Part 1
Asymmetric Iminophosphorane-catalyzed Reductive Coupling Reactions Between Benzylidene Pyruvates and Aldehydes
Coauthors: Matthew A. Horwitz and Jeffrey S. Johnson Abstract
A three-component reductive coupling reaction between dimethyl phosphite, benzylidene pyruvates, and aldehydes has been achieved via a chiral organocatalytic iminophosphorane. These mild catalytic conditions provide a new strategy for the diastereoselective and
enantioselective construction of vicinal polyfunctionalized stereocenters in high yield. Introduction
It is advantageous to have robust methods for both consonant (via normal reactivity) and dissonant (via umpolung reactivity)
disconnections in synthesis.1,2 The pinacol-type reductive coupling reaction is a subset of umpolung chemistry capable of
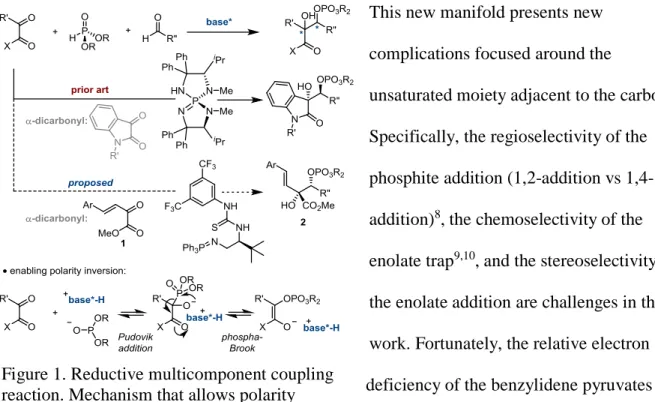
delivering vicinal diols.3 This motif is especially important in natural products (Figure 1).4 While the pinacol reaction has been extensively studied, several drawbacks
remain. Among the disadvantages are the use of stoichiometric amounts of low-valent metals and difficulties encountered in stereochemical control.4,5 We propose that many of these difficulties arise from the one-electron mechanism of the reaction. In order to address these issues, our laboratory recently developed a protocol that expanded the reaction into a two-electron manifold. The previous work relied on a base-catalyzed Pudovik addition of a dialkylphosphite to an isatin to trigger a phospha-Brook rearrangement. This rearrangement was vital for allowing polarity
inversion at the carbonyl carbon. Subsequent catalyst controlled trapping of the enolate with an aldehyde produced vicinal stereocenters (Figure 2).6 In this two-electron approach,
dialkylphosphite served as reductant. Inspired by this strategy and in collaboration with Matthew Horwitz and Blane Zavesky, we were able to apply and evaluate the reaction framework to benzylidene pyruvates.7
This new manifold presents new complications focused around the
unsaturated moiety adjacent to the carbonyl. Specifically, the regioselectivity of the phosphite addition (1,2-addition vs 1,4-addition)8, the chemoselectivity of the enolate trap9,10, and the stereoselectivity of the enolate addition are challenges in this work. Fortunately, the relative electron deficiency of the benzylidene pyruvates made the chemo-, stereo-, and regioselectivity issues manageable. The synthetic utility of accessing a phosphonate adjacent to complex functionality becomes clear when analyzing leustroducsin.
Results and Discussion
entry T (°C) Catalyst d.r. e.r. 2a:3a
1a 0 KOtBu 1.2:1 50:50 100:0
2b –60 C1 13:1 91:9 6:1
3b –60 C2 17:1 97:3 >20:1 Figure 1. Reductive multicomponent coupling
Figure 3. aReaction was conducted on 1.0 mmol scale, using 1.1 equiv. of dimethylphosphite and
5.0 equiv. of ArCHO; reaction was com-plete in minutes. bReactions were conducted on 0.2 mmol scale, using 1.1 equiv. of dimethylphosphite and 5.0 equiv. of ArCHO. Reactions were run for 24h.
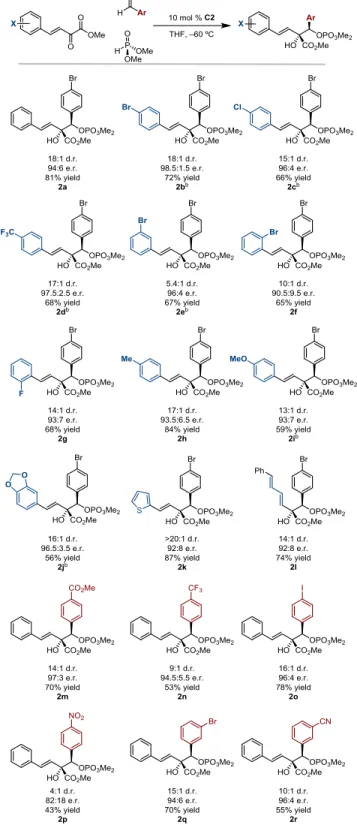
Initially, the dimethyl phosphite mediated reductive coupling with para-bromobenzaldehyde using 10 mol % KOtBu at 0 °C was studied. The reaction exclusively afforded hydroxy phosphate 2a (d.r. 1.2:1) (Figure 3). This early trial highlighted the possibility of obtaining the correct
chemoselectivity in phosphite addition and enolate trapping which led us to focus our efforts on the enantioselective variant.
Preliminary trials with cinchona alkaloid-derived thiourea catalysts proved not basic enough to permit the reaction to
proceed. We found adequate reactivity with the use of chiral tri-aryliminophosphoranes (C1 and C2) recently developed by Dixon and coworkers.11 The use of chiral
triaryliminophosphorane C1 revealed that after 48 h at 60 ºC, the starting material was completely consumed and a 6:1 ratio of
products was obtained arising from Figure 4.
aldehyde trapping (2a) relative to proton trapping (3a), the former with a diastereomer ratio of 13:1. Although product distribution was poor, this result led us to synthesize and evaluate catalyst C2, which gave a >20:1 ratio of 2a:3a, with >20:1 d.r. and 97:3 e.r. (Figure 3).With an adequate catalyst system, a broader range of reaction partners was undertaken (Figure 4). In particular, I helped with the synthesis of substrates and synthesis of racemic products for HPLC analysis and acquisition of characterization data that was required.
While electron deficient benzylidene pyruvates produced adequate yields and
stereoselectivities (2b-2d), we found that using a meta-bromo benzylidene pyruvate gave only 5.4:1 d.r. (2e). Additionally, while substrates with meta and para electron-withdrawing groups gave upward of 96:4 e.r., we observed enantioselectivities of 90.5:9.5 for 2f and 93:7 for 2g (o-bromo and o-fluoro, respectively). The use of electron rich benzylidene pyruvates (2i-2j) revealed the need for longer reaction times. Likely, the rate of Pudovik addition is slowed down by the strong electron-donating groups on the ring. Using the 2-thienylidene pyruvate gave 2k in >20:1 d.r., with 87% yield and 92:8 e.r., but extending the conjugation of the starting material as in 2l gave 14:1 d.r. and 92:8 e.r., with a 74% yield. In assessing the scope of aryl aldehydes, we found that the use electron deficient aryl aldehydes (2m−2r), either in the para or meta position, worked well. The only exception was para-nitrobenzaldehyde, where the d.r. drastically dropped (2p).
Conclusions
In summary, we have developed a highly chemoselective and stereoselective two electron multicomponent coupling reaction between benzylidene pyruvates and aldehydes. This
reaction provides a way for differentiating the alcohols that would normally be furnished by one-electron pinacol coupling by giving one phosphate bound oxygen and one free alcohol.
Experimental Details
General procedure for three-component reaction using chiral iminophosphorane: A test tube was charged sequentially with dimethyl phosphite (0.11 mmol, 1.1 equiv), aldehyde (0.5 mmol, 5.0 equiv), and –keto ester (0.1 mmol, 1.0 equiv), followed by THF (1.0 mL), which was used to
wash the residual solids and liquids on the sides of the test tube to the bottom. The reaction was stirred at -60 ºC in a cryogenic cooling apparatus for 30 minutes, then the iminophosphorane catalyst was added. The reaction was then stirred at -60 ºC and monitored by TLC. The crude reaction mixture was then flowed through a short silica plug and flushed through with diethyl ether, then concentrated in vacuo. The crude materials thus obtained were purified using flash column chromatography, with a gradient from 60:40 hexanes/EtOAc to 40:60 hexanes/EtOAc. References
1. Seebach, D. Angew. Chemie, Int. Ed. 1979, 18 (4), 239.
2. Shen, B.; Makley, D. M.; Johnston, J. N. Nature 2010, 465 (7301), 1027. 3. Chatterjee, A.; Joshi, N. N. Tetrahedron 2006, 62 (52), 12137.
4. Meyer, C.; Cossy, J. Tetrahedron 2010, 66 (33), 6358.
5. Shi, L.; Fan, C. A.; Tu, Y. Q.; Wang, M.; Zhang, F. M. Tetrahedron 2004, 60 (12), 2851. 6. Horwitz, M.; Tanaka, N.; Yokosaka, T.; Uraguchi, D.; Johnson, J. S.; Ooi, T. Chem. Sci.
2015, 6 (11), 6086.
7. Li, G.; Wang, L.; Yao, Z.; Xu, F. Tetrahedron Asymmetry 2014, 25 (13-14), 989.
8. Horwitz, M. A.; Zavesky, B. P.; Martinez-Alvarado, J. I.; Johnson, J. S. Org. Lett. 2016, 18 (1), 36.
9. Wang, S. R.; Radosevich, A. T. Org. Lett. 2015, 17 (15), 3810.
10. Hayashi, M.; Nakamura, S. Angew. Chemie - Int. Ed. 2011, 50 (10), 2249.
Part 2
The Importance of Field Strength in the Low Field Portion of a Differential Ion Mobility Spectrometry Waveform Coauthors: Brandon G. Santiago and Gary L. Glish
Introduction
Differential ion mobility spectrometry (DIMS) is a gas-phase separation technique that can be coupled to mass spectrometry to improve signal-to-noise ratios. DIMS separations use the dependence of ion mobility on electric field strength to separate ions.1 Through application of an asymmetric waveform alternating between “high” and “low” electric field strengths, the
difference between high and low field mobilities is sampled. Addition of solvent vapors to the DIMS carrier gas has been shown to alter the differential ion mobility of ions and enhance the selectivity of DIMS separations.2 The primary explanation in the literature to explain solvent effects is the dynamic cluster-decluster model. This model rationalizes the change in differential ion mobility, and thus the compensation field required to pass the ion through DIMS, by stating that an ion travels in a solvated state during a low field portion of the separation waveform and then undergoes rf heating during the high field portion of the waveform, causing it to travel as a bare ion.1,3 Herein, we investigate the dynamic cluster-decluster model by varying the amount of time spent in the low field portion of the DIMS waveform.
Experimental Methods
Experiments were performed using a Bruker HCT Quadrupole Ion Trap mass
to 5.0 L/min and either 200 or 300° C in the instrument control software. Solvent vapor from LC-MS grade methanol was introduced at a rate of 0.03 mL/min to the desolvation gas via a Swagelok tee connected to the output of a Hitachi LC 2100 pump. Electrospray ionization was used to ionize 0.001% triethylamine in 90/10 MeOH/EtOH at a flow rate of 2 μL/min. Results and Discussion
The dynamic cluster-decluster model is predicated around the idea that the ion travels in a solvated state during the low field portion of the separation waveform, and after undergoing rf heating travels as a bare ion during the high field portion of the waveform. In DIMS literature, “low” field is typically described as fields <10 kV/cm in strength (although some claim this value to be as low as 2.5 kV/cm).1,4 Due of the power requirements of using a rectangular waveform at the required voltages for DIMS, other alternatives are typically used. One such alternative is a bisinusoidal waveform, generated by coupling two sinusoidal waveforms. A drawback of using this type of waveform is that as the amplitude of the high field portion of the waveform is raised, the absolute amplitude of the low field portion of the waveform is also increased. It was observed that at field strengths where the “low” field portion of the waveform was >10 kV/cm the addition of methanol vapor still affected the differential ion mobility of various analyte ions.
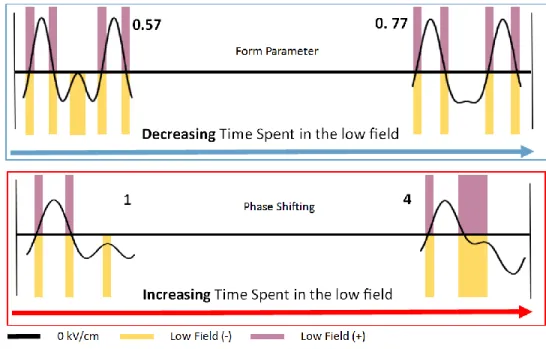
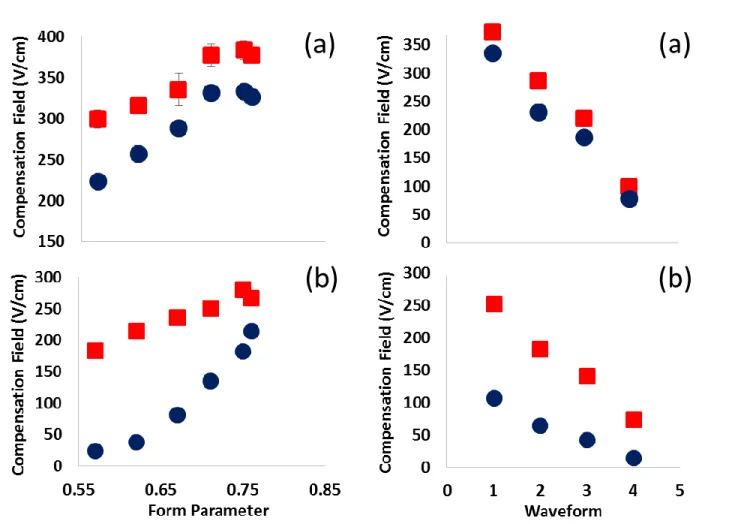
To further examine this previous observation, the form parameter of the bisinusoidal waveform was varied, with example waveforms shown in Figure 1.5 The form parameter is defined as the amplitude of the first harmonic divided by the summed amplitude of the two harmonics, with greater form parameter yielding less time spent in the low field. At a desolvation gas setting of 300 °C, it can be observed in Figure 2 that methanol changes the required
form parameter of 0.7 yields no net time in the low field at a dispersion field of 37.3 kV/cm, yet it is apparent that the differential ion mobility has been changed based on the shift in required compensation field. This shift is also observed at a desolvation gas setting of 200 °C (Figure 2). Additionally, Figure 2 shows that at both temperature settings form parameters generating greater time in the low field yield greater changes in the required compensation field. As expected, this effect is more prominent at 200 °C where the lower effective temperature of the ions makes clustering more favorable.
Further manipulation of the time spent in the low field was accomplished through varying the phase shift of the two sinusoidal waveforms relative to each other.4 Figure 1 depicts the changes to the waveform shape upon varying the phase shift and how time in the low field was increased. Although drawing conclusions from how the phase shift affected the required
compensation field is beyond the scope of this work, it can be observed that at both temperatures and all phase shifts the use of methanol led to a change in required compensation field. As with form parameter, a greater change in required compensation field was observed at a temperature setting of 200 °C, presumably due to the more favorable clustering kinetics present at lower ion effective temperatures.
Summary and Conclusions
The form parameter and phase shift of a bisinusoidal waveform used for DIMS
the low field showed greater changes in required compensation field. It was also shown that lower temperatures increased the shift in required compensation field caused by dopant vapors, a change that could be explained by more favorable ion kinetics for clustering at lower
temperatures. These results suggest another factor beyond the dynamic cluster-decluster theory plays a role in how solvent vapors affect differential ion mobility.
Appendix
References
1. Kolakowski, B. M.; Mester, Z. Analyst 2007, 132, 842.
2. Rorrer, L. C.; Yost, R. a. Int. J. Mass Spectrom. 2011, 300 (2-3), 173.
3. Kafle, A.; Coy, S. L.; Wong, B. M.; Fornace, A. J.; Glick, J. J.; Vouros, P. J. Am. Soc. Mass Spectrom. 2014, 25 (7), 1098.
4. Santiago, B. G.; Harris, R. a; Isenberg, S. L.; Glish, G. L. Analyst 2015, 140 (20), 6871. 5. Krylov, E. V.; Coy, S. L.; Vandermey, J.; Schneider, B. B.; Covey, T. R.; Nazarov, E. G.
Rev. Sci. Instrum. 2010, 81 (2).