N1R.Hdhdf4J/⫹males were also crossed toLmbr1ATG/Lmbr1ATG
females. Deletion progeny from this cross were identified by ggtacagactt-3⬘) and neo3 (5⬘-gcctcccctacccggtagaatt-3⬘) that
amplify a junction fragment created by homologous recombi- typing animals with primers ATGF and ATGR. The Hdhdf4J
deletion includesLmbr1exon 1, and inLmbr1ATG/Hdhdf4Jmice
nation between the 5⬘arm of the targeting construct and the
endogenousLmbr1 locus. PCR with primers LK3 and neo3 the exon 1 product amplified by ATGF and ATGR is not
was performed using the expand long template PCR system present (seeresults).
(Hoffmann La Roche, Basel, Switzerland) with cycle conditions Skeletal preparations:Alizarian red-stained skeletons were of 94⬚(2 min); 10 cycles of 94⬚(10 sec), 65⬚(30 sec), 68⬚(8 prepared as previously described (Green1952) from weaning min); followed by 25 cycles of 94⬚(10 sec), 65⬚(30 sec), 68⬚ age or adult mice.
(8 min with an additional 20-sec/cycle); followed by 68⬚(10 min). We refer to the allele created by targeting asLmbr1ATG. Generation and typing ofLmbr1ATGmutant mice:Two
inde-RESULTS pendently selected ES cell clones were injected into C57BL/
6J host blastocysts, and chimeric animals (agouti coat color) TheHxandHmmutations are gain of function:While were crossed to B6D2F1/J animals (The Jackson Laboratory).
the phenotypes and mode of inheritance of the mouse
Germline transmission was determined by coat color, and
HxandHm mutations are described in detail (Green
heterozygous progeny were identified. Homozygous animals
were generated by intercrossing heterozygotes, and pheno- 1964;KnudsenandKochhar1981;Zakeriet al. 1994;
typic analysis ofLmbr1ATG/⫹ and Lmbr1ATG/Lmbr1ATGanimals
Masuyaet al. 1995), it has not been clear whether these
as well as wild-type controls was performed on the 129SV/J⫻ dominant mutations produce limb abnormalities by
B6D2F1/J mixed genetic background. Initially, heterozygous
gain-of-function or loss-of-function mechanisms. A
dele-animals were typed with primers that were used to identify
tion of proximal mouse chromosome 5 (Hdhdf4J ) that
targeted ES clones (primers neo1, LK1, neo2, and LK2; see
above). Afterward, progeny were typed with duplexed primer does not produce obvious webbing or gross polydactyly
pairs that allowed all genotypes to be distinguished in single was previously reported (Schimenti et al. 2000). The
PCR reactions. One primer set amplifies a 174-bp fragment HxandHmmutations were previously mapped between
of Lmbr1 exon 1 that is deleted by the targeted mutation
theShhandIl6loci on proximal chromosome 5 (
Mar-[primers ATGF (5⬘-tcttgaaccgcttctccctgag-3⬘) and ATGR (5⬘
-tinet al.1990;Robertet al.1994;Marigoet al.1995;
cccttccatcctcctttcatacc-3⬘)], and another pair amplifies a
268-bp fragment from theneoresistance cassette inserted into the Clark et al. 2000) in a region likely to be included
locus by targeting [primers N1F (5⬘-acagacaatcggctgctctgatg- in theHdhdf4J deletion. PCR typing confirmed that this
3⬘) and N1R (5⬘-gatggatactttctcggcaggag-3⬘)]. PCR amplifica- deletion removesLmbr1coding sequences (see materi-tion condimateri-tions were 94⬚(2 min); 30 cycles of 94⬚ (30 sec),
als and methodsand Figure 3F), demonstrating that
63⬚(1 min), 72⬚(30 sec); followed by 72⬚(10 min).
Hdhdf4J/⫹mice carry a single copy of the
Lmbr1 candi-Northern blot analysis with wild-type and mutant RNA
sam-ples:Total brain RNA from wild-type and mutant mice was date region.
prepared using TRIzol (Invitrogen, Carlsbad, CA) according We examined limbs ofHdhdf4J/⫹mice to test whether
to the manufacturer’s instructions. Messenger RNA was iso- deletion of the candidate interval produces limb pheno-lated from total RNA using oligo(T) cellulose (FastTrack 2.0
types typical of those observed in eitherHm/⫹orHx/⫹
kit; Invitrogen). For Northern blot analysis, 2g of poly(A)
mice. No webbing was seen between digits, and Alizarin
RNA per lane was loaded and separated on 1% agarose/1.5%
formaldehyde gels by electrophoresis. RNA was then blotted red-stained skeletal preparations did not show extra
to Hybond N⫹ membrane (Amersham, Arlington Heights, skeletal elements characteristic of Hx/⫹ mice,
sug-IL). Blots were probed with a32P-labeledLmbr1cDNA probe.
gesting that the classicalHm andHxphenotypes arise
This probe contains the open reading frame (ORF) of the
by a gain-of-function mechanism rather than from loss-Lmbr1gene that encodes LMBR1L 3⬘of the sequence for exon
of-function mutations in a gene in the interval (Table
1. Blots were scanned on a model 425 E phosphor imager
(Molecular Dynamics, Sunnyvale, CA) and relative expression 1 and Figure 1). AlthoughHdhdf4J/⫹mice do not have
levels were calculated using a Gapdh probe to control for limb defects that resemble those inHxand Hmmice,
loading differences. minor coalitions of distal wrist bones were observed in
Hdhdf4J mice, crosses, and typing: A male mouse carrying
25% of wrists inHdhdf4J/⫹animals and included fusions
the proximal chromosome 5 deficiencyHdhdf4J(Schimentiet
of the central to either distal carpals 2 or 3 (dc2 and al. 2000) in trans to a Mus mus castaneous chromosome
(C57BL/6J⫻129/Jae⫻M. mus castaneousmixed-strain back- dc3, respectively; Table 2). Animals carrying theHdhdf4J
ground) was crossed to eitherHm/Hm(C3HeB/FeJLe strain) deletion were also smaller than wild-type mice as has or wild-type (B10.D2/nSn strain) females. To determine in- been observed for several other chromosomal deletions. heritance of the deletion, DNAs from progeny animals were
Dominant preaxial polydactyly mutations that map to
typed with primers that amplify the microsatellite locus
7q36 are gain of function:The critical region for human D5Mit148that is predicted to be removed by theHdhdf4J
defi-ciency. This locus is polymorphic betweenM. mus castaneous TPTPS mutations at 7q36 is homologous to the critical
and C3HeB/FeJLe and B10.D2/nSn strain DNA (data not region for the mouseHm andHxmutations and
con-shown), and progeny that inherited the deficiency that lacked tains the human ortholog of the mouse Lmbr1 gene
theM. mus castaneousallele were identified.Hm/Hdhdf4Jmale
(LMBR1;Heuset al. 1999;Clarket al. 2000). To
deter-progeny were crossed to wild-type B6D2F1/J females or to
mine whether the dominant human limb mutations Lmbr1ATG/⫹or Lmbr1ATG/Lmbr1ATGfemales and progeny that
TABLE 1
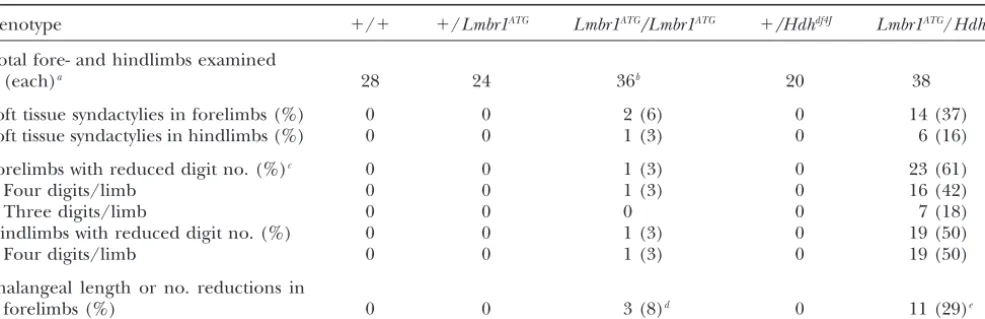
Frequency of defects affecting the digits
Genotype ⫹/⫹ ⫹/Lmbr1ATG Lmbr1ATG/Lmbr1ATG ⫹/Hdhdf4J Lmbr1ATG/Hdhdf4J
Total fore- and hindlimbs examined
(each)a 28 24 36b 20 38
Soft tissue syndactylies in forelimbs (%) 0 0 2 (6) 0 14 (37)
Soft tissue syndactylies in hindlimbs (%) 0 0 1 (3) 0 6 (16)
Forelimbs with reduced digit no. (%)c 0 0 1 (3) 0 23 (61)
Four digits/limb 0 0 1 (3) 0 16 (42)
Three digits/limb 0 0 0 0 7 (18)
Hindlimbs with reduced digit no. (%) 0 0 1 (3) 0 19 (50)
Four digits/limb 0 0 1 (3) 0 19 (50)
Phalangeal length or no. reductions in
forelimbs (%) 0 0 3 (8)d 0 11 (29)e
aAll limbs examined for both soft tissue and skeletal defects.
bTwo of 128Lmbr1ATGhomozygous animals (⬍1%) had obvious limb defects (syndactyly and/or oligodactyly) that were detected
as live mice. Limbs from these mice were prepared as skeletons and are included in this data set along with all other homozygotes that were also prepared as skeletons.
cDigit number reduction refers to instances of lateral digit fusions that produce a single distal-most phalanx (bony syndactylism)
or to cases where one or more digits are completely absent (oligodactyly).
dIn each case the most posterior digit (digit V) was affected. In one instance the second phalanx was reduced in size, while
in the other two affected digits a single bone replaced the first and second phalanges.
eIn each of the affected limbs the most posterior digit was affected, and in one limb a second digit was also affected. In 9 of
the 12 affected digits the first and second phalanges were replaced by a single bone. In the remaining affected digits the second phalanx was grossly reduced in length.
analysis to map the region surrounding theLMBR1in- gene (Roessler et al. 1997). None of these patients have limb defects that include preaxial polydactyly or terval in human patients with deletions or translocations
in the 7q36 region (Roessleret al.1997 and references polysyndactyly (Roessler et al. 1997). Therefore, the entire region containing the LMBR1 critical area can therein). A 135-kb human BAC clone that contains exon
1 of theLMBR1gene was isolated. This BAC produces be deleted without producing the TPTPS characteristics of the dominant limb mutations that were indepen-two chromosome 7q36-specific hybridization signals in
FISH analysis of control human cells (Figure 2A). FISH dently mapped to 7q36 (Figure 2E). These data suggest that, like the mouseHxandHm mutations, the domi-analysis of cells from a patient with a t(7;17)
transloca-tion (T4) shows two chromosome 7LMBR1 hybridiza- nant human limb mutations that map to 7q36 are likely to act by a gain-of-function rather than a loss-of-function tion signals [7 and der(7), Figure 2B], indicating that
LMBR1is proximal to the T4 translocation breakpoint. mechanism.
Generation of a loss-of-function allele of theLmbr1 FISH analysis of cells harboring a t(7;9) translocation
(T1) shows that oneLMBR1hybridization signal is asso- gene: The location of the Lmbr1 gene in the critical interval for both the mouse and human limb mutations ciated with the intact copy of chromosome 7 while the
other is on the chromosome 9 derivative [7 and der(9), and the altered expression of this gene in Hx mice suggest that regulatory mutations inLmbr1may be re-Figure 2C], indicating that LMBR1 is distal to the T1
translocation breakpoint. Only a single chromosome 7 sponsible for the dominant mouse and human limb defects. To examine the role of this gene during normal hybridization signal was seen in cells from a patient with
ade novo7q36 deletion (Figure 2D). These data suggest limb development, we created a loss-of-function muta-tion in the Lmbr1gene using embryonic stem cell tar-that the LMBR1 BAC maps precisely between the T1
and T4 translocations in a region completely covered geting.
TheLmbr1gene encodes a highly conserved product by the 7q36 deletion (Figure 2E). Physical estimates
based on assembly of genomic clones in the 7q36 region of 490 amino acids (LMBR1L) as well as a smaller prod-uct of 32 amino acids (LMBR1S) produced by an alter-suggest that the T1 and T4 translocations are located
ⵑ265 and 335 kb distal to theSHHgene (Roessleret natively spliced Lmbr1 transcript (Clark et al. 2000). The first 22 amino acids of both proteins are identical. al. 1997). The presentLmbr1mapping results, combined
with the earlier cytogenetic data, suggest that as many To create an allele of Lmbr1, we subcloned genomic DNA containing the 5⬘-most coding exon of theLmbr1 as 32 unrelated patients with cytogenetic deletions in
tion cassette (Figure 3, A–C). The deleted fragment contains 365 bp 5⬘of the predicted translational start site of exon 1 and 696 bp 3⬘of the exon 1 splice donor site. The targeted mutation therefore deletes the first known exon of theLmbr1gene that contains the transla-tional start site as well as coding sequence for both knownLmbr1products. The targeted mutation may also remove the endogenousLmbr1transcriptional start site. Two independent ES cell clones transmitted the Lmbr1mutation (Lmbr1ATG) through the germline. Mice heterozygous for the targeted mutation were inter-crossed to produceLmbr1ATG/
Lmbr1ATG mice, and PCR analysis confirmed that the coding exon containing the translational start site of both predictedLmbr1products was completely missing in DNA fromLmbr1ATG homozy-gotes (Figure 3D). Homozygous mice were present at normal Mendelian ratios (36⫹/⫹; 91Lmbr1ATG/⫹; 44
Lmbr1ATG/Lmbr1ATG;P⫽0.20, chi-square test). Both male and femaleLmbr1ATGhomozygous mice are fertile and we have been able to maintain theLmbr1ATGallele by in-tercrossing homozygotes. We also examined a variety of tissues fromLmbr1ATGhomozygotes by histology (includ-ing liver, kidney, spleen, testis, epididymus, and seminal vesicle) and did not detect significant abnormalities when compared to wild-type controls (data not shown). To assess whether the targeted mutation created a null allele of theLmbr1gene, we probed Northern blots of wild-type andLmbr1ATG/
Lmbr1ATGadult brain poly(A)
Figure1.—Mice hemizygous for theHx-Hminterval do not
RNA with aLmbr1cDNA probe. The normal 3- and 5-kb
have classicalHxorHmlimb phenotypes. For A–D, distal is
up, proximal is down, anterior is left, and posterior is right. messages that contain the first exon of theLmbr1gene
Digit number is indicated by Roman numerals. (A and B) (Clark et al. 2000) were greatly reduced or
undetect-Ventral views of adult forefeet showing soft tissues. InHm/⫹ able in Lmbr1ATG homozygotes (Figure 3E). However,
limbs (A) digits II–V are connected to each other by lateral
long exposures of Northern blots showed multiple
tran-soft tissue fusions (asterisks) as well as to the ventral surface
scripts in brain RNA from homozygotes (Figure 3E).
of the limb, and digits are flexed ventrally toward the viewer
(the “hammertoe” phenotype). In contrast, no soft tissue webs These transcripts were present at levels ofⵑ7% that of
are observed in Hdhdf4J/⫹ limbs that lack one copy of the
wild-typeLmbr1messages in brain. These molecular data
region that contains theHm-Hxinterval (B), and digits from suggest that the targeted mutation reducedLmbr1 func-these mice are fully extended (compare to Figure 4A). (C and
tion but that the mutation may not have created a null
D) Dorsal views of cleared forelimbs stained with alizarin red
allele (seediscussion).
to identify skeletal elements.Hx/⫹limbs (C) have preaxial
polydactyly in which digit I, which normally has two small Lmbr1ATG homozygous mice have low incidences of
phalanges (see Figure 4D), is replaced by extra triphalangeal limb defects:As expected from our studies of the chro-digits (asterisks). In contrast, limbs fromHdhdf4J/⫹mice (D)
mosome 5 deletion mice, the loss-of-function mutation
have normal digit morphology. These results demonstrate that
in Lmbr1 did not produce a phenocopy of either the
the classical webbing and polydactyly phenotypes result from
Hxor Hm mutations. We did, however, detect a very
gain-of-function mechanisms and not from haploinsufficiency.
low incidence of limb abnormalities inLmbr1ATG homozy-gous animals (Table 1). These phenotypes presented as digit loss or reduction rather than the gain-of-digit the 22 amino acids that are common to the N termini
of both the LMBR1L and LMBR1S proteins (Clarket al. number typically seen in Hxanimals. One mouse ap-peared to have only four digits on its right forefoot, 2000). Genomic sequence upstream of the translational
start site forLmbr1products is contiguous with sequence while a second homozygous mouse had only four digits on a hindfoot. The latter mouse also had mild soft tissue ofLmbr15⬘rapid amplification of cDNA ends (RACE)
products for 166 bp, at which point RACE products end syndactylies between central digits on its other three limbs (Table 1 and Figure 4B). Skeletal preparation (our unpublished data). This point may represent the
transcriptional start site for the Lmbr1 gene, and we revealed that reduction of digit number in the affected Lmbr1ATG/Lmbr1ATGforelimb arose from loss of the meta-refer to the exon we isolated asLmbr1exon 1. We used
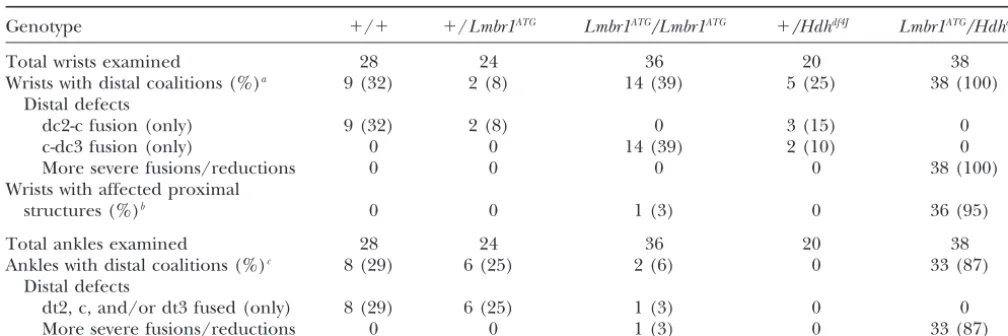
selec-TABLE 2
Frequency of wrist and ankle abnormalities in mice of different genotypes
Genotype ⫹/⫹ ⫹/Lmbr1ATG Lmbr1ATG/Lmbr1ATG ⫹/Hdhdf4J Lmbr1ATG/Hdhdf4J
Total wrists examined 28 24 36 20 38
Wrists with distal coalitions (%)a 9 (32) 2 (8) 14 (39) 5 (25) 38 (100)
Distal defects
dc2-c fusion (only) 9 (32) 2 (8) 0 3 (15) 0
c-dc3 fusion (only) 0 0 14 (39) 2 (10) 0
More severe fusions/reductions 0 0 0 0 38 (100)
Wrists with affected proximal
structures (%)b 0 0 1 (3) 0 36 (95)
Total ankles examined 28 24 36 20 38
Ankles with distal coalitions (%)c 8 (29) 6 (25) 2 (6) 0 33 (87)
Distal defects
dt2, c, and/or dt3 fused (only) 8 (29) 6 (25) 1 (3) 0 0
More severe fusions/reductions 0 0 1 (3) 0 33 (87)
adc2, dc3, and “c” are distal carpals 2 and 3 and the central, respectively. “More severe fusions/reductions” refers to fusions
that may involve dc2, dc3, and the central but also distal carpals 1 and 4/5 as well as reduced size of remaining elements.
bRefers to fusions involving the radiale, ulnar, and/or pisiforme and that were frequently accompanied by reduction in size
of remaining fused elements.
cdt2, dt3, and “c” are distal tarsals 2 and 3 and the central, respectively. “More severe fusions/reductions” refers to fusions
that may involve dt2, dt3, and the central but also dt4/5 and that were frequently accompanied by reduced size of remaining elements.
the missing digit is digit V (compare Figure 4E to 4D). ure 4M). However, the number of distal wrist bones has been observed to vary depending on strain background A small posterior element in this limb may be a rudiment
of the fifth metacarpal (see arrow in Figure 4E). In (DavisandCapecchi1994). In wild-type control mice from our cross, we observed a high frequency (32%) of contrast, in the affected hindlimb of the homozygous
mutant animal, reduction in digit number was caused forelimbs in which dc2 was partially or completely fused to the central (Table 2). InLmbr1ATGheterozygous mice, by fusion of digits III and IV at the level of the phalanges
to produce a single digit distally that ended in a single we observed a lower frequency (8%) of distal wrist coali-tions that involved fusion of the central to dc2 (Table third phalanx (Table 1 and Figure 4H).
Further examination of homozygous Lmbr1ATG ani- 2). In contrast, inLmbr1ATG/Lmbr1ATGwrists we observed a high frequency (39%) of distal coalitions that included mals revealed low incidences of additional digit defects
that included reduction in phalange length or number only fusions between the central and dc3 (Table 2 and Figure 4N). While no wild-type or heterozygous wrists that were never observed in wild-type or heterozygous
control limbs (Table 1). On both fore- and hindlimbs that we examined had defects in proximal carpal bones, in 1 of 36 homozygous wrists the ulnare and pisiforme digit I normally has two phalanges (P1 and P2, both of
which are very small in forelimbs), while digits II–V have were partially fused (Table 2).
We also observed coalitions of anklebones in wild-three phalanges each (P1, P2, and P3; Figure 4, D, G,
and J). While the fifth digits of mostLmbr1ATG/Lmbr1ATG type and heterozygous and homozygousLmbr1ATGmice (Table 2). Normally, six bones comprise the distal ankle forelimbs had three phalanges of normal relative
lengths, one forelimb had a fifth digit in which the size [distal tarsals 1 (dt1), 2 (dt2), 3 (dt3), and 4/5 (dt4/ 5); the pisiforme; and the central] while the talus and of P2 was greatly reduced (Table 1 and data not shown).
In two other limbs, the P1 and P2 elements were re- the calcaneus comprise the proximal ankle. While most ankles of each genotype that we examined had the ca-placed by a single element (Table 1 and Figure 4K). No
Lmbr1ATG/Lmbr1ATGhindlimbs had obvious reductions in nonical ankle organization, we observed coalitions of distal anklebones in wild-type andLmbr1ATG/⫹mice that the length of phalanges or any reductions in phalangeal
number (data not shown). included dt2, dt3, and/or the central (Table 2). In Lmbr1ATGhomozygous ankles we observed distal ankle-We also examined the organization of the wrists and
ankles in wild-type, heterozygous, and homozygous bone fusions that included coalitions involving dc2, dc3, dc4/5, and/or the central (Table 2). We did not observe mice. In general, five bones comprise the distal wrist
[distal carpals 1 (dc1), 2 (dc2), 3 (dc3), and 4/5 (dc4/ any defects in the proximal anklebones of wild-type or Lmbr1ATG heterozygous or homozygous mice (data not 5); and the central], while three bones comprise the
Defects in Lmbr1ATG mice appear to be confined to Hdhdf4Jmice were also produced by crossingHdhdf4J/Hm mice toLmbr1ATG/⫹animals (seematerials and meth-the wrist, ankle, and footplate regions, with no obvious
defects detected in more proximal limb structures or ods). As withHdhdf4J/⫹mice,Lmbr1ATG/Hdhdf4Jmice were structures outside the limb. typically smaller than littermates (eitherLmbr1ATG/⫹or
Lmbr1ATG/Hdhdf4Jmice have severe distal limb defects:
Lmbr1ATG/
Hm mice; data not shown). Whether the ob-To further test the role ofLmbr1 in development, we served decrease in survival ofLmbr1ATG/
Hdhdf4J relative generated micetrans-heterozygous for the Lmbr1ATG
al-toHdhdf4J/⫹mice results from a specific effect ofLmbr1 lele and the Hdhdf4J deletion that removes the region
or rather from differences in the interaction of the containing theLmbr1locus. If residualLmbr1function deletion with different stain backgrounds used in our is present in Lmbr1ATG homozygotes, we reasoned that
crosses is not known. Lmbr1ATG/Hdhdf4Jmice should have an even greater
re-Limbs from Lmbr1ATG/Hdhdf4J mice showed dramatic duction in Lmbr1 activity and therefore may display digit defects that include soft tissue webbing (Table 1 more severe phenotypes. Most Lmbr1ATG/
Hdhdf4J mice
and Figure 4C), reductions in the number of digits were produced by crossing eitherHdhdf4J/⫹or
Hdhdf4J/
(Table 1 and Figure 4, C, F, and I), and reductions in Hmmice toLmbr1ATG/Lmbr1ATGanimals. In these crosses,
the number or length of phalanges in digits (Table 1 progeny that inherited theHdhdf4J deletion (Lmbr1ATG/
and Figure 4L). These defects were more severe and Hdhdf4Jmice) were recovered at less than the expected
occurred at much greater frequencies than those in frequency of 50% at weaning (40 Lmbr1ATG/⫹ or
Lmbr1ATG/
Lmbr1ATG mice. For example, 55% of all
Lmbr1ATG/
Hmprogeny; 16Lmbr1ATG/
Hdhdf4Jprogeny;
P⫽ Lmbr1ATG/Hdhdf4Jlimbs had fewer than five digits, com-0.001, chi-square test). A small number of Lmbr1ATG/
pared to⬍1% inLmbr1ATGhomozygotes (Table 1). The digit reduction was also more severe inLmbr1ATG/Hdhdf4J animals, with one-third of the affected forelimbs show-ing only three remainshow-ing digits (Table 1 and Figure 4F). The reduction of digits appeared to be restricted to central or posterior digits on the basis of morphological criteria (compare Figure 4F and 4I to 4D and 4G).
Lmbr1ATG/Hdhdf4J mice also had high incidences of wrist and ankle defects (Table 2). While we observed minor distal wrist bone coalitions restricted to dc2, dc3, and/or the central inLmbr1ATG
homozygous mice and control mice, we observed dramatic coalitions/reduc-tions of distal wrist bones that involved dc2, dc3, and/or
Figure2.—Dominant TPTPS limb mutations at 7q36 are gain of function. (A–D) FISH analysis to chromosome meta-phase spreads of cells from patients with 7q36 rearrangements previously described byRoessleret al. (1997). In all cases, a BAC clone probe that contains exon 1 of theLMBR1gene was used for hybridization. Hybridization signal is red, and chromosome 7 and derivatives are as labeled (arrows). In normal cells (A) and in cells harboring the T4 (B) or the T1 (C) translocations theLMBR1probe recognizes two chromo-some 7 signals on either normal or translocated 7q36 segments as indicated. In contrast, in cells from a patient with ade novo
7q36 deletion (patient 30,Roessleret al. 1997) (D), only a single chromosome 7-specific hybridization signal was ob-served. These results localize a portion of the LMBR1gene between the T1 and T4 breakpoints in a region that was pre-viously shown by Roessleret al. (1997) to be distal to the
SHH locus on 7q36 (E). The LMBR1gene, which is within the TPTPS critical interval defined by Heuset al. (1999), is contained within the 7q36 deletion that also removes theSHH
Figure3.—Generation, typing, and characterization of animals harboring a targeted mutation in theLmbr1gene. (A) Genomic lo-cus and replacement construct are shown. B, M, and K denote sites recognized by BamHI, MluI, and
KpnI, respectively. A 1.1-kbMlu
I-KpnI fragment including the exon that contains the putative transla-tional start site of theLmbr1gene (ATG) was replaced by a PGKneo
selection cassette by homologous recombination in ES cells (dashed lines). Orientations of theLmbr1
and neo genes are as indicated (arrows). Primer sites used to de-tect homologous recombination or for subsequent typing experi-ments are indicated (split arrows). (B) Primers LK3 and neo3 amplify a 4.2-kb fragment generated by homologous recombination be-tween the 5⬘ arm of the replace-ment construct and theLmbr1 ge-nomic locus. (C) Primary PCR (1⬚ PCR) with primers neo1 and LK1 and secondary PCR (2⬚PCR) with primers neo2 and LK2 amplify a 3.1-kb fragment generated by homologous recombination be-tween the 3⬘ arm of the replace-ment construct and the endoge-nousLmbr1locus. (D) Genotyping by duplexed PCR with primer pairs that amplify (1) a 174-bp fragment (primers ATGF and ATGR) that is deleted from the
Lmbr1 locus by homologous re-combination and (2) a 268-bp fragment from theneogene that was inserted into theLmbr1locus by targeting (primers N1F and N1R). (E) Northern blot analysis of poly(A) brain RNA prepared from wild-type andLmbr1ATG/Lmbr1ATGmice as indicated. ALmbr1cDNA probe recognizes major
transcripts ofⵑ3 and 5 kb in wild-type RNA (these data andClarket al.2000).Lmbr1-specific signal is still observed inLmbr1ATG/ Lmbr1ATGRNA after long exposures (right), with most prominent transcripts (asterisks) ofⵑ3.5 and 5.5 kb along with additional
less abundant species (arrows). Remaining transcripts were present in homozygous mutant brain RNA atⵑ7% of wild-type levels as determined by quantification using aGapdhcontrol probe (bottom). (F) Typing of DNA from progeny from aHm/Hdhdf4J⫻ Lmbr1ATG/Lmbr1ATG cross by PCR as indicated. WhileLmbr1 exon 1 sequence was amplified from DNA of phenotypically Hm
progeny (left lane, bottom band), amplification was not observed from DNA from non-Hm(Hdhdf4J/Lmbr1ATG) progeny (right
lane, bottom band absent). This result demonstrates thatLmbr1is absent fromHdhdf4Jchromosomes. Amplification of theneo
product (top band) served as a positive control for PCR.
the central as well as other distal carpals in allLmbr1ATG/ and the ulna were sometimes narrow and the junction between the radius and ulna and abnormal proximal Hdhdf4Jwrists that we examined (Table 2, compare Figure
4O to 4N). Furthermore, 36 of 38Lmbr1ATG/Hdhdf4Jlimbs wrist bones was disorganized (data not shown). How-ever, lengths of the long bones of both the fore- and that we examined had coalitions/reductions of
proxi-mal wrist bones (Figure 4O) in contrast to only one hindlimbs were approximately identical to those from control wild-type andLmbr1ATG/⫹animals, and obvious observed proximal wrist bone coalition inLmbr1ATG
ho-mozygous mice (Table 2). skeletal defects outside limbs were not apparent. The abnormalities seen in both Lmbr1ATG/Lmbr1ATG The limb defects that we observed inLmbr1ATG/Hdhdf4J
mice are primarily restricted to the most distal structures andLmbr1ATG/Hdhdf4Jmice suggest that theLmbr1gene is required for normal development of the distal struc-of the limb (wrist/ankle and digits). In forelimbs with
DISCUSSION at 7q36 do not produce dominant limb phenotypes.
The dominant limb phenotypes previously associated Several dominant limb mutations that cause
polydac-with these regions thus must occur by gain-of-function tyly were recently shown to arise by haploinsufficiency.
mechanisms (see also Schimenti et al. 2000). As no For example, mice heterozygous for a loss-of-function
coding region mutations were found in any of the candi-mutation in Alx4 have preaxial polydactyly (Qu et al.
date genes within the mouseHx/Hmor human TPTPS 1998;Takahashiet al. 1998), and loss of a single copy of
critical regions (Heuset al. 1999;Clarket al. 2000), we theGli3gene in both mice and humans causes preaxial
think it likely that the dominant limb phenotypes arise polydactyly and syndactyly (Vortkampet al. 1991;Hui
by gain-of-function regulatory mutations that alter the andJoyner 1993). In contrast, our genetic data here
expression of one or more genes in the region. show that chromosome deletions covering either the
The most obvious candidate for theHxandHm muta-mouseHm/Hx interval or the human TPTPS interval
tions is the novelLmbr1gene.Lmbr1is one of only two known genes that are located entirely within the critical intervals for both the mouse and human limb mutations, and levels ofLmbr1transcripts are dramatically misregu-lated inHxlimbs at the exact time that theHxphenotype is first morphologically apparent (Clark et al. 2000). To test the requirement for the Lmbr1 gene during development, we created a mutation in theLmbr1gene that deletes the first known exon that contains the
pre-Figure 4.—Mice with reduced Lmbr1 function have dra-matic reductions in distal limb structures. Genotypes are indi-cated at the top of each column, and digit number is indiindi-cated by Roman numerals. Question marks denote instances where digit identity could not be assigned with certainty. For A–O, distal is up, proximal is down, anterior is left, and posterior is right. Soft tissues (A–C) and cleared skeletal preparations that were stained with alizarin red (D–O) are shown. (A–C) Ventral views of adult forefeet. (A) Lmbr1ATG heterozygous
forefoot with wild-type digit separations. (B) Lmbr1ATG/ Lmbr1ATGforefoot with mild soft tissue webbing between
cen-tral digits (asterisk). (C) Four-digitLmbr1ATG/Hdhdf4Jforefoot
displaying dramatic syndactyly of remaining central digits (as-terisk). (D–F) Dorsal views of forefeet. (D)Lmbr1ATG/⫹
hetero-zygotes have normal digit number and skeletal morphology. (E) ALmbr1ATGhomozygous limb with reduced digit number
that appeared to result from reduction of digit V (an arrow denotes what may be a rudimentary fifth metacarpal). (F) Severely affected Lmbr1ATG/Hdhdf4J forelimb with only three
remaining digits. (G–I) Dorsal views of hindfeet. (G)
Lmbr1ATG/⫹hindlimb of normal morphology. (H)
Homozy-gousLmbr1ATGhindlimb with reduced digit number resulting
from bony syndactyly of digits III and IV to produce a single digit distal to the site of fusion (asterisk). (I)Lmbr1ATG/Hdhdf4J
hindlimb with only four digits. ( J–L) Dorsal/lateral views high-lighting posteriormost digits of forelimbs. ( J) Lmbr1ATG/⫹
limbs have normal posterior digits consisting of metacarpals (m) and three phalanges (P1, P2, and P3). (K)Lmbr1ATG/ Lmbr1ATGlimb in which P1 and P2 elements of digit V have
been replaced by a single element. (L)Lmbr1ATG/Hdhdf4Jlimb
in which the most posterior digit (asterisk) is reduced in size and P1 and P2 are replaced by a single element. The base of posteriormost metacarpal was also fused to its nearest neigh-bor (arrow). (M–O) Dorsal or dorsal/lateral views of wrists. (M)Lmbr1ATG/⫹ wrist with canonical organization of distal
carpals 1, 2, 3, 4/5, and the central (c) in the distal wrist and the radiale (r), ulnare (u), and pisiforme (p) in the proximal wrist. (N) HomozygousLmbr1ATG wrist with minor fusion of
the central to distal carpal 3. (O) Severely affectedLmbr1ATG/ Hdhdf4Jwrist in which only a small single distal element
Figure 5.—Mutational and pheno-typic summary and model for role of
Lmbr1in causing limb phenotypes. (A) The humanLmbr1ortholog consists of 17 coding exons spread over 212 kb as determined by analysis of human geno-mic sequences (see alsoIanakievet al. 2001). While the exact size ofLmbr1in mouse has not been determined, the mouse ortholog is at least 100 kb in size (Clarket al. 2000). The targeted muta-tion that we created in the mouse deletes the first exon ofLmbr1and causes loss of distal limb structures.Ianakievet al.
(2001) showed that a small deletion
in-cluding exon 4 of the human LMBR1
gene causes ACHP. Patients with this dis-order have distal limb truncations that are more severe than those observed in mice. In humans the distance between exons 1 (deleted in mice) and 4 (deleted in ACHP patients) is 66 kb. The location of mutations at different sites in the
Lmbr1gene, each of which causes distal limb reductions, suggests a specific
re-quirement for Lmbr1 during normal
limb growth and patterning. (B) We pro-pose a model whereby differences in
Lmbr1 activity lead to reciprocal limb phenotypes. In gain-of-function (GOF) mutations, additional skeletal elements are formed. In contrast, loss-of-function (LOF) Lmbr1 mutations cause reduc-tions of the distal limb.
dicted site of translational initiation for both known of hands and feet) suggest that the humanLMBR1gene is also required for formation of distal limb structures. Lmbr1 products. Mice carrying this allele show greatly
reduced expression ofLmbr1transcripts but still express Acheiropodia (ACHP; OMIM 200500) is an autosomal recessive condition that has been mapped to a small low levels of novel transcripts. These transcripts may
initiate from an alternative promoter in the region or region on 7q36 that overlaps the region Heus et al. (1999) defined as containing TPTPS mutations (
Esca-from within the PGKneoselection cassette that was
in-serted into the gene to create theLmbr1ATG allele. On millaet al. 2000). Unlike the dominant TPTPS pheno-types, ACHP patients present with loss or truncation of the basis of the residual expression seen in Northern
blots and the genetic behavior of the mutation, we be- hands and feet, although more proximal limb skeletal elements are relatively spared (ToledoandSaldanha
lieve that theLmbr1ATGmutation is a hypomorphic rather
than a null mutation in theLmbr1gene. 1969; Toledo et al. 1972; Grimaldi et al. 1983). The molecular lesion for ACHP was recently identified as a Mice heterozygous or homozygous for the Lmbr1ATG
mutation are viable and fertile and do not show the small 4- to 6-kb deletion that eliminates exon 4 of the LMBR1gene (see Figure 5A;Ianakievet al. 2001). Loss typical limb defects that are induced by the classical
dominant gain-of-function mutations Hx and Hm. In- of this exon causes premature truncation of theLMBR1 open reading frame and likely generates a null allele stead, homozygous mutant mice show a very low
inci-dence of limb defects, including oligodactyly, reduction of the gene (Ianakiev et al. 2001). The phenotypes observed in ACHP patients clearly show a key role for in length or number of phalanges, and soft tissue or
bony syndactyly. The severity and penetrance of digit Lmbr1/LMBR1 during human limb development. Al-though the human hand and foot truncations are more defects is markedly increased when theLmbr1ATG allele
is placed over the deficiency mutation, and severe carpal severe than those seen in Lmbr1ATG homozygous and
LmbrATG/Hdhdf4Jmice, the human and mouse phenotypes and tarsal coalitions are also observed. The striking
dis-tal limb phenotypes seen in mice carrying theLmbr1ATG both involve distal limb truncations, loss of digits, and relative sparing of proximal skeletal structures. The dif-allele strongly argue that theLmbr1gene is required for
in affected mice. This possibility can be tested by gener- ing limb development are currently unknown. It is formally possible that the Lmbr1ATG mutation and the ating additionalLmbr1alleles.
The loss-of-function mouse and human phenotypes human ACHP mutation also disrupt distant cis -regula-tory interactions with theShhgene. Since the mouse and do not directly address whether the dominant limb
mu-tations are also due to mumu-tations in theLmbr1/LMBR1 human mutations cause small disruptions that remove single coding exons ofLmbr1/LMBR1 and are located gene. However, the types of defects caused by
loss-of-function Lmbr1 mutations (reductions of distal limb at two distinct locations within the gene, we think it is much more likely that they disrupt the function of the structures including digits) are reciprocal to those
caused by the gain-of-function mutations (extra distal Lmbr1/LMBR1gene itself and cause the limb defects. The predicted protein product of theLmbr1/LMBR1 limb structures including digits). Combined with
previ-ous expression data showing thatLmbr1is misregulated gene is a novel multipass transmembrane protein that does not fall into any known functional class but has in polydactylous limbs ofHxanimals (Clarket al. 2000),
it is likely that the reciprocal phenotypes are due to been highly conserved in different organisms (Clark
et al. 2000). Its structure suggests that it may encode a altered levels of expression of theLmbr1gene (see
Fig-ure 5B). An important goal for futFig-ure experiments will membrane anchoring protein, adhesion molecule, transporter, or cell surface receptor. An important goal be to identify the nature of the DNA alterations that
generate the dominant limb phenotypes in both mice for future studies will be to determine how this gene interacts with other pathways and how it acts to control and humans.
The reciprocal phenotypes seen in the current studies the development of distal skeletal structures in the verte-brate limb.
resemble the reciprocal effects of transplantation or ablation of zone of polarizing activity (ZPA) cells during normal limb patterning. The presence of an additional
source of ZPA activity on the anterior side of a devel- LITERATURE CITED oping limb induces preaxial polydactyly (Saunders
Belloni, E., M. Muenke, E. Roessler, G. Traverso, J. Siegel-
Bar-1948;HonigandSummerbell1985), while ablation of teltet al., 1996 Identification of Sonic hedgehog as a candidate the ZPA region from the posterior of the limb causes gene responsible for holoprosencephaly. Nat. Genet.14:353–
356. loss of distal limb structures (Pagan et al. 1996).Shhis
Canun, S., R. M. Lomeli, R. MartinezandA. Canevale, 1984 Ab-normally expressed along the posterior limb margin and sent tibiae, triphalangeal thumbs and polydactyly: description of is thought to mediate the proliferative and patterning a family and prenatal diagnosis. Clin. Genet.25:182–186.
Chang, D. T., A. Lopez, D. P. von Kessler, C. Chiang, B. K. Simandl
activity of the ZPA (Riddleet al. 1993). Previous studies
et al., 1994 Products, genetic linkage and limb patterning activity showed that theShhgene is ectopically expressed along of a murine hedgehog gene. Development120:3339–3353. the anterior of the limb margin ofHxmice in the region Clark, R. M., P. C. MarkerandD. M. Kingsley, 2000 A novel
candidate gene for mouse and human preaxial polydactyly with where extra digits form in affected limbs (Masuyaet al.
altered expression in limbs of Hemimelic extra-toes mutant mice. 1995). These phenotypes and the phenotypes seen in Genomics67:19–27.
the current studies could be explained if theLmbr1gene Cordeiro, I., H. SantosandP. Maroteaux, 1986 Congenital ab-sence of the tibiae and thumbs with polydactyly: a rare genetic normally acts as a positivetrans-acting regulator ofShh
disease (Werner’s syndrome). Ann. Genet.29:275–277. activity. Davis, A. P., andM. R. Capecchi, 1994 Axial homeosis and
appen-TheShhgene is also physically linked to theHx/Hm dicular skeleton defects in mice with a targeted disruption of hoxd-11. Development120:2187–2198.
and 7q36 critical regions, and it was previously proposed
Dobbs, M. B., F. R. Dietz, C. A. Gurnett, J. A. Morcuende, C. M.
that dominant human and mouse limb mutations may Steyerset al., 2000 Localization of dominantly inherited iso-be due to defects in cis-acting regulatory elements of lated triphalangeal thumb to chromosomal region 7q36. J.
Or-thop. Res.18:340–344. theShh gene (Changet al. 1994; Sharpeet al. 1999).
Escamilla, M. A., M. C. DeMille, E. Benavides, E. Roche, L. Almasy
The data reported here show that the human TPTPS et al., 2000 A minimalist approach to gene mapping: locating critical region containing theLMBR1 gene is located the gene for acheiropodia, by homozygosity analysis. Am. J. Hum.
Genet.66:1995–2000. ⵑ300 kb distal to SHH in the region between the T1
Green, M., 1952 A rapid method for clearing and staining specimens and T4 breakpoints. Patients carrying the T1 but not for the demonstration of bone. Ohio J. Sci.52:31–33. the T4 translocation show mild holoprosencephaly phe- Green, M. C., 1964 New mutation. Mouse News Lett.31:27.
Grimaldi, A., D. Masiero, A. Richieri-CostaandA. Freire-Maia, notypes thought to be caused by loss ofSHHin cranial
1983 Variable expressivity of the acheiropodia gene [letter]. midline structures (see Figure 2E;Roessleret al. 1997).
Am. J. Med. Genet.16:631–634.
These data suggest that control elements required for Heus, H. C., A. Hing, M. J. van Baren, M. Joosse, G. J. Breedveld
et al., 1999 A physical and transcriptional map of the preaxial normal expression ofShh during craniofacial
develop-polydactyly locus on chromosome 7q36. Genomics57:342–351. ment may be located far from the coding regions of
Heutink, P., J. Zguricas, L. van Oosterhout, G. J. Breedveld, L. SHH(Roessleret al. 1997). However, patients with the Testerset al., 1994 The gene for triphalangeal thumb maps to the subtelomeric region of chromosome 7q. Nat. Genet.6:
T1 and T4 translocations and numerous other patients
287–292. withLMBR1deletions do not have TPTPS limb
pheno-Hing, A. V., C. Helms, R. Slaugh, A. Burgess, J. C. Wanget al., types (Roessleret al. 1997), and the locations of regula- 1995 Linkage of preaxial polydactyly type 2 to 7q36. Am. J. Med.
dur-Honig, L. S., andD. Summerbell, 1985 Maps of strength of posi- Roessler, E., D. E. Ward, K. Gaudenz, E. Belloni, S. W. Scherer
et al., 1997 Cytogenetic rearrangements involving the loss of the tional signalling activity in the developing chick wing bud. J.
Embryol. Exp. Morphol.87:163–174. Sonic Hedgehog gene at 7q36 cause holoprosencephaly. Hum. Genet.100:172–181.
Hui, C. C., andA. L. Joyner, 1993 A mouse model of greig
cephalo-polysyndactyly syndrome: the extra-toesJ mutation contains an Saunders, Jr., J. W., 1948 The proximo-distal sequence of origin of the parts of the chick wing and the role of the ectoderm. J. intragenic deletion of the Gli3 gene. Nat. Genet. 3:241–246
(erratum: Nat. Genet.19(4): 404). Exp. Zool.108:363–403.
Schimenti, J. C., B. J. Libby, R. A. Bergstrom, L. A. Wilson, D. Ianakiev, P., M. J. van Baren, M. J. Daly, S. P. Toledo, M. G.
Cavalcantiet al., 2001 Acheiropodia is caused by a genomic Nafet al., 2000 Interdigitated deletion complexes on mouse chromosome 5 induced by irradiation of embryonic stem cells. deletion in C7orf2, the human orthologue of the Lmbr1 gene.
Am. J. Hum. Genet.68:38–45. Genome Res.10:1043–1050.
Sharpe, J., L. Lettice, J. Hecksher-Sorensen, M. Fox, R. Hillet
Joyner, A. L. (Editor), 1993 Gene Targeting: A Practical Approach.
Oxford University Press, New York. al., 1999 Identification of sonic hedgehog as a candidate gene responsible for the polydactylous mouse mutant Sasquatch. Curr.
Knudsen, T. B., andD. M. Kochhar, 1981 The role of
morphoge-netic cell death during abnormal limb bud outgrowth in mice Biol.9:97–100.
Sweet, H. O., 1982 Hm and Hx are not alleles. Mouse News Lett. heterozygous for the dominant mutation Hemimelic extra-toe
(Hm). J. Embryol. Exp. Morphol.65(Suppl.): 289–307. 66:66.
Takahashi, M., K. Tamura, D. Buscher, H. Masuya, S. Yonei
-Lichter, P., T. Cremer, J. Borden, L. ManuelidisandD. C. Ward,
Tamuraet al., 1998 The role of Alx-4 in the establishment 1988 Delineation of individual human chromosomes in
meta-of anteroposterior polarity during vertebrate limb development. phase and interphase cells by in situ suppression hybridization
Development125:4417–4425 (erratum: Development125(23): using recombinant DNA libraries. Hum. Genet.80:224–234.
preceding 4595).
Marigo, V., D. J. Roberts, S. M. K. Lee, O. Tsukurov, T. Leviet
Temtamy, S., andV. McKusick, 1978 The Genetics of Hand Malforma-al., 1995 Cloning, expression, and chromosomal location of
tions. A. R. Liss, New York. SHH and IHH: two human homologues of the Drosophila
seg-Toledo, S. P., andP. H. Saldanha, 1969 A radiological and genetic ment polarity gene hedgehog. Genomics28:44–51.
investigation of acheiropody in a kindred including six cases. J.
Martin, G. R., M. Richman, S. Reinsch, J. H. NadeauandA. Joyner,
Genet. Hum.17:81–94. 1990 Mapping of the two mouse engrailed-like genes: close
Toledo, S. P., P. H. Saldanha, A. BorelliandA. B. Cintra, 1972 linkage of En-1 to dominant hemimelia (Dh) on chromosome
Further data on acheiropody. J. Genet. Hum.20:253–258. 1 and of En-2 to hemimelic extra-toes (Hx) on chromosome 5.
Tsukurov, O., A. Boehmer, J. Flynn, J. P. Nicolai, B. C. Hamelet
Genomics6:302–308.
al., 1994 A complex bilateral polysyndactyly disease locus maps
Masuya, H., T. Sagai, S. Wakana, K. MoriwakiandT. Shiroishi,
to chromosome 7q36. Nat. Genet.6:282–286. 1995 A duplicated zone of polarizing activity in polydactylous
Vargas, F. R., R. L. Pontes, J. C. Llerena Juniorand J. C. de
mouse mutants. Genes Dev.9:1645–1653.
Almeida, 1995 Absent tibiae—olydactyly—triphalangeal thumbs
Pagan, S. M., M. A. Ros, C. TabinandJ. F. Fallon, 1996 Surgical
with fibular dimelia: variable expression of the Werner (McKusick removal of limb bud Sonic hedgehog results in posterior skeletal
188770) syndrome? Am. J. Med. Genet.55:261–264. defects. Dev. Biol.180:35–40.
Vargas, F. R., E. Roessler, K. Gaudenz, E. Belloni, A. S. Whitehead Pinkel, D., T. StraumeandJ. W. Gray, 1986 Cytogenetic analysis
et al., 1998 Analysis of the human Sonic Hedgehog coding and using quantitative, high-sensitivity, fluorescence hybridization.
promoter regions in sacral agenesis, triphalangeal thumb, and Proc. Natl. Acad. Sci. USA83:2934–2938.
mirror polydactyly. Hum. Genet.102:387–392.
Qu, S., S. C. Tucker, J. S. Ehrlich, J. M. Levorse, L. A. Flaherty Vortkamp, A., M. GesslerandK. H. Grzeschik, 1991 GLI3
zinc-et al., 1998 Mutations in mouse Aristaless-like4 cause Strong’s
finger gene interrupted by translocations in Greig syndrome fami-luxoid polydactyly. Development125:2711–2721. lies. Nature352:539–540.
Radhakrishna, U., J. L. Blouin, J. V. Solanki, G. M. Dhorianiand Zakeri, Z., D. QuaglinoandH. S. Ahuja, 1994 Apoptotic cell death
S. E. Antonarakis, 1996 An autosomal dominant triphalangeal in the mouse limb and its suppression in the hammertoe mutant. thumb: polysyndactyly syndrome with variable expression in a Dev. Biol.165:294–297.
large Indian family maps to 7q36. Am. J. Med. Genet.66:209–215. Zguricas, J., H. Heus, E. Morales-Peralta, G. Breedveld, B. Kuyt
Riddle, R. D., R. L. Johnson, E. LauferandC. Tabin, 1993 Sonic et al., 1999 Clinical and genetic studies on 12 preaxial polydac-hedgehog mediates the polarizing activity of the ZPA. Cell75: tyly families and refinement of the localisation of the gene
respon-1401–1416. sible to a 1.9 cM region on chromosome 7q36. J. Med. Genet.
Robert, B., X. Montagutelli, D. Houzelstein, L. Ferland, A. 36:32–40. Cohenet al., 1994 Msx1 is close but not allelic to either Hm