Epigenetic reprogramming of immune cells in injury, repair, and resolution
Full text
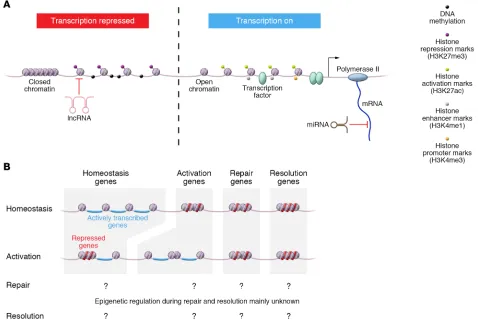
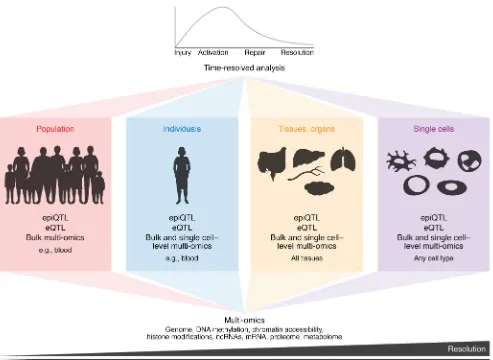
Figure

Related documents
Based on previous genetic studies, the results of this study demonstrate that increasing parental age, and in particular, increasing paternal age, in individuals diagnosed with
Indeed, we have defined the Lulu movements, fighting against mafia and corruption as territorial movements carrying on ‘anti-system’ struggles, because, while contrasting
In its present form Rule 11-9 (c) helps them in that regard. The 12-month-long certification period actually affords them somewhat of a safety net. It permits them the opportunity
Based on Sparse representation.To construct a base matrix whose columns represent features of fingerprint images ,referring the matrix dictionary whose columns are
• Out of more than 1300 procedures of adjuvant epinephrine in digital nerve block of the toes reviewed for this study, no cases were complicated with postoperative digital ischemia
According to general relativity (GR) the photon move in a curved trajectory in a gravitational field, although the magnitude of photon speed
From the above empirical evidence and discussion this paper concludes that there is no relationship between earnings management and aggregate stock market return, summing up all
